

Abstract
Anticholinergics are widely used for the treatment of COPD, and to a lesser extent for asthma. Primarily used as bronchodilators, they reverse the action of vagally derived acetylcholine on airway smooth muscle contraction. Recent novel studies suggest that the effects of anticholinergics likely extend far beyond inducing bronchodilation, as the novel anticholinergic drug tiotropium bromide can effectively inhibit accelerated decline of lung function in COPD patients. Vagal tone is increased in airway inflammation associated with asthma and COPD; this results from exaggerated acetylcholine release and enhanced expression of downstream signaling components in airway smooth muscle. Vagally derived acetylcholine also regulates mucus production in the airways. A number of recent research papers also indicate that acetylcholine, acting through muscarinic receptors, may in part regulate pathological changes associated with airway remodeling. Muscarinic receptor signalling regulates airway smooth muscle thickening and differentiation, both in vitro and in vivo. Furthermore, acetylcholine and its synthesizing enzyme, choline acetyl transferase (ChAT), are ubiquitously expressed throughout the airways. Most notably epithelial cells and inflammatory cells generate acetylcholine, and express functional muscarinic receptors. Interestingly, recent work indicates the expression and function of muscarinic receptors on neutrophils is increased in COPD. Considering the potential broad role for endogenous acetylcholine in airway biology, this review summarizes established and novel aspects of muscarinic receptor signaling in relation to the pathophysiology and treatment of asthma and COPD.
Introduction
Acetylcholine is the primary parasympathetic neurotransmitter in the airways, and is traditionally associated with inducing airway smooth muscle contraction and mucus secretion. Parasympathetic activity is increased in airway inflammation, which is the basis for the use of anticholinergic therapy in asthma and chronic obstructive pulmonary disease (COPD) [1]. Anticholinergics constitute a particularly important bronchodilator therapy in COPD, as vagal tone appears to be the only reversible component of airflow limitation in this condition [1]. Recent evidence indicates that acetylcholine production in the airways is not restricted to the parasympathetic nervous system: acetylcholine is also released from non-neuronal origins such as the bronchial epithelium and inflammatory cells [2]. Furthermore, accumulating evidence suggests acetylcholine (either neuronal or non-neuronal) may play an essential regulatory role in the mechanisms that drive the structural changes in the airways, called airway remodeling, that are associated with chronic airway inflammation [3,4]. These recent findings indicate that acetylcholine, acting on muscarinic receptors, may contribute to the pathophysiology and pathogenesis of asthma and COPD to a much larger extent than is currently appreciated. This concept is underscored by findings that the recently introduced long-acting anticholinergic agent, tiotropium bromide [5], markedly inhibits accelerated lung function decline in COPD patients [6]. This article will review the established and novel muscarinic receptor signaling mechanisms in airway physiology, and discuss their involvement in the pathophysiology of asthma and COPD. Though nicotinic cholinergic receptors are present throughout the airways (see [7] for review), their function will not be discussed in view of the muscarinic receptor specificity of the current clinically used anticholinergics.
Muscarinic receptor regulation of airway smooth muscle tone
Airway smooth muscle expresses abundant muscarinic M2 and M3 receptors, roughly in a 4:1 ratio [8]. Despite its lower expression levels, the Gq coupled muscarinic M3 receptor is the primary subtype responsible for bronchial and tracheal smooth muscle contraction; this is evident from the functional affinities of a variety of subtype selective antagonists in airway tissues from diverse species, including humans [8-11] (Table 1). In addition, muscarinic M3 receptor -, but not M2 receptor-knockout mice lack both methacholine and vagally induced bronchoconstriction in vivo [12]. Nonetheless, some pharmacological studies have suggested a small role for Gi – coupled M2 receptors in mediating airway smooth muscle contraction in the peripheral airways [13,14]. Muscarinic receptor regulation of airway smooth muscle tone is enhanced in asthma and COPD by two major mechanisms: first, increased expression and enhanced function of signaling molecules essential for muscarinic receptor mediated airway smooth muscle contraction; and second, exaggerated release of neuronal acetylcholine due to neuronal mechanisms associated with inflammation.
Table 1.
Affinity profiles of selective and nonselective muscarinic receptor antagonists for muscarinic M2 and M3 receptors.
| M2* | M3# | ||
| 4-DAMP | 7.8 | 9.0 | [8, 11] |
| AQ-RA 741 | 8.3 | 7.5–6.6 | [135] |
| Gallamine | 6.5 | 4.3 | [8] |
| DAU 5884 | 6.6 | 8.7 | [14] |
| Methoctramine | 7.5 | 6.5 | [8] |
| Pirenzepine | 6.2 | 6.8 | [8, 11] |
| AF-DX 116 | 7.0 | 5.6–6.3 | [8, 11] |
| Ipratropium | 9.7† | 9.7† | [5] |
| Tiotropium | 10.7† | 11.0† | [5] |
* Data represent binding affinities (pKi) for cardiac muscarinic M2 receptors
# Data represent functional affinities (pA2) to methacholine-induced contraction of tracheal and bronchial preparations.
† Data represent binding affinities (pKi) to cloned human muscarinic receptor subtypes.
Intracellular signaling in airway smooth muscle
Muscarinic receptors induce airway smooth muscle contraction through a number of intracellular signaling mechanisms; most of these are well described and have been reviewed extensively [4,15,16]. These include a number of recently identified cascades that are of specific interest to airway inflammation in asthma and COPD (Figure 1). Several researchers have postulated that enhanced Ca2+ signaling underpins the genesis of obstructive airways diseases that are associated with airway hyperreactivity [17,18]. In part, this is based on observations from animal models, for example airway smooth muscle cells obtained from hyperresponsive Fisher rats show elevated Ca2+ responses when compared to the less responsive Lewis rats [19]. Several studies have also shown that isolated airway smooth muscle preparations from asthma and COPD patients respond with increased maximal force generation to contractile stimulation in vitro [20-24]. Thus, intrinsic abnormalities that contribute to cholinergic hyperreactivity may exist in at least a proportion of asthmatics and COPD patients.
Figure 1.
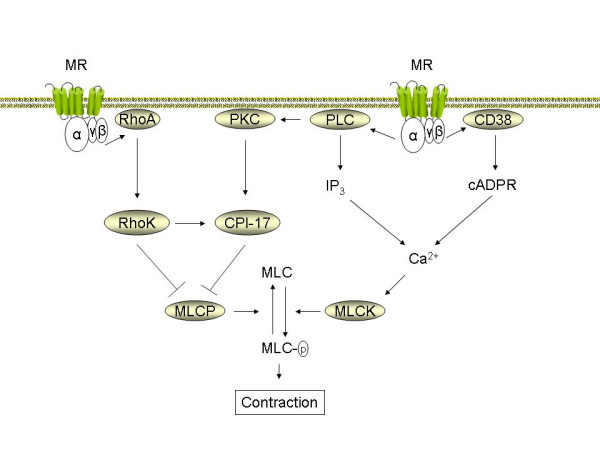
Pathways central in muscarinic receptor mediated airway smooth muscle contraction. Muscarinic receptor (MR) agonists induce contraction of airway smooth muscle by Ca2+ dependent and Ca2+ independent pathways. Through associated Gq alpha subunits, the muscarinic M3 receptor activates phospholipase C (PLC), which releases inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) after hydrolytic conversion of phosphatidylinositol-4,5-bisphosphate (PIP2). IP3 induces the release of Ca2+ from internal sarcoplasmatic reticulum (SR) stores. Coupling of M3 receptor to CD38 through as yet undefined mechanisms contributes to the production of cyclic ADP ribose (cADPR) and the release of Ca2+ through ryanodine receptor channels in the SR. Ca2+ release increases free cytosolic Ca2+ and promotes calmodulin-dependent activation of myosin light chain kinase (MLCK). MLCK mediated phosphorylation of 20 kDa regulatory myosin light chain (MLC) in the contractile apparatus is an obligatory event to induce smooth muscle contraction. MLC phosphorylation level is also controlled by pathways that inhibit myosin light chain phosphatase (MLCP) and, thus enhance Ca2+ sensitivity. PLC-derived DAG activates protein kinase C (PKC), leading to CPI-17 phosphorylation and downstream MLCP inhibition. Rho-kinase, which is activated by the monomeric G protein RhoA, both phosphorylates CPI-17 and inhibits MLCP directly. The expression and function of RhoA, CPI-17 and CD38 are increased by pro-inflammatory cytokines in vitro and in animal models of asthma and COPD ex vivo (see text).
Although the altered expression of the postjunctional muscarinic M3 receptor on airway smooth muscle cells is not a feature of airway hyperreactivity to inhaled methacholine, changes in downstream signaling from these receptors may be a contributing factor [16]. In addition to activating phospholipase Cβ1 (PLC), which leads to inositol 1,4,5-trisphosphate (IP3) production necessary for triggering release of intracellular Ca2+ stores, muscarinic receptors also regulate signaling pathways involving CD38, cyclic ADP ribose (cADPR) and ryanodine receptor channels that can play an important role in airway smooth muscle Ca2+ homeostasis [25] (Figure 1). The CD38/cADPR pathway contributes significantly to muscarinic receptor mediated changes in lung compliance and resistance, as evident in CD38 knockout mice [26]. Initial studies suggest that this pathway may be activated selectively by muscarinic M2 receptors [27], though other studies suggest that muscarinic M3, rather than M2 receptors are coupled to cADPR production [28]. Several pro-inflammatory cytokines, including IL-1β [29], IL-13 [30], TNF-α [31] and IFN-γ [32] can increase CD38 expression, ADP-ribosyl cyclase activity, and Ca2+ responses to cholinergic agonists in airway smooth muscle. TNF-α and IL-1β also increase Gi and Gq alpha protein expression in airway smooth muscle, which could account for increased Ca2+ responses and contraction [33,34]. Furthermore, treatment of airway smooth muscle strips with IL-13 or TNF-α for extended periods, induces hyperresponsiveness to cholinergic agonists [35,36].
Contraction of airway smooth muscle is regulated by Ca2+ dependent and Ca2+ independent mechanisms. Ca2+ independent mechanisms are characterized by augmented contraction at a fixed Ca2+ concentration; this phenomenon is referred to as Ca2+ sensitization [37]. Regulation of Ca2+ sensitivity by cholinergic agonists is an important step in airway smooth muscle contraction (Figure 1). The RhoA/Rho-kinase cascade, a key regulatory pathway of Ca2+ sensitivity in airway smooth muscle, can be activated by both muscarinic M2 and muscarinic M3 receptors [38,39]. RhoA and Rho-kinase augment agonist-induced contraction primarily by inactivating myosin light chain phosphatase (MLCP), although direct effects on myosin light chain phosphorylation and on actin cytoskeletal dynamics have also been described [40]. MLCP is inhibited by the direct phosphorylation of its regulatory myosin binding subunit by Rho kinase. MLCP is also inhibited by binding to the phosphoprotein CPI-17, which is targeted for phosphorylation by both Rho kinase and PKC (see [37] for detailed review on the role of Rho-kinase in airway hyperresponsiveness). The anti-spasmogenic effects of Rho-kinase inhibition are distinctly smaller than their relaxant effects on a pre-established cholinergic contraction, indicating that the RhoA/Rho-kinase pathway is particularly important in maintaining a sustained contraction to cholinergic agonists [41-43].
In experimental models of inflammatory airway disease, muscarinic receptor-linked signaling pathways that regulate Ca2+ sensitivity of airway smooth muscle cells appear to be enhanced. Both RhoA and CPI-17 expression are increased in rats exposed to repeated allergen challenge [44,45]. Furthermore, allergic sensitization by itself, without subsequent allergen exposure, appears to be sufficient to induce an increase in RhoA expression [46]. Cytokines, including TNFα, have been identified as contributors to increased RhoA abundance [47]. In line with these observations, cholinergic agonist-induced RhoA translocation to the membrane, RhoA-mediated Ca2+ sensitization, and contraction are increased in bronchial smooth muscle from rats and mice exposed to repeated allergen challenge [45,48,49]. Recent observations indicate that the same is true for cigarette smoke induced airway hyperresponsiveness in rat bronchial smooth muscle [50], which could be of significant importance to the pathophysiology of COPD. Effects of lipopolysaccharide (LPS) on cholinergic reactivity of airway smooth muscle have also been described [51]; however, it is yet to be established what mechanisms exactly mediate this change.
Neuronal mechanisms
In addition to postjunctional mechanisms that involve muscarinic receptor signaling in airway smooth muscle cells, neuronal mechanisms are important, and they also appear to be affected in inflammatory airways disease (Figure 2). Neuronal acetylcholine is synthesized by the enzyme choline acetyl transferase (ChAT), stored in vesicles, and released upon membrane depolarization. Once released, the functional effects of acetylcholine are terminated primarily by acetylcholinesterase (AChE) in the synaptic cleft. AChE activity is decreased in tracheal smooth muscle homogenates from ragweed pollen sensitized dogs [52]; this represents a mechanism to increase and prolong the action of acetylcholine on postjunctional target cells, such as airway smooth muscle cells, in allergic airways diseases. In addition, mediators of inflammation can enhance the release of acetylcholine from vagal nerve endings, an effect mediated through prejunctional facilitatory receptors. Examples include tachykinins, prostaglandins and thromboxane A2 [53]. Furthermore, the autoinhibitory prejunctional muscarinic M2 receptor, that limits acetylcholine release under normal conditions (Figure 2), is dysfunctional in several experimental models of airways disease including allergen exposure, viral infection, and ozone exposure [54,55]. M2 autoreceptors have also been reported to be dysfunctional in some, but not all asthmatics [56,57]. In addition, asthmatics with active viral infections show greater bronchodilator responses to inhaled anticholinergics, suggesting an increased vagal tone [58]. Nonetheless, in patients with stable COPD the M2 autoreceptor appears to function normally [59]. Distinct mechanisms underlie the M2 autoreceptor dysfunction. In guinea pigs, ozone and allergen-induced M2 dysfunction is mediated by eosinophils that are recruited to airway nerves, and secrete major basic protein, that acts as an allosteric muscarinic M2 receptor antagonist [55]. Viral infections, which may play a role in both asthma and COPD, induce M2 dysfunction through neuraminidases that cleave portions of the M2 receptor, and through as yet incompletely characterized mechanisms involving macrophages, CD8+ lymphocytes, and possibly IFN-γ [60].
Figure 2.
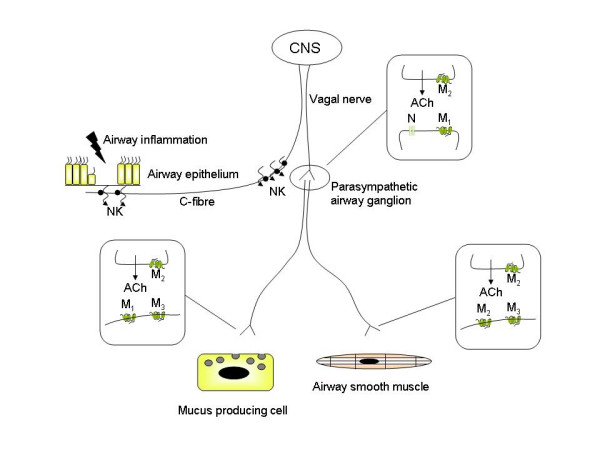
Cholinergic receptors involved in neuronal acetylcholine release and function. Neuronal acetylcholine release is regulated by a network of afferent and efferent airway nerves that interact with their surrounding cells. Afferent C-fibers project to the subepithelial region where they can be activated by inflammatory mediators and non-specific stimuli. In asthma, epithelial damage can expose sensory nerve endings to the airway lumen, potentiating their activation. Activated C-fibers secrete neurokinins (NK) that exert local effects and facilitate ganglionic neurotransmission (peripheral reflex arc). In addition, the activated C-fiber increases the output of the vagal nerve through regulation in the central nervous system (CNS) (central reflex arc). Neurotransmission in parasympathetic ganglia of the airway is mediated by acetylcholine through nicotinic (N) and muscarinic M1 receptors and can be markedly facilitated by inflammatory mediators (see text). Presynaptic muscarinic M2 autoreceptors inhibit acetylcholine release and are dysfunctional in airway inflammation. The postganglionic neurons project primarily to mucus producing cells and airway smooth muscle, where neurotransmission is regulated by muscarinic M1, M2 and M3 receptors, as indicated. As in the ganglia, prejunctional acetylcholine release is autoinhibited by muscarinic M2 receptors that are dysfunctional in airway inflammation. Acetylcholine release is augmented further by direct effects of inflammatory mediators on facilitatory presynaptic receptors. See the text for further detail.
Cholinergic neurotransmission in the parasympathetic ganglia is regulated by nicotinic receptors in conjunction with muscarinic M1 receptors, whereas ganglionic release of acetylcholine from preganglionic nerves is under regulation of M2 autoreceptors [61,62] (Figure 2). Though the M2 autoreceptor can be dysfunctional in allergic airway inflammation, as described above, currently no evidence suggests that ganglionic muscarinic M1 receptor expression is altered [63]. However, several inflammatory mediators facilitate ganglionic neurotransmission, including tachykinins, histamine, bradykinin and prostaglandins [64]. Airway ganglia function to filter the signals from the rapidly firing preganglionic neurons; therefore faciliation of ganglionic transmission by inflammatory mediators is likely of significance in the regulation of airway tone [53].
Most of the afferent nerve fibres in the airways are C-fibers, which are present throughout the airways, from the larynx down to the lung parenchyma. C-fibers respond to stimuli such as heat and cold, but can also be activated by inflammatory mediators, resulting in reflex bronchoconstriction, mucus production and cough [65]. The localization of the reflex mechanism can be central and local, and may contribute considerably to the increased vagal tone in COPD, and to airway hyperreactivity in asthma and COPD [65,66]. Several inflammatory mediators, including histamine, prostanoids, thromboxane A2, bradykinin, serotonin and tachykinins are known to stimulate sensory nerve fibres [67]. Afferent sensory nerve endings project to the subepithelial layer in healthy airways, but may be exposed to the airway lumen upon the induction of epithelial damage by mediators such as eosinophil-derived major basic protein [68]. This is considered an important mechanism in the regulation of vagally mediated airway hyperrresponsiveness.
Muscarinic receptor regulation of mucus hypersecretion
The production of airway mucus in the central airways is under cholinergic control, and plays an important role in asthma and COPD [69,70]. Airway mucus is a protective film that serves to prevent inhaled particles from damaging the airway epithelium. It is composed of electrolytes, water and contains high amounts of mucins [69]. Mucins are glycoproteins that are responsible for the high viscosity of mucus; the primary mucins found in airway mucus are of the MUC5AC and MUC5B isoforms. Mucus secreting cells in the central airways include goblet cells, which are embedded in the epithelium, and submucosal glands that are in connection to the airway lumen. Acetylcholine is the dominant neurotransmiter involved in mucus secretion in the central airways [71] (Figure 2). Thus, electrical field stimulation increases mucus production in bronchial preparations, which is sensitive to atropine and tetrodotoxin [72]. Airway submucosal glands are likely the primary source of this vagally regulated mucus production. Submucosal glands are innervated, and express functional muscarinic M1 and M3 receptors, roughly in a 1:2 ratio [73,74]. The muscarinic M3 receptor is the predominant receptor that mediates mucus secretion, whereas electrolyte and water secretion are probably mediated by muscarinic M3 receptors in cooperation with M1 receptors [72,75]. Goblet cells can also produce mucus in response to muscarinic receptor stimulation, albeit at relatively high concentrations of agonist [71].
Mucus hypersecretion is a pathological feature seen in both asthma and COPD that contributes significantly to airflow limitation by obstructing the airways [76]. The composition of mucus in asthma and COPD is generally altered, with higher expression of the low charge isoform MUC5B, particularly in COPD, and with the expression of small amounts of the insoluble MUC2; in COPD the ratio of mucus cells to serous cells in the submucosal glands is also increased [69]. Since mucus production in the central airways is, to a large extent, vagally mediated, acute airway inflammation can regulate mucus hypersecretion by augmenting acetylcholine release in the same way as described in the previous section. In addition, cholinergic receptor stimulation interacts synergistically with epidermal growth factor (EGF) on mucus cell activation in airway submucosal glands [77]. Since EGF is thought to regulate goblet cell hyperplasia and mucus gland hypertrophy in asthma and COPD [78,79], this may have additional implications for the effects of muscarinic receptors on this pathology. Indeed, muscarinic receptor stimulation transactivates the EGF receptor in conjunctival goblet cells, which is involved in mucin production by these cells [80]. In addition, based on observations in experimental animal models, repeated administration of the muscarinic agonists pilocarpine and methacholine can promote goblet cell hyperplasia and mucus gland hypertrophy [81]. This raises the real possibility that excessive release of endogenous acetylcholine could promote remodeling of mucus secreting cells in asthma and COPD, but this still needs to be assessed in human subjects.
Muscarinic receptor regulation of airway inflammation
Traditionally, acetylcholine is not considered to regulate airway inflammation. Recruited inflammatory cells distribute throughout the lung, and are not primarily localized to vagal nerves. Early studies suggested that blood lymphocytes and peripheral polymorphonuclear leukocytes do not express functional muscarinic receptors [82]. This view is changing, however. Accumulating evidence demonstrates that acetylcholine and its synthesizing enzyme choline acetyltransferase (ChAT) are present not only in airway nerves, but localize to epithelial and endothelial cells, smooth muscle cells, lymphocytes, macrophages, mast cells, eosinophils and neutrophils as well [2,62]. Furthermore, rigorous investigation has now revealed that most inflammatory cells express functional muscarinic receptors (Table 2). These findings suggest that acetylcholine can regulate inflammatory processes by paracrine and/or autocrine mechanisms [83-86]. Notably, elevated levels of acetylcholine have been noted in skin biopsies from patients with atopic dermatitis, a condition often associated with bronchial asthma [87].
Table 2.
Muscarinic regulation of (airway) inflammation.
| Cell type | Presence of muscarinic receptors, ChAT and/or acetylcholine | Functional effects of acetylcholine | |
| T lymphocyte | Muscarinic receptors (M1-M5 *) ChAT Acetylcholine |
Increased cytotoxicity Cytokine production Proliferation |
[2, 83, 92] |
| B lymphocyte | Muscarinic receptors (M1-M5 *) ChAT Acetylcholine |
Proliferation | [2, 83] |
| Mast cell | Muscarinic receptors (M1) ChAT Acetylcholine |
Inhibition of histamine release | [2, 136, 137] |
| Neutrophil | Muscarinic receptors (M1/M2/M3) ChAT |
Chemotaxis LTB4 production # |
[93, 138] |
| Eosinophil | Muscarinic receptors (M1) ChAT |
unknown | [93, 138] |
| Macrophage/monocyte | Muscarinic receptors (M1/M2/M3) ChAT Acetylcholine |
LTB4 production # | [2, 83, 93] |
| Bronchial epithelium | Muscarinic receptors (M1/M3) ChAT Acetylcholine |
Release of monocyte, eosinophil and neutrophil chemotactic factors | [95, 96, 98] |
| Airway smooth muscle | Muscarinic receptors (M2/M3) ChAT |
Pro-inflammatory gene expression | [2, 8, 99] |
* Further characterization is necessary: putative presence of muscarinic M1 – M5 receptors shows high variability and is based on their presence in mononuclear leukocytes [83].
# Presence of muscarinic receptors on macrophages and neutrophils suggests their involvement in LTB4 production by sputum cells [93].
Mild asthma and stable COPD show distinct patterns in the nature of airway inflammation. Stable COPD is characterized by pulmonary infiltration of neutrophils, cytotoxic (CD8+) T lymphocytes, monocytes and macrophages, whereas in mild asthma, Th2 (CD4+) lymphocytes and eosinophils show a distinct increase. The nature and extent of the airway inflammation are, however, dependent on severity of the disease, as in COPD and acute severe asthma both neutrophils and CD8+ T lymphocytes are key contributors to disease pathology [88].
There is considerable evidence that the non-neuronal cholinergic system plays a role in lymphocytes, although its relative importance to airway physiology is not yet established. Mononuclear leukocytes, consisting mainly of lymphocytes, express ChAT mRNA and protein, contain ACh and express muscarinic and nicotinic receptors. The expression profile of muscarinic receptors shows high individual variability, although all 5 classes (M1-M5) of muscarinic receptors have been detected [83]. Muscarinic receptor agonists increase cytosolic Ca2+ both in human T- and B-cell lines in an atropine-sensitive manner, and increase c-fos mRNA expression in response to the muscarinic agonist oxotremorine [89]. The latter effect is sensitive to 4-diphenylacetoxy-N-methylpiperidine (4-DAMP) methobromide, but not to pirenzepine or AF-DX 116, which is consistent with the involvement of muscarinic M3 receptors (Table 1). Furthermore, phytohemagglutinin (a T-cell activator) increased ChAT mRNA [90] and muscarinic M5 receptor expression [91] in stimulated mononuclear leukocytes. The significance of these observations is that muscarinic receptors and non-neuronal acetylcholine could contribute to lymphocyte proliferation and cytokine release, with obvious implications for airway inflammation in asthma and COPD. Furthermore, it is established that muscarinic receptors play an important role in regulating cytotoxicity of T lymphocytes [92]. Future studies are needed, however, to characterize the expression and function of the non-neuronal cholinergic system in lymphocytes that have infiltrated the lungs and in T lymphocytes that adopted a specific CD8+ or CD4+ phenotype
A recent study by Profita et al. [93] investigated the expression of muscarinic M1, M2 and M3 receptors in sputum cells obtained from healthy controls, smokers, and patients with COPD. In this study, all three subtypes of muscarinic receptors were observed in macrophages and neutrophils of all patient groups. M1 receptors were expressed in low abundance in eosinophils from COPD patients, but not from healthy controls. Importantly, the expression of muscarinic M3 receptors on macrophages is significantly increased in COPD patients, whereas muscarinic M2 receptor expression is decreased. The expression of muscarinic M1 receptors on macrophages, and the expression of M1 and M3 receptors on neutrophils tended to be increased, though this did not reach statistical significance. Functional studies showed that acetylcholine induced the release of significant amounts of leukotriene B4 and activated the p42/p44 MAP kinase pathway in sputum cells from COPD patients [93]. Neutrophil chemotactic activity induced by acetylcholine was also increased in COPD. These results are entirely consistent with a study demonstrating that bovine alveolar macrophages release eosinophil, monocyte and neutrophil chemotactic activities in response to acetylcholine, with probably a predominant involvement of leukotriene B4 [94]. These observations clearly reveal that regulated expression of muscarinic receptor subtypes is a feature of inflammatory cells that migrate to the airways, though the precise functional impact of dynamic receptor expression on these cells needs to be elucidated.
In addition to its direct effects on inflammatory cells, acetylcholine may also trigger chemokine and cytokine release from structural cells. Bronchial epithelial cells release eosinophil, monocyte and neutrophil chemotactic activity in response to acetylcholine [95,96]. Consistent with the previously mentioned findings, there appears to be an important role for leukotriene B4 in these effects. Acetylcholine is also known to induce the release of GM-CSF from human bronchial epithelial cells by a mechanism that involves nicotinic receptors [97]. Since the expression of non-neuronal acetylcholine is relatively high in bronchial epithelial cells [98], these results could implicate a role for epithelial acetylcholine in initiating inflammatory responses.
Muscarinic receptors on airway smooth muscle cells may play a profound role in regulating airway inflammation: a recent study demonstrates that the muscarinic receptor agonist carbachol increases inflammatory gene transcription in bovine tracheal smooth muscle strips [99]; quantitative RT-PCR analysis demonstrates that carbachol can modulate expression of a number of genes, including IL-8, cyclo-oxygenase (COX) 1 and 2 and urokinase type plasminogen activator (PLAU); and, carbachol markedly augments pro-inflammatory gene expression induced by sinusoidal length oscillation, with synergistic effects on IL-6, IL-8 and COX 2 and to a lesser extent PLAU and CCL-2 [99]. Collectively these studies suggest acetylcholine is an autocrine or paracrine hormone that may be involved in regulating inflammation at a number of cellular sites in the airways (Table 2). At this point, evidence is lacking however to indicate a direct involvement of non-neuronal acetylcholine in the pathophysiology of asthma and COPD and future studies are clearly warranted within this area.
Muscarinic receptor regulation of airway remodeling
Chronic inflammatory conditions of the airways are usually associated with the development of structural changes of the airways; a phenomenon commonly referred to as airway remodeling. Airway remodeling is seen in both asthma and COPD, albeit the nature, localization and extent of the remodeling are variable (Table 3). Airway remodeling is progressive, both in asthma and COPD, and the extent of structural change correlates with disease severity [100-102]. Based on these considerations, it is believed that most structural changes, e.g. increased airways smooth muscle mass and mucus gland hypertrophy, contribute to a progressive increase in disease severity over time and to the irreversible decline in lung function in patients with chronic disease. Some structural changes on the other hand, including matrix deposition in the airway wall, are not necessarily detrimental, but may actually protect the diseased airway from airway closure by increasing airway wall stiffness [103,104]. Clearly a complex relationship exists between airway structure and function. Indeed, at present there is considerable ongoing research effort using in vitro, ex vivo, and in vivo systems to clarify key structural determinants of airway and lung function in health and disease.
Table 3.
Airway remodeling in asthma and COPD. Muscarinic receptors and acetylcholine play significant roles in airway smooth muscle remodeling, and possibly in goblet cell hyperplasia and mucus gland hypertrophy. Their involvement in other aspects of airway remodeling is less well explored.
| Asthma | COPD | |||
| Airway smooth | Hyperplasia | [121, 139] | Increased ASM mass | [140] |
| muscle (ASM) | Hypertrophy | [101, 139] | Hypercontractility | [23] |
| Hypercontractility | [20-22, 24] | |||
| Mucus production | Goblet cell hyperplasia Mucus gland hypertrophy |
[141] | Goblet cell hyperplasia Mucus gland hypertrophy |
[142] |
| Vasculature | Pulmonary vascular remodeling | [143] | Pulmonary vascular remodeling | [144-146] |
| Basement membrane | Thickened | [76] | Not thickened | [76] |
| Extracellular matrix | Subepithelial collagen deposition (Myo)fibroblast accumulation |
[101, 147] | Airway wall fibrosis Loss of alveolar walls |
[76] |
Contractile agonists acting on G-protein coupled receptors are increasingly being recognized as key contributors to airway remodeling in asthma. Cysteinyl leukotrienes have received significant attention in this regard: the capacity for anti-leukotrienes to prevent allergen-induced airway inflammation, mucus production and occlusion, goblet cell hyperplasia, and most notably airway fibrosis and airway smooth muscle thickening have been described [105-109]. Acetylcholine, on the other hand, has not generally been considered to be a crucial determinant of structural changes in the airways. However, recent findings are changing this view. Indeed, there may be a prominent regulatory role for endogenous acetylcholine in promoting allergen-induced airway remodeling [4,110]. In the following sections the potential contribution of acetylcholine to specific components of airway remodelling are being discussed.
Mesenchymal cell proliferation
Muscarinic receptor stimulation induces profound proliferation of primary cultured human lung fibroblasts [111]. In addition, though stimulation of muscarinic receptors is not sufficient to induce airway smooth muscle proliferation; muscarinic receptor agonists augment responses to epidermal growth factor (EGF) and platelet-derived growth factor (PDGF), in both human and bovine airway smooth muscle [112,113]. This augmentation is considerable: the dose-response curve to PDGF is shifted both upward and leftward, indicating that muscarinic receptors increase the mitogenic response to any PDGF concentration. Moreover, muscarinic agonists can potentiate the mitogenic response of myocytes in response to low concentrations of PDGF that would otherwise be insufficient to stimulate cell growth. The inhibitory profile of a range of subtype-selective antagonists (4-DAMP and DAU5884, but not gallamine) demonstrates the exclusive involvement of muscarinic M3 receptors in this effect [113] (Table 1). The importance of this potentiating effect is demonstrated in vivo: repeated exposures of sensitized guinea pigs to ovalbumin increases airway smooth muscle mass in the small, non-cartilaginous airways, which is largely inhibited by treatment with the anticholinergic agent tiotropium bromide [3]. In contrast, tiotropium bromide itself had no effect on airway smooth muscle mass, either in the cartilaginous or non-cartilaginous airways, which corroborates the in vitro findings that muscarinic receptor stimulation by itself is not sufficient to induce mitogenic responses.
The mechanisms that underlie the pro-mitogenic effects of muscarinic receptor stimulation have not yet been studied in detail. However, several intracellular signaling pathways have been identified that regulate the synergistic mitogenic interaction of other GPCR agonists with growth factors in airway smooth muscle (Figure 3). These pathways are not necessarily the same for every GPCR agonist; however, those studies do offer some important clues. The GPCR agonist thrombin augments EGF-induced proliferation through a pathway involving Gβγ, phosphatidylinositol-3-kinase, Akt and p70S6kinase [114]. Although thrombin is mitogenic by itself and therefore presumably differs in its signaling profile from muscarinic receptor agonists, this pathway is of interest as synergistic activation of p70S6kinase by carbachol and EGF has been noted in human airway smooth muscle cells [112]. This mitogenic synergism might also involve PKC; this enzyme is responsible for the potentiating effects of the GPCR agonist bradykinin with EGF in airway smooth muscle [115], and regulates p70S6kinase activity [116,117]. PKC also regulates p42/p44 MAP kinase activation by muscarinic receptor agonists in airway smooth muscle [4]. The involvement of the small G protein RhoA in synergism induced by GPCR agonists and growth factors should also be considered [118], which is interesting given that RhoA expression is increased in animal models of asthma and COPD, as discussed before. Clearly, further studies are required to unravel in detail the signaling pathways involved in the potentiating effects of muscarinic receptor agonists on growth factor-induced airway smooth muscle proliferation.
Figure 3.
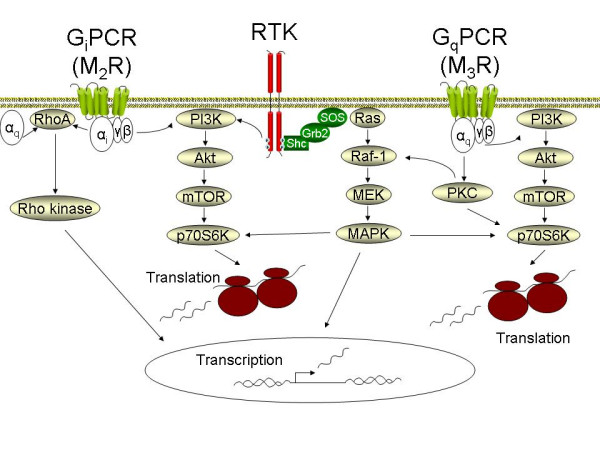
Pathways involved in mesenchymal cell proliferation and differentiation induced by G protein coupled receptors (GPCRs). G protein coupled muscarinic receptors activate signaling cascades resulting in p42/p44 MAP kinase (MAPK), Rho-kinase and phosphatidyl-inositol-3-kinase (PI3K) activity. In addition, the signaling output of receptor tyrosine kinases (RTKs) is enhanced. Activation of the PI3K pathway appears to be particularly important in mesenchymal cell proliferation and differentiation. With Akt and mammalian target of rapamycin (mTOR) as signaling intermediates, PI3K activates p70S6K, which is involved in ribosome mediated protein translation. p42/p44 MAPK, activated by the sequential activation of Ras, Raf and MEK, also activates p70S6K and plays an important role in the induction of transcription factors involved in cell cycle progression. Rho-kinase activated transcription factors also play a central role in smooth muscle specific gene transcription, ultimately mediating the accumulation of contractile and contraction regulatory proteins. See the text for further detail.
Mesenchymal cell differentiation
Mediators that drive contractile protein expression (e.g. TGF-β) are thought to play an important role in the differentiation of proliferating mesenchymal cells into mature airway smooth muscle cells, in airway smooth muscle cell hypertrophy, and in myofibroblast accumulation [119,120]. These responses, together with smooth muscle cell proliferation, contribute to increased airway smooth muscle mass in asthma, and possibly in COPD [101,119,121,122]. Increased expression of contractile apparatus associated proteins likely plays an important role in determining both airway bronchoconstrictor responsiveness and the extent of airway remodeling in asthma and COPD (Table 3) [110].
Mesenchymal cells from all individual airway wall compartments (adventitial fibroblasts, airway smooth muscle cells, mucosal fibroblasts) can be induced to acquire a more contractile phenotype, characterized by increases in smooth muscle specific protein expression, such as smooth muscle (sm)-α-actin, sm-myosin heavy chain (MHC) and desmin [123-125]. Induction of contractile protein gene transcription and protein translation in airway smooth muscle cells and fibroblasts is regulated by at least two pathways: the RhoA/Rho-kinase pathway and the phosphatidylinositol-3-kinase (PI3K)/mammalian target of rapamycin (mTOR)/p70S6kinase pathway [122,126] (Figure 3). There also appears to be a parallel and significant role for PKC as a modulator of the contribution of these pathways in the control of smooth muscle specific gene transcription, and protein accumulation [110,127]. Both the RhoA and PI3K pathways can be activated by muscarinic receptor agonists [43,112], and may be involved in mediating effects of muscarinic receptor agonists on SM22 and sm-MHC promoter activity [128]. Muscarinic receptor stimulation also leads to increased levels of sm-α-actin and sm-MHC mRNA in intact bovine tracheal smooth muscle strips, an effect that was also linked to mechanical strain applied to the strips [129]. Inhibition of PKC leads to an increase in RhoA-dependent transcription of SM22 and smMHC promoters [127]. Since PKC is strongly activated by muscarinic receptor agonists, therefore the contribution of this signaling pathway in the control of smooth muscle gene transcription needs to be established more clearly. In addition, Ca2+ dependent pathways, induced by high concentrations of muscarinic receptor agonists, modulate smooth muscle specific contractile protein expression and contractility in organ-cultured bovine tracheal smooth muscle strips [130]. This indicates that the effects of muscarinic receptors are under tight control of multiple pathways. Future studies are clearly warranted in this area to better understand the interplay between the multiple pathways induced by muscarinic receptors, and their significance as determinants of airway smooth muscle differentiation and cellular hypertrophy.
Repeated exposures of sensitized guinea pigs to ovalbumin cause a ~4-fold increase in pulmonary sm-MHC expression with little effect on sm-α-actin expression [3]. Since sm-MHC is a far more stringent marker for mature airway smooth muscle cells than sm-α-actin, which is a more general marker for lung cells of mesenchymal origin [125], these results indicate that maturation of differentiated mesenchymal cells may have occurred in this model. Indeed, the contractile response of tracheal smooth muscle strips to methacholine was increased in the allergen challenged animals, whereas muscle mass in the large airways had not changed. Treatment of these animals with tiotropium bromide significantly inhibited the ovalbumin-induced sm-MHC expression and increases in tracheal contractility, indicating that endogenous acetylcholine contributes to these effects [3]. Collectively, these studies point to an important role for acetylcholine and muscarinic receptors in mesenchymal cell remodeling in allergic airways disease. The effects of muscarinic receptor antagonists on airway wall remodeling in animal models of COPD have not yet been investigated.
Other aspects of airway remodeling
Evidence of the involvement of muscarinic receptor stimulation in other aspects of airway remodeling in asthma and COPD is scarce, mainly because this has not yet received sufficient attention to date. Since G-protein coupled receptor signaling has been associated with extracellular matrix production [131] and pulmonary vascular smooth muscle cell proliferation [132], effects of muscarinic receptor agonists on extracellular matrix remodeling and pulmonary vascular remodeling could be envisaged, though entirely speculative at this point. In addition, a role for muscarinic receptors in goblet cell hyperplasia and mucus gland hypertrophy has been postulated (see section on mucus hypersecretion). Future studies are clearly required to investigate the effects of muscarinic receptors on these aspects of airway remodeling.
Therapeutic implications
Collectively, the observations we have discussed in this review suggest significant hitherto unexpected therapeutic implications. Thus, anticholinergic therapy could achieve far reaching and significant controller effects for chronic asthma and COPD that extend its capacity as reliever medications to promote bronchodilation. Based on the findings and considerations presented above, it could be envisaged that anticholinergics inhibit airway inflammation and limit airway remodeling, retarding the progressive decline in lung function in asthma and COPD patients.
Though previous studies using ipratropium bromide indicate no improvement in the annual decline in lung function in patients with obstructive airways diseases [132], these therapeutic outcomes may relate to limitations of this drug. Ipratropium bromide is short-acting, whereas the recently introduced anticholinergic agent tiotropium bromide is long-acting and more potent. In addition, tiotropium bromide has a considerably longer relative half-life of dissociation from muscarinic M3 and M1 receptors than from muscarinic M2 receptors, making the drug 'kinetically selective' for M3 and M1 receptors [5,133,134]. It is still debatable as to whether this kinetic selectivity is clinically important; however, since M2 autoreceptor blockade is associated with enhanced acetylcholine outflow from the vagal nerve, whereas muscarinic M3 receptor blockade inhibits most of the postjunctional effects of acetylcholine (as described above) some beneficial effects of this property could be envisaged.
Indeed, a recent study indicates that tiotropium bromide induces a marked reduction in lung function decline of COPD patients [6]. Although this study was retrospective, the results of this study were remarkable: the mean decline in FEV1 in one year was 58 ml in the placebo group vs. 12 ml in the tiotropium group. This reduction has not been observed with ipratropium bromide in COPD patients [133]. Tiotropium bromide is also superior to ipratropium bromide on other aspects, both with respect to spirometry, health related quality of life and number of exacerbations in COPD patients [134]. In view of our own recent findings using a guinea pig model of ongoing allergic asthma [3], showing that tiotropium bromide protects against allergen-induced increases in airway smooth muscle thickening, contractile protein accumulation and tracheal hypercontractility, the drug tiotropium bromide might also be effective in slowing or preventing airway remodeling in chronic asthma. Future studies are required to translate these findings to asthma patients, however.
Conclusion
Acetylcholine is a parasympathetic neurotransmitter and an autocrine or paracrine hormone that regulates airway smooth muscle contraction, mucus production, airway inflammation and airway remodeling. The release of acetylcholine, and the expression of several effector systems central in muscarinic regulation of airway function are enhanced in asthma and COPD, suggesting that the effects of acetylcholine could contribute significantly to the pathophysiology of these obstructive airways diseases. Recent clinical and experimental findings support this hypothesis, suggesting that anticholinergics, most notably the long-acting tiotropium bromide, could achieve reductions in airway remodeling and lung function decline in addition to its effects as a bronchodilator.
Authors' contributions
RG participated in the design of the article and drafted the manuscript. AJH, JZ and HM participated in the design of the article, assisted in drafting the manuscript and revised it critically for important intellectual content. All authors approved the final manuscript.
Acknowledgments
Acknowledgements
The Manitoba institute of Child Health (MICH), National Training Program in Allergy and Asthma (NTPAA), Sick Kids Foundation/Institute of Human Development, Child and Youth Health (#XG05-011), Canadian Institutes of Health Research, and the Netherlands Asthma Foundation are all greatly acknowledged for their financial contributions. RG is the recipient of a Marie Curie Outgoing International Fellowship (MOIF-2005-008823). AJH is supported by a Canadian Institutes of Health Research New Investigator Award.
Contributor Information
Reinoud Gosens, Email: rgosens@mich.ca.
Johan Zaagsma, Email: j.zaagsma@rug.nl.
Herman Meurs, Email: h.meurs@rug.nl.
Andrew J Halayko, Email: ahalayk@cc.umanitoba.ca.
References
- Gross NJ, Skorodin MS. Role of the parasympathetic system in airway obstruction due to emphysema. N Engl J Med. 1984;311:421–425. doi: 10.1056/NEJM198408163110701. [DOI] [PubMed] [Google Scholar]
- Wessler IK, Kirkpatrick CJ. The non-neuronal cholinergic system: an emerging drug target in the airways. Pulm Pharmacol Ther. 2001;14:423–434. doi: 10.1006/pupt.2001.0313. [DOI] [PubMed] [Google Scholar]
- Gosens R, Bos IS, Zaagsma J, Meurs H. Protective effects of tiotropium bromide in the progression of airway smooth muscle remodeling. Am J Respir Crit Care Med. 2005;171:1096–1102. doi: 10.1164/rccm.200409-1249OC. [DOI] [PubMed] [Google Scholar]
- Gosens R, Zaagsma J, Grootte Bromhaar M, Nelemans A, Meurs H. Acetylcholine: a novel regulator of airway smooth muscle remodelling? Eur J Pharmacol. 2004;500:193–201. doi: 10.1016/j.ejphar.2004.07.025. [DOI] [PubMed] [Google Scholar]
- Disse B, Speck GA, Rominger KL, Witek TJ, Jr, Hammer R. Tiotropium (Spiriva): mechanistical considerations and clinical profile in obstructive lung disease. Life Sci. 1999;64:457–464. doi: 10.1016/S0024-3205(98)00588-8. [DOI] [PubMed] [Google Scholar]
- Anzueto A, Tashkin D, Menjoge S, Kesten S. One-year analysis of longitudinal changes in spirometry in patients with COPD receiving tiotropium. Pulm Pharmacol Ther. 2005;18:75–81. doi: 10.1016/j.pupt.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Racke K, Juergens UR, Matthiesen S. Control by cholinergic mechanisms. Eur J Pharmacol. 2006;533:57–68. doi: 10.1016/j.ejphar.2005.12.050. [DOI] [PubMed] [Google Scholar]
- Roffel AF, Elzinga CR, Van Amsterdam RG, De Zeeuw RA, Zaagsma J. Muscarinic M2 receptors in bovine tracheal smooth muscle: discrepancies between binding and function. Eur J Pharmacol. 1988;153:73–82. doi: 10.1016/0014-2999(88)90589-4. [DOI] [PubMed] [Google Scholar]
- Ten Berge RE, Roffel AF, Zaagsma J. The interaction of selective and non-selective antagonists with pre- and postjunctional muscarinic receptor subtypes in the guinea pig trachea. Eur J Pharmacol. 1993;233:279–284. doi: 10.1016/0014-2999(93)90062-M. [DOI] [PubMed] [Google Scholar]
- van Nieuwstadt RA, Henricks PA, Hajer R, van der Meer van Roomen WA, Breukink HJ, Nijkamp FP. Characterization of muscarinic receptors in equine tracheal smooth muscle in vitro. Vet Q. 1997;19:54–57. doi: 10.1080/01652176.1997.9694740. [DOI] [PubMed] [Google Scholar]
- Roffel AF, Elzinga CR, Zaagsma J. Muscarinic M3 receptors mediate contraction of human central and peripheral airway smooth muscle. Pulm Pharmacol. 1990;3:47–51. doi: 10.1016/0952-0600(90)90009-8. [DOI] [PubMed] [Google Scholar]
- Fisher JT, Vincent SG, Gomeza J, Yamada M, Wess J. Loss of vagally mediated bradycardia and bronchoconstriction in mice lacking M2 or M3 muscarinic acetylcholine receptors. FASEB J. 2004;18:711–713. doi: 10.1096/fj.03-0648fje. [DOI] [PubMed] [Google Scholar]
- Struckmann N, Schwering S, Wiegand S, Gschnell A, Yamada M, Kummer W, Wess J, Haberberger RV. Role of muscarinic receptor subtypes in the constriction of peripheral airways: studies on receptor-deficient mice. Mol Pharmacol. 2003;64:1444–1451. doi: 10.1124/mol.64.6.1444. [DOI] [PubMed] [Google Scholar]
- Roffel AF, Elzinga CR, Zaagsma J. Cholinergic contraction of the guinea pig lung strip is mediated by muscarinic M2-like receptors. Eur J Pharmacol. 1993;250:267–279. doi: 10.1016/0014-2999(93)90391-T. [DOI] [PubMed] [Google Scholar]
- Roffel AF, Meurs H, Zaagsma J. Identification, localization and function of muscarinic receptor subtypes in the airways. In: Zaagsma J, Meurs H, Roffel AF, editor. Muscarinic Receptors in Airways Diseases. Basel: Birkhauser Verlag; 2001. pp. 63–87. [Google Scholar]
- Zaagsma J, Roffel AF, Meurs H. Muscarinic control of airway function. Life Sci. 1997;60:1061–1068. doi: 10.1016/S0024-3205(97)00048-9. [DOI] [PubMed] [Google Scholar]
- Parameswaran K, Janssen LJ, O'Byrne PM. Airway hyperresponsiveness and calcium handling by smooth muscle: a "deeper look". Chest. 2002;121:621–624. doi: 10.1378/chest.121.2.621. [DOI] [PubMed] [Google Scholar]
- Amrani Y, Panettieri RA., Jr Modulation of calcium homeostasis as a mechanism for altering smooth muscle responsiveness in asthma. Curr Opin Allergy Clin Immunol. 2002;2:39–45. doi: 10.1097/00130832-200202000-00007. [DOI] [PubMed] [Google Scholar]
- Tao FC, Tolloczko B, Eidelman DH, Martin JG. Enhanced Ca(2+) mobilization in airway smooth muscle contributes to airway hyperresponsiveness in an inbred strain of rat. Am J Respir Crit Care Med. 1999;160:446–453. doi: 10.1164/ajrccm.160.2.9811098. [DOI] [PubMed] [Google Scholar]
- Bramley AM, Thomson RJ, Roberts CR, Schellenberg RR. Hypothesis: excessive bronchoconstriction in asthma is due to decreased airway elastance. Eur Respir J. 1994;7:337–341. doi: 10.1183/09031936.94.07020337. [DOI] [PubMed] [Google Scholar]
- Ma X, Cheng Z, Kong H, Wang Y, Unruh H, Stephens NL, Laviolette M. Changes in biophysical and biochemical properties of single bronchial smooth muscle cells from asthmatic subjects. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1181–1189. doi: 10.1152/ajplung.00389.2001. [DOI] [PubMed] [Google Scholar]
- de Jongste JC, Mons H, Bonta IL, Kerrebijn KF. In vitro responses of airways from an asthmatic patient. Eur J Respir Dis. 1987;71:23–29. [PubMed] [Google Scholar]
- Opazo Saez AM, Seow CY, Pare PD. Peripheral airway smooth muscle mechanics in obstructive airways disease. Am J Respir Crit Care Med. 2000;161:910–917. doi: 10.1164/ajrccm.161.3.9903138. [DOI] [PubMed] [Google Scholar]
- Bai TR. Abnormalities in airway smooth muscle in fatal asthma. Am Rev Respir Dis. 1990;141:552–557. doi: 10.1164/ajrccm/141.3.552. [DOI] [PubMed] [Google Scholar]
- Deshpande DA, White TA, Dogan S, Walseth TF, Panettieri RA, Kannan MS. CD38/cyclic ADP-ribose signaling: role in the regulation of calcium homeostasis in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2005;288:L773–788. doi: 10.1152/ajplung.00217.2004. [DOI] [PubMed] [Google Scholar]
- Deshpande DA, White TA, Guedes AG, Milla C, Walseth TF, Lund FE, Kannan MS. Altered airway responsiveness in CD38-deficient mice. Am J Respir Cell Mol Biol. 2005;32:149–156. doi: 10.1165/rcmb.2004-0243OC. [DOI] [PubMed] [Google Scholar]
- White TA, Kannan MS, Walseth TF. Intracellular calcium signaling through the cADPR pathway is agonist specific in porcine airway smooth muscle. FASEB J. 2003;17:482–484. doi: 10.1096/fj.02-0622fje. [DOI] [PubMed] [Google Scholar]
- Higashida H, Yokoyama S, Hashii M, Taketo M, Higashida M, Takayasu T, Ohshima T, Takasawa S, Okamoto H, Noda M. Muscarinic receptor-mediated dual regulation of ADP-ribosyl cyclase in NG108-15 neuronal cell membranes. J Biol Chem. 1997;272:31272–31277. doi: 10.1074/jbc.272.50.31272. [DOI] [PubMed] [Google Scholar]
- Deshpande DA, Walseth TF, Panettieri RA, Kannan MS. CD38/cyclic ADP-ribose-mediated Ca2+ signaling contributes to airway smooth muscle hyper-responsiveness. Faseb J. 2003;17:452–454. doi: 10.1096/fj.02-0450fje. [DOI] [PubMed] [Google Scholar]
- Deshpande DA, Dogan S, Walseth TF, Miller SM, Amrani Y, Panettieri RA, Kannan MS. Modulation of calcium signaling by interleukin-13 in human airway smooth muscle: role of CD38/cyclic adenosine diphosphate ribose pathway. Am J Respir Cell Mol Biol. 2004;31:36–42. doi: 10.1165/rcmb.2003-0313OC. [DOI] [PubMed] [Google Scholar]
- Tliba O, Panettieri RA, Jr, Tliba S, Walseth TF, Amrani Y. Tumor necrosis factor-alpha differentially regulates the expression of proinflammatory genes in human airway smooth muscle cells by activation of interferon-beta-dependent CD38 pathway. Mol Pharmacol. 2004;66:322–329. doi: 10.1124/mol.104.001040. [DOI] [PubMed] [Google Scholar]
- Tliba O, Cidlowski J, Amrani Y. CD38 expression is insensitive to steroid action in cells treated with TNFα and IFNγ by a mechanism involving the upregulation of glucocorticoid receptor β isoform. Mol Pharmacol. 2005 doi: 10.1124/mol.105.019679. [DOI] [PubMed] [Google Scholar]
- Hotta K, Emala CW, Hirshman CA. TNF-alpha upregulates Gialpha and Gqalpha protein expression and function in human airway smooth muscle cells. Am J Physiol. 1999;276:L405–411. doi: 10.1152/ajplung.1999.276.3.L405. [DOI] [PubMed] [Google Scholar]
- Hakonarson H, Herrick DJ, Serrano PG, Grunstein MM. Mechanism of cytokine-induced modulation of beta-adrenoceptor responsiveness in airway smooth muscle. J Clin Invest. 1996;97:2593–2600. doi: 10.1172/JCI118708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Tliba O, Van Besien CR, Panettieri RA, Jr, Amrani Y. TNF-[alpha] modulates murine tracheal rings responsiveness to G-protein-coupled receptor agonists and KCl. J Appl Physiol. 2003;95:864–872. doi: 10.1152/japplphysiol.00140.2003. discussion 863. [DOI] [PubMed] [Google Scholar]
- Tliba O, Deshpande D, Chen H, Van Besien C, Kannan M, Panettieri RA, Jr, Amrani Y. IL-13 enhances agonist-evoked calcium signals and contractile responses in airway smooth muscle. Br J Pharmacol. 2003;140:1159–1162. doi: 10.1038/sj.bjp.0705558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosens R, Schaafsma D, Nelemans SA, Halayko AJ. Rho-kinase as a drug target for the treatment of airway hyperresponisveness in asthma. Mini Rev Med Chem. 2006;6:339–348. doi: 10.2174/138955706776073402. [DOI] [PubMed] [Google Scholar]
- Hirshman CA, Lande B, Croxton TL. Role of M2 muscarinic receptors in airway smooth muscle contraction. Life Sci. 1999;64:443–448. doi: 10.1016/S0024-3205(98)00586-4. [DOI] [PubMed] [Google Scholar]
- Lutz S, Freichel-Blomquist A, Yang Y, Rumenapp U, Jakobs KH, Schmidt M, Wieland T. The guanine nucleotide exchange factor p63RhoGEF, a specific link between Gq/11-coupled receptor signaling and RhoA. J Biol Chem. 2005;280:11134–11139. doi: 10.1074/jbc.M411322200. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83:1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- Yoshii A, Iizuka K, Dobashi K, Horie T, Harada T, Nakazawa T, Mori M. Relaxation of contracted rabbit tracheal and human bronchial smooth muscle by Y-27632 through inhibition of Ca2+ sensitization. Am J Respir Cell Mol Biol. 1999;20:1190–1200. doi: 10.1165/ajrcmb.20.6.3441. [DOI] [PubMed] [Google Scholar]
- Janssen LJ, Wattie J, Lu-Chao H, Tazzeo T. Muscarinic excitation-contraction coupling mechanisms in tracheal and bronchial smooth muscles. J Appl Physiol. 2001;91:1142–1151. doi: 10.1152/jappl.2001.91.3.1142. [DOI] [PubMed] [Google Scholar]
- Gosens R, Schaafsma D, Meurs H, Zaagsma J, Nelemans SA. Role of Rho-kinase in maintaining airway smooth muscle contractile phenotype. Eur J Pharmacol. 2004;483:71–78. doi: 10.1016/j.ejphar.2003.10.027. [DOI] [PubMed] [Google Scholar]
- Sakai H, Chiba Y, Hirano T, Misawa M. Possible involvement of CPI-17 in augmented bronchial smooth muscle contraction in antigen-induced airway hyper-responsive rats. Mol Pharmacol. 2005;68:145–151. doi: 10.1124/mol.104.004325. [DOI] [PubMed] [Google Scholar]
- Chiba Y, Takada Y, Miyamoto S, MitsuiSaito M, Karaki H, Misawa M. Augmented acetylcholine-induced, Rho-mediated Ca2+ sensitization of bronchial smooth muscle contraction in antigen-induced airway hyperresponsive rats. Br J Pharmacol. 1999;127:597–600. doi: 10.1038/sj.bjp.0702585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaafsma D, Gosens R, Bos IS, Meurs H, Zaagsma J, Nelemans SA. Allergic sensitization enhances the contribution of Rho-kinase to airway smooth muscle contraction. Br J Pharmacol. 2004;143:477–484. doi: 10.1038/sj.bjp.0705903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai H, Otogoto S, Chiba Y, Abe K, Misawa M. TNF-alpha augments the expression of RhoA in the rat bronchus. J Smooth Muscle Res. 2004;40:25–34. doi: 10.1540/jsmr.40.25. [DOI] [PubMed] [Google Scholar]
- Chiba Y, Ueno A, Shinozaki K, Takeyama H, Nakazawa S, Sakai H, Misawa M. Involvement of RhoA-mediated Ca2+ sensitization in antigen-induced bronchial smooth muscle hyperresponsiveness in mice. Respir Res. 2005;6:4. doi: 10.1186/1465-9921-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba Y, Sakai H, Misawa M. Augmented acetylcholine-induced translocation of RhoA in bronchial smooth muscle from antigen-induced airway hyperresponsive rats. Br J Pharmacol. 2001;133:886–890. doi: 10.1038/sj.bjp.0704137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba Y, Murata M, Ushikubo H, Yoshikawa Y, Saitoh A, Sakai H, Kamei J, Misawa M. Effect of cigarette smoke exposure in vivo on bronchial smooth muscle contractility in vitro in rats. Am J Respir Cell Mol Biol. 2005;33:574–581. doi: 10.1165/rcmb.2005-0177OC. [DOI] [PubMed] [Google Scholar]
- Yamawaki I, Tamaoki J, Kanemura T, Horii S, Takizawa T. Effects of lipopolysaccharide from Pseudomonas aeruginosa on airway smooth muscle functions in guinea pigs. Respiration. 1990;57:268–274. doi: 10.1159/000195854. [DOI] [PubMed] [Google Scholar]
- Mitchell RW, Kelly E, Leff AR. Reduced activity of acetylcholinesterase in canine tracheal smooth muscle homogenates after active immune-sensitization. Am J Respir Cell Mol Biol. 1991;5:56–62. doi: 10.1165/ajrcmb/5.1.56. [DOI] [PubMed] [Google Scholar]
- Undem BJ, Myers AC. Cholinergic and noncholinergic parasympathetic control of airway smooth muscle. In: Zaagsma J, Meurs H, Roffel AF, editor. Muscarinic receptors in airways diseases. Basel: Birkhauser; 2001. pp. 1–24. [Google Scholar]
- ten Berge RE, Santing RE, Hamstra JJ, Roffel AF, Zaagsma J. Dysfunction of muscarinic M2 receptors after the early allergic reaction: possible contribution to bronchial hyperresponsiveness in allergic guinea-pigs. Br J Pharmacol. 1995;114:881–887. doi: 10.1111/j.1476-5381.1995.tb13286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulson FR, Fryer AD. Muscarinic acetylcholine receptors and airway diseases. Pharmacol Ther. 2003;98:59–69. doi: 10.1016/S0163-7258(03)00004-4. [DOI] [PubMed] [Google Scholar]
- Minette PA, Lammers JW, Dixon CM, McCusker MT, Barnes PJ. A muscarinic agonist inhibits reflex bronchoconstriction in normal but not in asthmatic subjects. J Appl Physiol. 1989;67:2461–2465. doi: 10.1152/jappl.1989.67.6.2461. [DOI] [PubMed] [Google Scholar]
- Okayama M, Shen T, Midorikawa J, Lin JT, Inoue H, Takishima T, Shirato K. Effect of pilocarpine on propranolol-induced bronchoconstriction in asthma. Am J Respir Crit Care Med. 1994;149:76–80. doi: 10.1164/ajrccm.149.1.8111602. [DOI] [PubMed] [Google Scholar]
- Kanazawa H, Hirata K, Yoshikawa J. Increased responses to inhaled oxitropium bromide in asthmatic patients with active hepatitis C virus infection. Chest. 2004;125:1368–1371. doi: 10.1378/chest.125.4.1368. [DOI] [PubMed] [Google Scholar]
- On LS, Boonyongsunchai P, Webb S, Davies L, Calverley PM, Costello RW. Function of pulmonary neuronal M(2) muscarinic receptors in stable chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;163:1320–1325. doi: 10.1164/ajrccm.163.6.2002129. [DOI] [PubMed] [Google Scholar]
- Belmonte KE. Cholinergic pathways in the lungs and anticholinergic therapy for chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2005;2:297–304. doi: 10.1513/pats.200504-043SR. discussion 311–292. [DOI] [PubMed] [Google Scholar]
- ten Berge RE, Roffel AF, Zaagsma J. Conditional involvement of muscarinic M1 receptors in vagally mediated contraction of guinea-pig bronchi. Naunyn Schmiedebergs Arch Pharmacol. 1995;352:173–178. doi: 10.1007/BF00176771. [DOI] [PubMed] [Google Scholar]
- Racke K, Matthiesen S. The airway cholinergic system: physiology and pharmacology. Pulm Pharmacol Ther. 2004;17:181–198. doi: 10.1016/j.pupt.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Yang ZJ, Biggs DF. Muscarinic receptors and parasympathetic neurotransmission in guinea-pig trachea. Eur J Pharmacol. 1991;193:301–308. doi: 10.1016/0014-2999(91)90143-E. [DOI] [PubMed] [Google Scholar]
- Myers AC. Transmission in autonomic ganglia. Respir Physiol. 2001;125:99–111. doi: 10.1016/S0034-5687(00)00207-3. [DOI] [PubMed] [Google Scholar]
- Undem BJ, Kollarik M. The role of vagal afferent nerves in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2005;2:355–360. doi: 10.1513/pats.200504-033SR. discussion 371-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spina D, Shah S, Harrison S. Modulation of sensory nerve function in the airways. Trends Pharmacol Sci. 1998;19:460–466. doi: 10.1016/S0165-6147(98)01261-9. [DOI] [PubMed] [Google Scholar]
- Undem BJ, Carr MJ. Pharmacology of airway afferent nerve activity. Respir Res. 2001;2:234–244. doi: 10.1186/rr62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleich GJ, Flavahan NA, Fujisawa T, Vanhoutte PM. The eosinophil as a mediator of damage to respiratory epithelium: a model for bronchial hyperreactivity. J Allergy Clin Immunol. 1988;81:776–781. doi: 10.1016/0091-6749(88)90931-1. [DOI] [PubMed] [Google Scholar]
- Rogers DF. Airway mucus hypersecretion in asthma: an undervalued pathology? Curr Opin Pharmacol. 2004;4:241–250. doi: 10.1016/j.coph.2004.01.011. [DOI] [PubMed] [Google Scholar]
- Rogers DF. Mucus hypersecretion in chronic obstructive pulmonary disease. Novartis Found Symp. 2001;234:65–77. doi: 10.1002/0470868678.ch5. discussion 77–83. [DOI] [PubMed] [Google Scholar]
- Rogers DF. Motor control of airway goblet cells and glands. Respir Physiol. 2001;125:129–144. doi: 10.1016/S0034-5687(00)00209-7. [DOI] [PubMed] [Google Scholar]
- Ramnarine SI, Haddad EB, Khawaja AM, Mak JC, Rogers DF. On muscarinic control of neurogenic mucus secretion in ferret trachea. J Physiol. 1996;494:577–586. doi: 10.1113/jphysiol.1996.sp021515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak JC, Barnes PJ. Autoradiographic visualization of muscarinic receptor subtypes in human and guinea pig lung. Am Rev Respir Dis. 1990;141:1559–1568. doi: 10.1164/ajrccm/141.6.1559. [DOI] [PubMed] [Google Scholar]
- Laitinen A, Partanen M, Hervonen A, Laitinen LA. Electron microscopic study on the innervation of the human lower respiratory tract: evidence of adrenergic nerves. Eur J Respir Dis. 1985;67:209–215. [PubMed] [Google Scholar]
- Ishihara H, Shimura S, Satoh M, Masuda T, Nonaka H, Kase H, Sasaki T, Sasaki H, Takishima T, Tamura K. Muscarinic receptor subtypes in feline tracheal submucosal gland secretion. Am J Physiol. 1992;262:L223–228. doi: 10.1152/ajplung.1992.262.2.L223. [DOI] [PubMed] [Google Scholar]
- Jeffery PK. Remodeling and inflammation of bronchi in asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2004;1:176–183. doi: 10.1513/pats.200402-009MS. [DOI] [PubMed] [Google Scholar]
- Iwase N, Sasaki T, Oshiro T, Tamada T, Nara M, Sasamori K, Hattori T, Shirato K, Maruyama Y. Differential effect of epidermal growth factor on serous and mucous cells in porcine airway submucosal gland. Respir Physiol Neurobiol. 2002;132:307–319. doi: 10.1016/S1569-9048(02)00118-0. [DOI] [PubMed] [Google Scholar]
- Amishima M, Munakata M, Nasuhara Y, Sato A, Takahashi T, Homma Y, Kawakami Y. Expression of epidermal growth factor and epidermal growth factor receptor immunoreactivity in the asthmatic human airway. Am J Respir Crit Care Med. 1998;157:1907–1912. doi: 10.1164/ajrccm.157.6.9609040. [DOI] [PubMed] [Google Scholar]
- O'Donnell RA, Richter A, Ward J, Angco G, Mehta A, Rousseau K, Swallow DM, Holgate ST, Djukanovic R, Davies DE, Wilson SJ. Expression of ErbB receptors and mucins in the airways of long term current smokers. Thorax. 2004;59:1032–1040. doi: 10.1136/thx.2004.028043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno H, Horikawa Y, Hodges RR, Zoukhri D, Shatos MA, Rios JD, Dartt DA. Cholinergic agonists transactivate EGFR and stimulate MAPK to induce goblet cell secretion. Am J Physiol Cell Physiol. 2003;284:C988–998. doi: 10.1152/ajpcell.00582.2001. [DOI] [PubMed] [Google Scholar]
- Rogers DF. Muscarinic control of airway mucus secretion. In: Zaagsma J, Meurs H, Roffel AF, editor. Muscarinic receptors in airways diseases. Basel: Birkhauser; 2001. pp. 175–201. [Google Scholar]
- Meurs H, Timmermans A, de Monchy JG, Zaagsma J, Kauffman HF. Lack of coupling of muscarinic receptors to phosphoinositide metabolism and adenylyl cyclase in human lymphocytes and polymorphonuclear leukocytes: studies in healthy subjects and allergic asthmatic patients. Int Arch Allergy Immunol. 1993;100:19–27. doi: 10.1159/000236382. [DOI] [PubMed] [Google Scholar]
- Fujii T, Kawashima K. An independent non-neuronal cholinergic system in lymphocytes. Jpn J Pharmacol. 2001;85:11–15. doi: 10.1254/jjp.85.11. [DOI] [PubMed] [Google Scholar]
- Kawashima K, Fujii T. Extraneuronal cholinergic system in lymphocytes. Pharmacol Ther. 2000;86:29–48. doi: 10.1016/S0163-7258(99)00071-6. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick CJ, Bittinger F, Nozadze K, Wessler I. Expression and function of the non-neuronal cholinergic system in endothelial cells. Life Sci. 2003;72:2111–2116. doi: 10.1016/S0024-3205(03)00069-9. [DOI] [PubMed] [Google Scholar]
- Wessler I, Kilbinger H, Bittinger F, Unger R, Kirkpatrick CJ. The non-neuronal cholinergic system in humans: expression, function and pathophysiology. Life Sci. 2003;72:2055–2061. doi: 10.1016/S0024-3205(03)00083-3. [DOI] [PubMed] [Google Scholar]
- Wessler I, Reinheimer T, Kilbinger H, Bittinger F, Kirkpatrick CJ, Saloga J, Knop J. Increased acetylcholine levels in skin biopsies of patients with atopic dermatitis. Life Sci. 2003;72:2169–2172. doi: 10.1016/S0024-3205(03)00079-1. [DOI] [PubMed] [Google Scholar]
- Tillie-Leblond I, Gosset P, Tonnel AB. Inflammatory events in severe acute asthma. Allergy. 2005;60:23–29. doi: 10.1111/j.1398-9995.2005.00632.x. [DOI] [PubMed] [Google Scholar]
- Fujii T, Kawashima K. Ca2+ oscillation and c-fos gene expression induced via muscarinic acetylcholine receptor in human T- and B-cell lines. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:14–21. doi: 10.1007/s002100000251. [DOI] [PubMed] [Google Scholar]
- Fujii T, Yamada S, Watanabe Y, Misawa H, Tajima S, Fujimoto K, Kasahara T, Kawashima K. Induction of choline acetyltransferase mRNA in human mononuclear leukocytes stimulated by phytohemagglutinin, a T-cell activator. J Neuroimmunol. 1998;82:101–107. doi: 10.1016/S0165-5728(97)00195-1. [DOI] [PubMed] [Google Scholar]
- Fujii T, Watanabe Y, Inoue T, Kawashima K. Upregulation of mRNA encoding the M5 muscarinic acetylcholine receptor in human T- and B-lymphocytes during immunological responses. Neurochem Res. 2003;28:423–429. doi: 10.1023/A:1022840416292. [DOI] [PubMed] [Google Scholar]
- Strom TB, Deisseroth A, Morganroth J, Carpenter CB, Merrill JP. Alteration of the cytotoxic action of sensitized lymphocytes by cholinergic agents and activators of adenylate cyclase. Proc Natl Acad Sci USA. 1972;69:2995–2999. doi: 10.1073/pnas.69.10.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Profita M, Giorgi RD, Sala A, Bonanno A, Riccobono L, Mirabella F, Gjomarkaj M, Bonsignore G, Bousquet J, Vignola AM. Muscarinic receptors, leukotriene B4 production and neutrophilic inflammation in COPD patients. Allergy. 2005;60:1361–1369. doi: 10.1111/j.1398-9995.2005.00892.x. [DOI] [PubMed] [Google Scholar]
- Sato E, Koyama S, Okubo Y, Kubo K, Sekiguchi M. Acetylcholine stimulates alveolar macrophages to release inflammatory cell chemotactic activity. Am J Physiol. 1998;274:L970–979. doi: 10.1152/ajplung.1998.274.6.L970. [DOI] [PubMed] [Google Scholar]
- Koyama S, Rennard SI, Robbins RA. Acetylcholine stimulates bronchial epithelial cells to release neutrophil and monocyte chemotactic activity. Am J Physiol. 1992;262:L466–471. doi: 10.1152/ajplung.1992.262.4.L466. [DOI] [PubMed] [Google Scholar]
- Koyama S, Sato E, Nomura H, Kubo K, Nagai S, Izumi T. Acetylcholine and substance P stimulate bronchial epithelial cells to release eosinophil chemotactic activity. J Appl Physiol. 1998;84:1528–1534. doi: 10.1152/jappl.1998.84.5.1528. [DOI] [PubMed] [Google Scholar]
- Klapproth H, Racke K, Wessler I. Acetylcholine and nicotine stimulate the release of granulocyte-macrophage colony stimulating factor from cultured human bronchial epithelial cells. Naunyn Schmiedebergs Arch Pharmacol. 1998;357:472–475. doi: 10.1007/PL00005195. [DOI] [PubMed] [Google Scholar]
- Proskocil BJ, Sekhon HS, Jia Y, Savchenko V, Blakely RD, Lindstrom J, Spindel ER. Acetylcholine is an autocrine or paracrine hormone synthesized and secreted by airway bronchial epithelial cells. Endocrinology. 2004;145:2498–2506. doi: 10.1210/en.2003-1728. [DOI] [PubMed] [Google Scholar]
- Kanefsky J, Lenburg M, Hai CM. Cholinergic Receptor and Cyclic Stretch-Mediated Inflammatory Gene Expression in Intact ASM. Am J Respir Cell Mol Biol. 2006;34:417–425. doi: 10.1165/rcmb.2005-0326OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai TR, Cooper J, Koelmeyer T, Pare PD, Weir TD. The effect of age and duration of disease on airway structure in fatal asthma. Am J Respir Crit Care Med. 2000;162:663–669. doi: 10.1164/ajrccm.162.2.9907151. [DOI] [PubMed] [Google Scholar]
- Benayoun L, Druilhe A, Dombret MC, Aubier M, Pretolani M. Airway structural alterations selectively associated with severe asthma. Am J Respir Crit Care Med. 2003;167:1360–1368. doi: 10.1164/rccm.200209-1030OC. [DOI] [PubMed] [Google Scholar]
- Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, Cherniack RM, Rogers RM, Sciurba FC, Coxson HO, Pare PD. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:2645–2653. doi: 10.1056/NEJMoa032158. [DOI] [PubMed] [Google Scholar]
- McParland BE, Macklem PT, Pare PD. Airway wall remodeling: friend or foe? J Appl Physiol. 2003;95:426–434. doi: 10.1152/japplphysiol.00159.2003. [DOI] [PubMed] [Google Scholar]
- Bai TR, Knight DA. Structural changes in the airways in asthma: observations and consequences. Clin Sci (Lond) 2005;108:463–477. doi: 10.1042/CS20040342. [DOI] [PubMed] [Google Scholar]
- Salmon M, Walsh DA, Huang TJ, Barnes PJ, Leonard TB, Hay DW, Chung KF. Involvement of cysteinyl leukotrienes in airway smooth muscle cell DNA synthesis after repeated allergen exposure in sensitized Brown Norway rats. Br J Pharmacol. 1999;127:1151–1158. doi: 10.1038/sj.bjp.0702669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihaku D, Cameron L, Suzuki M, Molet S, Martin J, Hamid Q. Montelukast, a leukotriene receptor antagonist, inhibits the late airway response to antigen, airway eosinophilia, and IL-5-expressing cells in Brown Norway rats. J Allergy Clin Immunol. 1999;104:1147–1154. doi: 10.1016/S0091-6749(99)70006-0. [DOI] [PubMed] [Google Scholar]
- Wang CG, Du T, Xu LJ, Martin JG. Role of leukotriene D4 in allergen-induced increases in airway smooth muscle in the rat. Am Rev Respir Dis. 1993;148:413–417. doi: 10.1164/ajrccm/148.2.413. [DOI] [PubMed] [Google Scholar]
- Holgate ST, Peters-Golden M, Panettieri RA, Henderson WR., Jr Roles of cysteinyl leukotrienes in airway inflammation, smooth muscle function, and remodeling. J Allergy Clin Immunol. 2003;111:S18–34. doi: 10.1067/mai.2003.25. discussion S34-16. [DOI] [PubMed] [Google Scholar]
- Henderson WR, Jr, Tang LO, Chu SJ, Tsao SM, Chiang GK, Jones F, Jonas M, Pae C, Wang H, Chi EY. A role for cysteinyl leukotrienes in airway remodeling in a mouse asthma model. Am J Respir Crit Care Med. 2002;165:108–116. doi: 10.1164/ajrccm.165.1.2105051. [DOI] [PubMed] [Google Scholar]
- Halayko AJ, Tran T, Ji SY, Yamasaki A, Gosens R. Airway smooth muscle phenotype and function: interaction with current asthma therapies. Inflamm Allergy Drug Targets. 2006. [DOI] [PubMed]
- Matthiesen S, Kempkens S, Bahulayan A, Juergens UR, Racke K. Muscarinic Stimulation of Human Lung Fibroblast Proliferation. Proceeedings of the British Pharmacological Society. 2005;3(4):37. http://www.pa2online.org/abstracts/Vol3Issue4abst037P.pdf [Google Scholar]
- Krymskaya VP, Orsini MJ, Eszterhas AJ, Brodbeck KC, Benovic JL, Panettieri RA, Jr, Penn RB. Mechanisms of proliferation synergy by receptor tyrosine kinase and G protein-coupled receptor activation in human airway smooth muscle. Am J Respir Cell Mol Biol. 2000;23:546–554. doi: 10.1165/ajrcmb.23.4.4115. [DOI] [PubMed] [Google Scholar]
- Gosens R, Nelemans SA, Grootte Bromhaar MM, McKay S, Zaagsma J, Meurs H. Muscarinic M3-receptors mediate cholinergic synergism of mitogenesis in airway smooth muscle. Am J Respir Cell Mol Biol. 2003;28:257–262. doi: 10.1165/rcmb.2002-0128OC. [DOI] [PubMed] [Google Scholar]
- Billington CK, Kong KC, Bhattacharyya R, Wedegaertner PB, Panettieri RA, Jr, Chan TO, Penn RB. Cooperative Regulation of p70S6 Kinase by Receptor Tyrosine Kinases and G Protein-Coupled Receptors Augments Airway Smooth Muscle Growth. Biochemistry. 2005;44:14595–14605. doi: 10.1021/bi0510734. [DOI] [PubMed] [Google Scholar]
- Gosens R, Grootte Bromhaar MM, Maarsingh H, ten Damme A, Meurs H, Zaagsma J, Nelemans SA. Bradykinin augments EGF-induced airway smooth muscle proliferation by activation of conventional protein kinase C iso-enzymes. Eur J Pharmacol. 2006;535:253–262. doi: 10.1016/j.ejphar.2006.01.065. [DOI] [PubMed] [Google Scholar]
- Romanelli A, Martin KA, Toker A, Blenis J. p70 S6 kinase is regulated by protein kinase Czeta and participates in a phosphoinositide 3-kinase-regulated signalling complex. Mol Cell Biol. 1999;19:2921–2928. doi: 10.1128/mcb.19.4.2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yutsudo Y, Kido Y, Okabayashi Y, Matsumoto M, Ogawa W, Ohba M, Kuroki T, Kasuga M. Protein kinase Calpha is implicated in cholecystokinin-induced activation of 70-kd S6 kinase in AR42J cells. Pancreas. 2005;30:50–53. [PubMed] [Google Scholar]
- Ediger TL, Schulte NA, Murphy TJ, Toews ML. Transcription factor activation and mitogenic synergism in airway smooth muscle cells. Eur Respir J. 2003;21:759–769. doi: 10.1183/09031936.03.00075702. [DOI] [PubMed] [Google Scholar]
- Goldsmith AM, Bentley JK, Zhou L, Jia Y, Bitar KN, Fingar DC, Hershenson MB. Transforming Growth Factor-β Induces Airway Smooth Muscle Hypertrophy. Am J Respir Cell Mol Biol. 2006;34:247–254. doi: 10.1165/rcmb.2005-0166OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima Y, Nomura A, Uchida Y, Noguchi Y, Sakamoto T, Ishii Y, Goto Y, Masuyama K, Zhang MJ, Hirano K, Mochizuki M, Ohtsuka M, Sekizawa K. Triggering the induction of myofibroblast and fibrogenesis by airway epithelial shedding. Am J Respir Cell Mol Biol. 2001;24:1–11. doi: 10.1165/ajrcmb.24.1.4040. [DOI] [PubMed] [Google Scholar]
- Woodruff PG, Dolganov GM, Ferrando RE, Donnelly S, Hays SR, Solberg OD, Carter R, Wong HH, Cadbury PS, Fahy JV. Hyperplasia of smooth muscle in mild to moderate asthma without changes in cell size or gene expression. Am J Respir Crit Care Med. 2004;169:1001–1006. doi: 10.1164/rccm.200311-1529OC. [DOI] [PubMed] [Google Scholar]
- Halayko AJ, Kartha S, Stelmack GL, McConville J, Tam J, Camoretti-Mercado B, Forsythe SM, Hershenson MB, Solway J. Phophatidylinositol-3 kinase/mammalian target of rapamycin/p70S6K regulates contractile protein accumulation in airway myocyte differentiation. Am J Respir Cell Mol Biol. 2004;31:266–275. doi: 10.1165/rcmb.2003-0272OC. [DOI] [PubMed] [Google Scholar]
- Halayko AJ, Solway J. Molecular mechanisms of phenotypic plasticity in smooth muscle cells. J Appl Physiol. 2001;90:358–368. doi: 10.1152/jappl.2001.90.1.358. [DOI] [PubMed] [Google Scholar]
- Yamasaki A, Voros A, McNeill K, Stelmack GL, Unruh H, Soussi-Gounni A, Halayko AJ. Phenotype diversity of mesenchymal cells from compartments of the human airway wall. Am J Respir Crit Care Med. 2004;169:A266. [Google Scholar]
- Halayko AJ, Salari H, Ma X, Stephens NL. Markers of airway smooth muscle cell phenotype. Am J Physiol. 1996;270:L1040–1051. doi: 10.1152/ajplung.1996.270.6.L1040. [DOI] [PubMed] [Google Scholar]
- Liu HW, Halayko AJ, Fernandes DJ, Harmon GS, McCauley JA, Kocieniewski P, McConville J, Fu Y, Forsythe SM, Kogut P, Bellam S, Dowell M, Churchill J, Lesso H, Kassiri K, Mitchell RW, Hershenson MB, Camoretti-Mercado B, Solway J. The RhoA/Rho kinase pathway regulates nuclear localization of serum response factor. Am J Respir Cell Mol Biol. 2003;29:39–47. doi: 10.1165/rcmb.2002-0206OC. [DOI] [PubMed] [Google Scholar]
- Wang L, Liu HW, McNeill KD, Stelmack G, Scott JE, Halayko AJ. Mechanical strain inhibits airway smooth muscle gene transcription via protein kinase C signaling. Am J Respir Cell Mol Biol. 2004;31:54–61. doi: 10.1165/rcmb.2003-0240OC. [DOI] [PubMed] [Google Scholar]
- Liu HW, Kassiri K, Voros A, Hillier CT, Wang L, Solway J, Halayko AJ. Gaq-receptor coupled signaling induces RHO-dependent transcription of smooth muscle specific genes in cultured canine airway myocytes. Am J Respir Crit Care Med. 2002;165:A670. [Google Scholar]
- Wahl M, Eddinger TJ, Hai CM. Sinusoidal length oscillation- and receptor-mediated mRNA expression of myosin isoforms and alpha-SM actin in airway smooth muscle. Am J Physiol Cell Physiol. 2004;287:C1697–1708. doi: 10.1152/ajpcell.00214.2004. [DOI] [PubMed] [Google Scholar]
- Gosens R, Bromhaar MM, Tonkes A, Schaafsma D, Zaagsma J, Nelemans SA, Meurs H. Muscarinic M(3) receptor-dependent regulation of airway smooth muscle contractile phenotype. Br J Pharmacol. 2004;141:943–950. doi: 10.1038/sj.bjp.0705709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asakura T, Ishii Y, Chibana K, Fukuda T. Leukotriene D4 stimulates collagen production from myofibroblasts transformed by TGF-beta. J Allergy Clin Immunol. 2004;114:310–315. doi: 10.1016/j.jaci.2004.04.037. [DOI] [PubMed] [Google Scholar]
- Mandegar M, Fung YC, Huang W, Remillard CV, Rubin LJ, Yuan JX. Cellular and molecular mechanisms of pulmonary vascular remodeling: role in the development of pulmonary hypertension. Microvasc Res. 2004;68:75–103. doi: 10.1016/j.mvr.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Anthonisen NR, Connett JE, Kiley JP, Altose MD, Bailey WC, Buist AS, Conway WAb, Jr, Enright PL, Kanner RE, O'Hara P, Owens GR, Scanlon PD, Tashkin DP, Wise RA. Effects of smoking intervention and the use of an inhaled anticholinergic bronchodilator on the rate of decline of FEV1. The Lung Health Study. JAMA. 1994;272:1497–1505. doi: 10.1001/jama.272.19.1497. [DOI] [PubMed] [Google Scholar]
- Vincken W, van Noord JA, Greefhorst AP, Bantje TA, Kesten S, Korducki L, Cornelissen PJ. Improved health outcomes in patients with COPD during 1 yr's treatment with tiotropium. Eur Respir J. 2002;19:209–216. doi: 10.1183/09031936.02.00238702. [DOI] [PubMed] [Google Scholar]
- Roffel AF, Hamstra JJ, Elzinga CR, Zaagsma J. Selectivity profile of some recent muscarinic antagonists in bovine and guinea-pig trachea and heart. Arch Int Pharmacodyn Ther. 1994;328:82–98. [PubMed] [Google Scholar]
- Reinheimer T, Mohlig T, Zimmermann S, Hohle KD, Wessler I. Muscarinic control of histamine release from airways. Inhibitory M1-receptors in human bronchi but absence in rat trachea. Am J Respir Crit Care Med. 2000;162:534–538. doi: 10.1164/ajrccm.162.2.9911094. [DOI] [PubMed] [Google Scholar]
- Reinheimer T, Baumgartner D, Hohle KD, Racke K, Wessler I. Acetylcholine via muscarinic receptors inhibits histamine release from human isolated bronchi. Am J Respir Crit Care Med. 1997;156:389–395. doi: 10.1164/ajrccm.156.2.96-12079. [DOI] [PubMed] [Google Scholar]
- Hagforsen E, Einarsson A, Aronsson F, Nordlind K, Michaelsson G. The distribution of choline acetyltransferase- and acetylcholinesterase-like immunoreactivity in the palmar skin of patients with palmoplantar pustulosis. Br J Dermatol. 2000;142:234–242. doi: 10.1046/j.1365-2133.2000.03290.x. [DOI] [PubMed] [Google Scholar]
- Ebina M, Takahashi T, Chiba T, Motomiya M. Cellular hypertrophy and hyperplasia of airway smooth muscles underlying bronchial asthma. A 3-D morphometric study. Am Rev Respir Dis. 1993;148:720–726. doi: 10.1164/ajrccm/148.3.720. [DOI] [PubMed] [Google Scholar]
- Saetta M, Di Stefano A, Turato G, Facchini FM, Corbino L, Mapp CE, Maestrelli P, Ciaccia A, Fabbri LM. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;157:822–826. doi: 10.1164/ajrccm.157.3.9709027. [DOI] [PubMed] [Google Scholar]
- Shimura S, Andoh Y, Haraguchi M, Shirato K. Continuity of airway goblet cells and intraluminal mucus in the airways of patients with bronchial asthma. Eur Respir J. 1996;9:1395–1401. doi: 10.1183/09031936.96.09071395. [DOI] [PubMed] [Google Scholar]
- Rogers DF. Mucociliary dysfunction in COPD: effect of current pharmacotherapeutic options. Pulm Pharmacol Ther. 2005;18:1–8. doi: 10.1016/j.pupt.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Charan NB, Baile EM, Pare PD. Bronchial vascular congestion and angiogenesis. Eur Respir J. 1997;10:1173–1180. doi: 10.1183/09031936.97.10051173. [DOI] [PubMed] [Google Scholar]
- Santos S, Peinado VI, Ramirez J, Morales-Blanhir J, Bastos R, Roca J, Rodriguez-Roisin R, Barbera JA. Enhanced expression of vascular endothelial growth factor in pulmonary arteries of smokers and patients with moderate chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2003;167:1250–1256. doi: 10.1164/rccm.200210-1233OC. [DOI] [PubMed] [Google Scholar]
- Kranenburg AR, de Boer WI, Alagappan VK, Sterk PJ, Sharma HS. Enhanced bronchial expression of vascular endothelial growth factor and receptors (Flk-1 and Flt-1) in patients with chronic obstructive pulmonary disease. Thorax. 2005;60:106–113. doi: 10.1136/thx.2004.023986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranenburg AR, De Boer WI, Van Krieken JH, Mooi WJ, Walters JE, Saxena PR, Sterk PJ, Sharma HS. Enhanced expression of fibroblast growth factors and receptor FGFR-1 during vascular remodeling in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2002;27:517–525. doi: 10.1165/rcmb.4474. [DOI] [PubMed] [Google Scholar]
- Brewster CE, Howarth PH, Djukanovic R, Wilson J, Holgate ST, Roche WR. Myofibroblasts and subepithelial fibrosis in bronchial asthma. Am J Respir Cell Mol Biol. 1990;3:507–511. doi: 10.1165/ajrcmb/3.5.507. [DOI] [PubMed] [Google Scholar]