

Abstract
Context
Alzheimer disease (AD) is a major public health issue with a prediction of 12 million Americans being affected by 2025 from the present 4 million. Molecular and genetic findings have provided significant insights into the roles that amyloid, tau, and apolipoprotein E isoforms have in the causation of AD. A central issue in AD pathogenesis is the amyloid cascade hypothesis. It states that abnormal amyloid processing and accumulation is the primary causative factor of AD and other associated neuropathologic abnormalities are of secondary consequence. It is presented to provide the rationale for novel drug and vaccination therapeutic strategies. Future research directed at prediction and prevention of AD through a genomic and proteomic analysis with identification of multiple polymorphic genes that interact, resulting in increased risk for late-onset AD, are the realistic and ultimate goals. A new approach for drug development is required, one that will emphasize a genomic and proteomic analysis to identify at-risk gene sets whose genetic expression is sufficient to cause late onset, sporadic AD. Prediction and prevention of disease prior to clinical signs and symptoms are the goals.
Objective
A review and analysis from electronic literature databases and subsequent reference searches of the molecular genetic data including pertinent genetic mutations and abnormal biochemical findings causal of AD, are cited. The amyloid cascade hypothesis, the contributions of apolipoprotein E, and hyperphosphorylated tau are discussed as to their roles in pathogenesis. Molecular targets for potential drug and vaccination therapies are cited from a critical assessment of the molecular and biomedical data. These data form the basis for rational, target-specific drug and vaccination therapies currently employed and planned for the near future. Phase 2 and 3 clinical trial results of drug and vaccination therapies are cited.
Conclusions
A new approach is needed as current pharmacologic therapy directed at symptomatic relief has proved to be marginally effective. The genomic and proteomic basis of AD will be defined in the near future, and corresponding molecular therapeutic targets will be identified. Genomic neurology has arrived and its application to resolving AD is our best hope.
INTRODUCTION
The molecular and genetic data related to early-onset Alzheimer disease (AD) causation are in strong support of the amyloid cascade hypothesis. In support of the amyloid cascade hypothesis is that amyloid vaccination therapy in selected patients may have shown slowing of cognitive loss and resolution of amyloid burden in autopsied brain. However, current pharmacologic therapy has been of limited benefit to slow effectively the cognitive loss in patients with AD. It is proposed that a genomic and proteomic analysis may be of value to identify at-risk gene sets whose genetic expression is sufficient to cause late-onset, sporadic AD. This information would provide essential data for the prediction of disease prior to the appearance of clinical signs and symptoms and serve to direct genomic and proteomic research strategies to prevent it.
PATHOGENESIS OF DISEASE
Research in AD is proceeding at a rapid pace. Clinicians now have drugs that marginally ameliorate the cognitive and behavioral symptoms of AD. More effective therapies directed at the biological basis of disease pathogenesis are needed. Advances in knowledge of the molecular and genetic aspects of AD are providing therapeutic targets to attack more directly the molecular processes of disease.1 Much of this knowledge comes from study of familial AD. Although the early-onset autosomal-dominant form of AD clearly results from specific genetic mutations, late-onset sporadic AD, representing 90% of patients, appears to result from multifactorial environmental events and genetic influences, as inheritance of the apolipoprotein E ɛ4 allele and other late-onset acting polymorphic genes.1 The importance of nongenetic factors in AD is underscored by the fact that only one third of identical twins are concordant of the disease.2 The environmental risk factors have remained elusive, but progress in finding genes causal of AD as well as increasing risk for it has been steady and impressive. Mutations in the amyloid precursor protein (APP) gene on chromosome 21q and of the presenilin 1 (PS1) and presenilin 2 (PS2) genes on chromosomes 14q and 1q, respectively, account for approximately one half of early-onset forms of autosomal-dominant inherited disease and about 3% to 5% of AD overall (Table). Additional gene loci that may increase the risk for AD have been identified on chromosomes 9, 10, 12, and 19.
Table.
| Chromosome | Gene Effects | Functional Effects |
|---|---|---|
| 1: aulosomal dominant, early onset | Missense mutations of presenilin 2 gene | Increased synthesis and release of Aβ42 |
| 14: autosomal dominant, early onset | Missense mutations of presenilin 1 gene | Increased synthesis and release of Aβ42 |
| 19: risk factor, late onset | Inheritance of APOE ɛ4 allele | Increased β amyloid aggregation |
| 21; Down syndrome | Reduplication of APP gene | Increased APP processing; plaques and tangles by age 40 years |
| 21: autosomal dominant, early onset | Missense mutations of APP gene | Increased synthesis and release of Aβ40, Aβ42 |
Abbreviations: Aβ40, 40amino acid length β amyloid protein: Aβ42 42 amino acid length β-amyloid protein; APOE, apolipoprotein E; APP, amyloid precursor protein.
Alzheimer disease may be considered as a form of amyloidosis resulting from the abnormal processing and intramembranous proteolysis of APP, a transmembrane protein whose function is unknown. The primary role of altered amyloidogenesis in the causation of AD has been convincingly supported.1–2,12–24 An increased synthesis of β amyloid peptide (Aβ) from APP in the early-onset forms of AD due to APP and PS1 and PS2 mutations is a central point in support of the amyloid hypothesis, which states that increased amyloidogenesis and/or decreased amyloid clearance with increased amyloid fibrillization are primarily causal of the pathogenesis of AD.25–27 Other molecular pathologic abnormalities, such as tangles composed of hyperphosphorylated tau, are of secondary importance. A more cautious view would be to say that amyloid deposition is necessary but not sufficient to cause the dementia of AD. Increased tau deposition facilitates Aβ toxicity.
Amyloid precursor protein processing involves 3 classes of enzymes: α-, β-, and γ-secretase. As shown in Figure 1, APP is first cut enzymatically by γ- or β-secretase. The products of these first cleavages are cut again by γ-secretase, yielding a soluble fragment from the portion of the molecule produced by an α- γ cleavage and a self-aggregating fragment (β amyloid 40–42) from the portion of the molecule produced by the β-γ cleavage.
Figure 1.
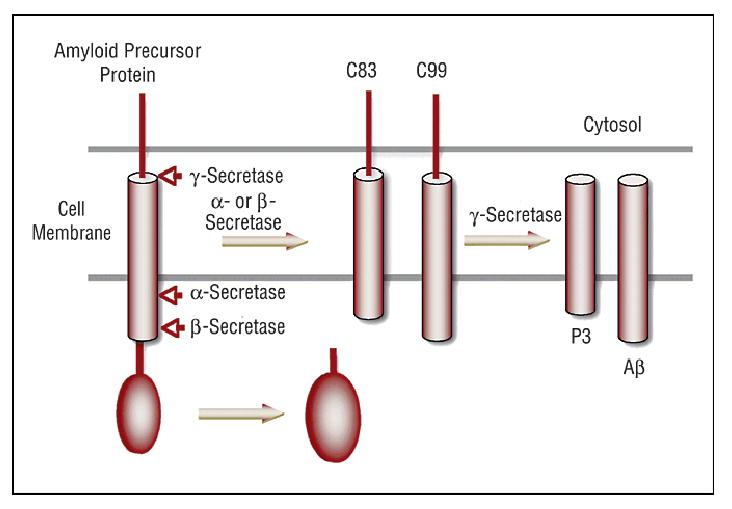
Amyloid precursor protein processing. Amyloid precursor protein is processed by α- or β-secretase to products C83 or C99, respectively. γ-Secretase processes C83 and C99 to P3 and Aβ, respectively. βAmyloid then can polymerize to form oligomers (Aβ)n in the amyloid plaque.
Environmental or other nongenetic factors can activate α secretase in sporadic AD, reducing levels of Aγ .28 The insulin-like degrading enzyme insulysin, neprolysin, and other enzymes as cathepsins are important for β amyloid degradation and clearance.29 A balance between rates of Aβ synthesis and its degradation and removal from brain is a central research issue in late-onset AD. B amyloid increases selectively in brain with AD and not in other organs relates to the fact that β-secretase 2, which produces nonamyloidogenic fragments from APP, is active in nonneural tissue, thus reducing APP as a substrate for β-secretase in nonneural tissue to form Aβ.30
Mutations in the APP, PS1, and PS2 genes causal of AD (Table) have been studied in transgenic mouse lines and have provided important insights into the processes of amyloid deposition.31 The development of amyloid plaques in the APP 717 transgenic mouse depends on the expression of apolipoprotein E.32
AMYLOID PLATELET BIOMARKERS
Amyloid precursor protein processing by platelets in patients with AD is different from that of control subjects.33 There was partial normalization of the mean ratio of the 120- to 130-kD APP isoforms to the 110-kD isoforms in patients treated for 6 weeks with a statin drug, suggesting possible use of this biomarker in clinical trials in evaluating statins and other drugs.34
PROTECTIVE GENES
Just as the apolipoprotein E ɛ2 allele appears to delay the onset of AD, there may be other genetically determined factors that protect against AD. As the genetic degree of Cherokee Indian ancestry increases, the representation of AD decreases.1 Choctaw Native American people have a later age at onset of AD with Mini-Mental Status Examination scores equivalent to those of white people, suggesting again a protective genetic influence. Increased vascular disease is a major factor for adding to the disease burden in the Choctaw patient and the emergence of AD.35 These data indicate that genetic protective and vascular risk factors are involved in a variable manner resulting in AD in the Native American patient.
From the molecular and genetic data reviewed emerges a standard model for AD that includes its central pathogenetic features (Figure 2) (modified from another article1).
Figure 2.

Standard model of Alzheimer disease: a synthesis of pathogenetic mechanisms in support of the amyloid cascade hypothesis.36–37 Aβ indicates βamyloid; APOE, apolipoprotein E; APP, amyloid precursor protein; CSF, cerebrospinal fluid; LRP, lipoprotein receptor-related protein; VLDLR, very low-density lipoprotein receptor.
SYMPTOMATIC THERAPY
CHOLINESTERASE INHIBITORS
Cholinergic basal forebrain neurons that provide cholinergic innervation diffusely throughout the neocortex are especially vulnerable to the neuropathologic process of AD. Loss of cholinergic input due to neuronal forebrain degeneration is thought to be an important contribution to cognitive loss in patients with AD. These observations led to the development of cholinesterase inhibitors to increase acetylcholine levels in brain by inhibiting the enzymes that metabolize it. Four cholinesterase inhibitors have been approved as therapy: tacrine hydrochloride, donepezil hydrochloride, rivastigmine tartrate, and galantamine hydrobromide.
The regimens of the 3 widely used cholinesterase inhibitors follow: donepezil hydrochloride, 5 mg daily at first and then 10 mg daily; rivastigmine tartrate, 1.5 mg twice daily at first and then 6 mg twice daily; galantamine hydrobromide, 4 mg twice daily and then 12 mg twice daily. Cognitive assessments of all 3 cholinesterase inhibitors have shown similar efficacy. There is a statistically significant slowing of cognitive loss using the Alzheimer’s Disease Assessment Scale–Cognitive Subscale, and the 3 cholinesterase inhibitors have similar levels of response.38
However, in a comprehensive assessment of donepezil by the AD 2000 consortium in 2004, it was found that donepezil produced no measurable reduction in rate of institutionalization or progress of disability. It was not found to be cost-effective because donepezil did not delay institutionalization sufficiently to offset the cost of the medication. The AD 2000 consortium found no evidence that costs of caring for patients with AD in the community are reduced by donepezil.39
Presented at the Ninth International Conference on Alzheimer Disease and Related Diseases (July 2004), a study reported on the effects of donepezil compared with placebo to delay the time that patients with mild cognitive impairment developed AD. Seven hundred ninety patients were randomized, and 769 patients had initial, baseline evaluations. Patients were followed up for 3 years and about 30% dropped out prior to the end of the study. The important finding was that donepezil appeared to delay the onset of AD for about 6 months after which there were no significant differences between donepezil and placebo groups. Clearly, donepezil had positive effects on overall cognition, memory, and language tests related to those of the placebo group.40
These studies, in general, indicate that donepezil reduces memory and cognitive loss during the prodromal or early phase of AD for several months.38–40 Behavioral and psychological symptoms associated with mild to moderate AD improved by 6.2 points on the Neuropsychiatric Inventory for patients receiving 10 mg of donepezil daily for 12 weeks compared with patients receiving placebo in a recently completed study in 2004.41 Cholinesterase inhibitor therapy provides clinical benefit to patients in the early phases of AD by slowing the rate of cognitive and memory abilities, and it is recommended for appropriate patients.
MEMANTINE
The Food and Drug Administration has approved memantine, an N-methyl-D-aspartate antagonist, for treatment of moderately advanced AD. It is proposed that its mechanism of action is to reduce potential glutamatergic excitotoxicity.
A study of memantine in moderate to severe AD showed its effectiveness in the Activities of Daily Living Inventory and the Severe Impairment Battery but not the Global Deterioration Scale as compared with placebo.42
It is prescribed at a dose of 5 mg daily and increased to 5 mg twice daily and then to 10 mg twice daily. In patients with moderate to severe AD receiving stable doses of donepezil, memantine resulted in significantly better outcomes than placebo on measures of cognition, activities of daily living, global outcome, and behavior and was well tolerated. It is suggested by these positive data that memantine offers a new approach for therapy of patients with more advanced disease43 and may be administered together with cholinesterase inhibitor therapy in appropriate patients.
HORMONE REPLACEMENT THERAPY
The Women’s Health Initiative Study of estrogen and medroxyprogesterone demonstrated an increased occurrence of dementia in postmenopausal women.44 Estrogen therapy alone in postmenopausal women did not reduce dementia or incidence of mild cognitive impairment and increased risk for both conditions combined.45 Further for women aged 65 years or older, estrogen therapy had an adverse effect on cognition.46 It is now clear that hormone replacement therapy has adverse effects, increasing the risk for dementia and impaired cognition. Use of hormone therapy to prevent dementia or cognitive decline in women 65 years of age or older is not recommended.44–46
ANTIOXIDANTS
One significant study is a randomized, placebo-controlled clinical trial using selegiline (N,2-dimethyl-N-2 propynyl phenethylamine hydrochloride, an irreversible inhibitor of monoamine oxidase B), vitamin E, and both agents together compared with placebo in patients with AD. The one positive result was that the time to nursing home placement, the time to death, and the time to severe dementia were extended in selegiline, in vitamin E, and in combined groups compared with patients receiving placebo.47 Vitamin E at 2000 international units a day was recommended based as a modest means to slow the progress of the disease.47
However, the Cochrane Dementia Group Register of Clinical Trials was searched for effectiveness of vitamin E in AD in 2000. It was concluded that there is insufficient evidence of efficacy of vitamin E in the treatment of AD.48
Use of vitamin E and vitamin C supplements in combination in the Cache County Study was associated with reduced prevalence and incidence of AD and thus merits further study for the primary prevention of AD.49
ANTI-INFLAMMATORY DRUGS
Increased acute phase reactants including cytokines and interleukins and minor signs of cellular inflammation in patients with AD have resulted in clinical trials to study the effectiveness of anti-inflammatory drugs. Naproxen (a cyclooxygenase-1 and -2 inhibitor) and rofecoxib (a cyclooxygenase-2 inhibitor) have been studied. A multicenter, randomized double-blind, placebo-controlled parallel group trial with 1-year exposure to rofecoxib or naproxen was conducted with patients with mild to moderate AD. The primary outcome measure was the 1-year change in the Alzheimer Disease Assessment Scale–Cognitive Subscale score. The results of this study indicated that rofecoxib or low-dose naproxen did not slow cognitive decline.50 These drugs do not reduce the rate of cognitive loss nor functional decline in patients with AD.50 A prospective, placebo-controlled, double-blind prevention trial with an anti-inflammatory agent has not been completed.
CHOLESTEROL LOWERING THERAPY
Statins, 3-hydroxy-3-methylglutaryl coenzyme A (HMG CoA) reductase inhibitors, may prevent AD. Evidence for a protective effect of HMG CoA reductase inhibitors has been provided by comparing the prevalence of the diagnosis of AD in 3 groups of individuals from Veterans Administration hospital records. It was found that the cohort of individuals receiving HMG CoA reductase inhibiting drugs had a prevalence 60% to 73% lower than that of either the total patient population or patients taking other medications typically used to treat hypertension or cardiovascular disease.51 In another retrospective study, persons aged 50 years or older who were prescribed HMG CoA reductase inhibitors had substantially lower risk of developing dementia, independent of the presence or absence of untreated hyperlipidemia or exposure to non–HMG CoA reductase lipid-lowering agents. The adjusted risk ratio for those receiving HMG CoA reductase inhibitors was 0.29 (confidence interval, 0.13–0.63; P = .002).52 These data from retrospective medical-record reviews need to be validated with prospective, double-blind, placebo-controlled trials with statins.
Recently, a prospective, randomized, dose-finding, 36-week treatment trial with statins (simvastatin or atorvastatin) was conducted with 39 patients with hypercholesterolemia. Plasma levels of Aβ40 and Aβ42 were measured. Both statins reduced total plasma cholesterol levels by 56%, but levels of Aβ40, Aβ42, and total Aβ were unchanged. Thus, this study does not support the effect of statins on altering the processing of APP in humans.53
The Heart Protection Study Collaborative Group54 and the Prospective Study of Pravastatin in the Elderly at Risk (PROSPER)55 have both recently reported that neither simvastatin nor pravastatin appeared to slow cognitive decline in the elderly during 5 years of treatment in the Heart Protection Study and 3.2 years in the PROSPER. These findings suggest that lower rates of dementia found in the medical record reviews51–52 among persons taking statins may have been due to other factors that were actually responsible for the lower risks.56
VACCINATION
Monthly immunization for 11 months with injections of Aβ42 was found to prevent the development of Aβ plaque formation, neuritic dystrophy, and astrogliosis in mice transfected with the V717F mutant human APP gene.57 Partial regression and clearance of plaques were seen in animals that had already developed them. Substantial reduction of reactive gliosis also occurred.
Based on the impressive reduction in brain amyloid burden in the immunized transgenic mice, safety and efficacy trials of the injectable vaccine were initiated in patients with AD. It was shown that patients immunized with a primary injection of preaggregate Aβ42 followed by 1 booster injection in a placebo-controlled study did generate antibodies that recognized Aβ plaques, diffuse Aβ deposits, and vascular Aβ in brain blood vessels. Thus, Aβ42 vaccination in patients with AD induces antibodies that have a high degree of selectivity for the pathogenic target structures.58
Some patients who generated such antibodies showed slower rates of decline of cognitive functions and activities of daily living.59–60 The Aβ42 immunization was apparently having a positive therapeutic effect, but unfortunately, the clinical trial had to be terminated because 6% of immunized patients developed autoimmune meningoencephalitis.61 The brains of 4 people with mild to moderate AD who had been vaccinated and died from unrelated causes were examined. Each brain showed an almost complete lack of Aβ with maintenance of neurofibrillary tangles.62–63 The vaccine targeted normal myelin in addition to Aβ as some patients developed acute demyelinating lesions.61–62,64 The specificity of the Aβ peptide vaccine must be reviewed because the role of microglial nonspecific activation by any vaccine reducing the accumulation of Aβ remains an unresolved issue.
A program using passive immunization with preformed Aβ antibodies is currently being developed to prevent secondary encephalitis. Genetic vaccination using the complementary DNA for Aβ in transgenic mice generated antibodies against Aβ without activating cytotoxic T cells causal of autoimmune meningoencephalitis and thus might be a form of active immunization to develop for future clinical trials.65 Immunization therapy for AD should be viewed optimistically because it has shown a positive response in lowering the Aβ burden in selected patients with AD. The significant adverse effects need to be reviewed carefully and new immune strategies designed, including active immunization with the Aβ gene, which shows some promise.64–65
SECRETASE INHIBITORS
Of great interest are the reports1 of the cloning, isolation, and characterization of the membrane-bound aspartyl protease that cleaves full-length APP at the β-secretase cleavage site and finds it to be the predominant β-cleavage activity in human brain. Overexpression of the β-secretase gene increased the amount of β-secretase cleavage products. Demonstration of the specific A β site APP-cleaving enzyme provides a direct approach to develop pharmacologic agents that can specifically inhibit this gene’s expression. γ-secretase inhibition by a specific inhibitor is also a potential target to inhibit Aβ synthesis. Inhibition of β- or γ-secretase would prevent Aβ synthesis and affect the rate of plaque production.1 Efforts to achieve a clinically useful secretase inhibitor form an area of active research.
CLIOQUINOL
Metal chelation using clioquinol has been reported in a pilot study with 36 patients with AD to reduce the rate of cognitive loss in a double-blind, placebo-controlled, phase 2 clinical trial. Clioquinol’s effect in this preliminary study is due to its ability to chelate zinc and copper associated with amyloid plaques. The mobilization and removal of brain amyloid is believed to be basis of its therapeutic effect.66 Clioquinol increased serum zinc and copper levels, which is explained only in part by the chelation model.
INSULYSIN
It has been recently demonstrated that the peptidase insulysin has a quantitatively significant and rate-limiting role in degrading brain Aβ peptides in mice in vivo. Because insulysin also has a prominent role in insulin degradation, decreased or aberrant insulysin expression is not only a likely risk factor for AD but also a reasonable mechanism to explain a high incidence of abnormalities in glucose and insulin metabolism in AD. Insulysin activation is a possible therapeutic approach to reduce brain levels of Aβ.29
Cholinergic basal forebrain neurons that provide cholinergic innervation diffusely throughout the neocortex are especially vulnerable to the neuropathologic process of AD. Loss of cholinergic input due to neuronal forebrain degeneration is thought to be an important contribution to cognitive loss in patients with AD. These observations led to the development of cholinesterase inhibitors to increase acetylcholine levels in brain by inhibiting the enzymes that metabolize it. Four cholinesterase inhibitors have been approved as therapy: tacrine hydrochloride, donepezil hydrochloride, rivastigmine tartrate, and galantamine hydrobromide.
The regimens of the 3 widely used cholinesterase inhibitors follow: donepezil hydrochloride, 5 mg daily at first and then 10 mg daily; rivastigmine tartrate, 1.5 mg twice daily at first and then 6 mg twice daily; galantamine hydrobromide, 4 mg twice daily and then 12 mg twice daily. Cognitive assessments of all 3 cholinesterase inhibitors have shown similar efficacy. There is a statistically significant slowing of cognitive loss using the Alzheimer’s Disease Assessment Scale–Cognitive Subscale, and the 3 cholinesterase inhibitors have similar levels of response.38
However, in a comprehensive assessment of donepezil by the AD 2000 consortium in 2004, it was found that donepezil produced no measurable reduction in rate of institutionalization or progress of disability. It was not found to be cost-effective because donepezil did not delay institutionalization sufficiently to offset the cost of the medication. The AD 2000 consortium found no evidence that costs of caring for patients with AD in the community are reduced by donepezil.39
Presented at the Ninth International Conference on Alzheimer Disease and Related Diseases (July 2004), a study reported on the effects of donepezil compared with placebo to delay the time that patients with mild cognitive impairment developed AD. Seven hundred ninety patients were randomized, and 769 patients had initial, baseline evaluations. Patients were followed up for 3 years and about 30% dropped out prior to the end of the study. The important finding was that donepezil appeared to delay the onset of AD for about 6 months after which there were no significant differences between donepezil and placebo groups. Clearly, donepezil had positive effects on overall cognition, memory, and language tests related to those of the placebo group.40
These studies, in general, indicate that donepezil reduces memory and cognitive loss during the prodromal or early phase of AD for several months.38–40 Behavioral and psychological symptoms associated with mild to moderate AD improved by 6.2 points on the Neuropsychiatric Inventory for patients receiving 10 mg of donepezil daily for 12 weeks compared with patients receiving placebo in a recently completed study in 2004.41 Cholinesterase inhibitor therapy provides clinical benefit to patients in the early phases of AD by slowing the rate of cognitive and memory abilities, and it is recommended for appropriate patients.
MEMANTINE
The Food and Drug Administration has approved memantine, an N-methyl-D-aspartate antagonist, for treatment of moderately advanced AD. It is proposed that its mechanism of action is to reduce potential glutamatergic excitotoxicity.
A study of memantine in moderate to severe AD showed its effectiveness in the Activities of Daily Living Inventory and the Severe Impairment Battery but not the Global Deterioration Scale as compared with placebo.42
It is prescribed at a dose of 5 mg daily and increased to 5 mg twice daily and then to 10 mg twice daily. In patients with moderate to severe AD receiving stable doses of donepezil, memantine resulted in significantly better outcomes than placebo on measures of cognition, activities of daily living, global outcome, and behavior and was well tolerated. It is suggested by these positive data that memantine offers a new approach for therapy of patients with more advanced disease43 and may be administered together with cholinesterase inhibitor therapy in appropriate patients.
HORMONE REPLACEMENT THERAPY
The Women’s Health Initiative Study of estrogen and medroxyprogesterone demonstrated an increased occurrence of dementia in postmenopausal women.44 Estrogen therapy alone in postmenopausal women did not reduce dementia or incidence of mild cognitive impairment and increased risk for both conditions combined.45 Further for women aged 65 years or older, estrogen therapy had an adverse effect on cognition.46 It is now clear that hormone replacement therapy has adverse effects, increasing the risk for dementia and impaired cognition. Use of hormone therapy to prevent dementia or cognitive decline in women 65 years of age or older is not recommended.44–46
ANTIOXIDANTS
One significant study is a randomized, placebo-controlled clinical trial using selegiline (N,2-dimethyl-N-2 propynyl phenethylamine hydrochloride, an irreversible inhibitor of monoamine oxidase B), vitamin E, and both agents together compared with placebo in patients with AD. The one positive result was that the time to nursing home placement, the time to death, and the time to severe dementia were extended in selegiline, in vitamin E, and in combined groups compared with patients receiving placebo.47 Vitamin E at 2000 international units a day was recommended based as a modest means to slow the progress of the disease.47
However, the Cochrane Dementia Group Register of Clinical Trials was searched for effectiveness of vitamin E in AD in 2000. It was concluded that there is insufficient evidence of efficacy of vitamin E in the treatment of AD.48
Use of vitamin E and vitamin C supplements in combination in the Cache County Study was associated with reduced prevalence and incidence of AD and thus merits further study for the primary prevention of AD.49
ANTI-INFLAMMATORY DRUGS
Increased acute phase reactants including cytokines and interleukins and minor signs of cellular inflammation in patients with AD have resulted in clinical trials to study the effectiveness of anti-inflammatory drugs. Naproxen (a cyclooxygenase-1 and -2 inhibitor) and rofecoxib (a cyclooxygenase-2 inhibitor) have been studied. A multicenter, randomized double-blind, placebo-controlled parallel group trial with 1-year exposure to rofecoxib or naproxen was conducted with patients with mild to moderate AD. The primary outcome measure was the 1-year change in the Alzheimer Disease Assessment Scale–Cognitive Subscale score. The results of this study indicated that rofecoxib or low-dose naproxen did not slow cognitive decline.50 These drugs do not reduce the rate of cognitive loss nor functional decline in patients with AD.50 A prospective, placebo-controlled, double-blind prevention trial with an anti-inflammatory agent has not been completed.
CHOLESTEROL LOWERING THERAPY
Statins, 3-hydroxy-3-methylglutaryl coenzyme A (HMG CoA) reductase inhibitors, may prevent AD. Evidence for a protective effect of HMG CoA reductase inhibitors has been provided by comparing the prevalence of the diagnosis of AD in 3 groups of individuals from Veterans Administration hospital records. It was found that the cohort of individuals receiving HMG CoA reductase inhibiting drugs had a prevalence 60% to 73% lower than that of either the total patient population or patients taking other medications typically used to treat hypertension or cardiovascular disease.51 In another retrospective study, persons aged 50 years or older who were prescribed HMG CoA reductase inhibitors had substantially lower risk of developing dementia, independent of the presence or absence of untreated hyperlipidemia or exposure to non–HMG CoA reductase lipid-lowering agents. The adjusted risk ratio for those receiving HMG CoA reductase inhibitors was 0.29 (confidence interval, 0.13–0.63; P = .002).52 These data from retrospective medical-record reviews need to be validated with prospective, double-blind, placebo-controlled trials with statins.
Recently, a prospective, randomized, dose-finding, 36-week treatment trial with statins (simvastatin or atorvastatin) was conducted with 39 patients with hypercholesterolemia. Plasma levels of Aβ40 and Aβ42 were measured. Both statins reduced total plasma cholesterol levels by 56%, but levels of Aβ40, Aβ42, and total Aβ were unchanged. Thus, this study does not support the effect of statins on altering the processing of APP in humans.53
The Heart Protection Study Collaborative Group54 and the Prospective Study of Pravastatin in the Elderly at Risk (PROSPER)55 have both recently reported that neither simvastatin nor pravastatin appeared to slow cognitive decline in the elderly during 5 years of treatment in the Heart Protection Study and 3.2 years in the PROSPER. These findings suggest that lower rates of dementia found in the medical record reviews51–52 among persons taking statins may have been due to other factors that were actually responsible for the lower risks.56
VACCINATION
Monthly immunization for 11 months with injections of Aβ42 was found to prevent the development of Aβ plaque formation, neuritic dystrophy, and astrogliosis in mice transfected with the V717F mutant human APP gene.57 Partial regression and clearance of plaques were seen in animals that had already developed them. Substantial reduction of reactive gliosis also occurred.
Based on the impressive reduction in brain amyloid burden in the immunized transgenic mice, safety and efficacy trials of the injectable vaccine were initiated in patients with AD. It was shown that patients immunized with a primary injection of preaggregate Aβ42 followed by 1 booster injection in a placebo-controlled study did generate antibodies that recognized Aβ plaques, diffuse Aβ deposits, and vascular Aβ in brain blood vessels. Thus, Aβ42 vaccination in patients with AD induces antibodies that have a high degree of selectivity for the pathogenic target structures.58
Some patients who generated such antibodies showed slower rates of decline of cognitive functions and activities of daily living.59–60 The Aβ42 immunization was apparently having a positive therapeutic effect, but unfortunately, the clinical trial had to be terminated because 6% of immunized patients developed autoimmune meningoencephalitis.61 The brains of 4 people with mild to moderate AD who had been vaccinated and died from unrelated causes were examined. Each brain showed an almost complete lack of Aβ with maintenance of neurofibrillary tangles.62–63 The vaccine targeted normal myelin in addition to Aβ as some patients developed acute demyelinating lesions.61–62,64 The specificity of the Aβ peptide vaccine must be reviewed because the role of microglial nonspecific activation by any vaccine reducing the accumulation of Aβ remains an unresolved issue.
A program using passive immunization with preformed Aβ antibodies is currently being developed to prevent secondary encephalitis. Genetic vaccination using the complementary DNA for Aβ in transgenic mice generated antibodies against Aβ without activating cytotoxic T cells causal of autoimmune meningoencephalitis and thus might be a form of active immunization to develop for future clinical trials.65 Immunization therapy for AD should be viewed optimistically because it has shown a positive response in lowering the Aβ burden in selected patients with AD. The significant adverse effects need to be reviewed carefully and new immune strategies designed, including active immunization with the Aβ gene, which shows some promise.64–65
SECRETASE INHIBITORS
Of great interest are the reports1 of the cloning, isolation, and characterization of the membrane-bound aspartyl protease that cleaves full-length APP at the β-secretase cleavage site and finds it to be the predominant β-cleavage activity in human brain. Overexpression of the β-secretase gene increased the amount of β-secretase cleavage products. Demonstration of the specific βsite APP-cleaving enzyme provides a direct approach to develop pharmacologic agents that can specifically inhibit this gene’s expression. γ-secretase inhibition by a specific inhibitor is also a potential target to inhibit Aβ synthesis. Inhibition of β- or γ-secretase would prevent Aβ synthesis and affect the rate of plaque production.1 Efforts to achieve a clinically useful secretase inhibitor form an area of active research.
CLIOQUINOL
Metal chelation using clioquinol has been reported in a pilot study with 36 patients with AD to reduce the rate of cognitive loss in a double-blind, placebo-controlled, phase 2 clinical trial. Clioquinol’s effect in this preliminary study is due to its ability to chelate zinc and copper associated with amyloid plaques. The mobilization and removal of brain amyloid is believed to be basis of its therapeutic effect.66 Clioquinol increased serum zinc and copper levels, which is explained only in part by the chelation model.
INSULYSIN
It has been recently demonstrated that the peptidase insulysin has a quantitatively significant and rate-limiting role in degrading brain Aβ peptides in mice in vivo. Because insulysin also has a prominent role in insulin degradation, decreased or aberrant insulysin expression is not only a likely risk factor for AD but also a reasonable mechanism to explain a high incidence of abnormalities in glucose and insulin metabolism in AD. Insulysin activation is a possible therapeutic approach to reduce brain levels of Aβ.29
CONCLUSIONS
It is clear from the molecular and genetic data that AD is a clinical syndrome with a common set of clinical features due to several genotypes causal of early-onset autosomal-dominant disease (APP, PS1, and PS2 mutations) or, more significantly, in the vast majority of patients with late-onset disease, due to the complex interaction of polygenetic influences and environmental risk factors. The present view is that a subset of risk-producing polymorphic genes are expressed and result in the neuropathologic abnormalities and clinical dementia of AD. The future resolution of its molecular pathogenesis and subsequent pharmacologic therapies will depend on a genomic and proteomic analysis of patients, their nonaffected family members, and control subjects. Genomic and proteomic profiling as shown on DNA and protein microarrays, it is believed, will show a pattern of expression that correlates with a high-risk state for subsequent AD. Genomic and proteomic analyses for prediction and prevention of AD may supplant current clinical diagnosis and symptomatic treatment.
Pharmacogenomic therapy designed to prevent progression of preclinical to overt clinical disease with dementia based on the genomic or proteomic profile of the individual patient is the intent and hope. It is premature to predict exactly what therapeutic effect will result from a genomic/proteomic approach. Gene linkage and sequencing studies for finding at-risk genes for AD have found some promising leads, including insulysin (chromosome 10) and possibly α2-macroglobulin (chromosome 12). A recent genomic analysis suggested that patients with AD show about 3 to 5 times higher genetic variation than a control population.67 There are data indicating that the therapeutic response in patients with AD to cholinesterase inhibitors is genotype specific.68 A growing view is that genomics provide the potential to offer insights into the molecular and genetic basis of pathogenesis of complex diseases like AD.68–70 This approach may eventually prove to be productive in finding a cohort of genes and expressed products, relatively small in number, whose polymorphic profile will identify high-risk states for AD. In this regard, a proteomic analysis of AD brains has shown quantitative differences in the expression of proteins in 6 areas of brain. The molecular identity of 37 proteins with significantly altered expression was determined.70 Identification of altered expression of genes and proteins primarily causal of AD from those showing secondary and reactive change is the challenge. When this goal is achieved, it will be possible to consider pharmacogenomic therapy at an early point in the disease process with careful clinical diagnostic criteria for AD before irreversible neuropathologic changes result.71
A new approach is needed as current pharmacologic therapy directed at symptomatic relief has proved to be marginally effective. The genomic and proteomic basis of AD will be defined in the near future, and corresponding molecular therapeutic targets will be identified. Genomic neurology has arrived and its application to resolving AD is our best hope.30
Footnotes
Funding/Support: This study was supported by grant P30AG12300-12 for the Alzheimer’s Disease Center, University of Texas Southwestern Medical Center at Dallas (Dallas), from the National Institute on Aging (Bethesda, Md) and grant U01 AG16976 from the National Alzheimer’s Coordinating Center (Seattle, Wash). Dr Rosenberg has received educational grants from Pfizer, New York, NY; Novartis, Basel, Switzerland; Janssen Pharmaceutica, Beerse, Belgium; Eisai, Inc, Teaneck, NJ; and Forest Laboratories, New York. He has been a consultant and a participant in a clinical trial for Elan, Dublin, Ireland.
References
- 1.Rosenberg RN. The molecular and genetic basis of AD: the end of the beginning: the 2000 Wartenberg Lecture. Neurology. 2000;54:2045–2054. doi: 10.1212/wnl.54.11.2045. [DOI] [PubMed] [Google Scholar]
- 2.Jin YP, Gatz M, Johansson B, Pedersen N. Sensitivity and specificity of dementia coding in two Swedish disease registries. Neurology. 2004;63:739–741. doi: 10.1212/01.wnl.0000134604.48018.97. [DOI] [PubMed] [Google Scholar]
- 3.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 4.Mullan M, Crawford F, Axelman K, Houlden H, Lilius L, Winblad B, Lannfelt L. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet. 1992;1:345–347. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- 5.St George-Hyslop PH, Tanzi RE, Polinsky RJ, Haines JL, Nee L, Watkins PC, Myers RH, Feldman RG, Pollen D, Drachman D. The genetic defect causing familial Alzheimer’s disease maps on chromosome 21. Science. 1987;235:885–890. doi: 10.1126/science.2880399. [DOI] [PubMed] [Google Scholar]
- 6.Zabar Y, Kawas C. Epidemiology and clinical genetics of Alzheimer disease. In: Clark CM, Trojanowski JQ, eds. Neurodegenerative Dementias New York, NY: McGraw-Hill; 2000:79–94.
- 7.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 8.Bird TD, Lampe TH, Nemens EJ, Miner GW, Sumi SM, Schellenberg GD. Familial Alzheimer’s disease in American descendants of the Volga Germans: probable genetic founder effect. Ann Neurol. 1988;23:25–31. doi: 10.1002/ana.410230106. [DOI] [PubMed] [Google Scholar]
- 9.Corder E, Saunders A, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Genetic dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 10.Saunders AM, Strittmatter WJ, Schmechel D, St George-Hyslop PH, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-MacLachlan DR, Alberts MJ. Association of apolipoprotein E allele ɛ4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 11.Weiner MF, Vega G, Risser RC, Honig LS, Cullum CM, Crumpacker D, Rosenberg RN. Apolipoprotein E epsilon 4, other risk factors, and course of Alzheimer’s disease. Biol Psychiatry. 1999;45:633–638. doi: 10.1016/s0006-3223(98)00222-4. [DOI] [PubMed] [Google Scholar]
- 12.Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 13.Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- 14.Dickson DW, Lee SC, Mattiace LA, Yen SH, Brosnan C. Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer’s disease. Glia. 1993;7:75–83. doi: 10.1002/glia.440070113. [DOI] [PubMed] [Google Scholar]
- 15.Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW, Mrak RE. Glial-neuronal interactions in Alzheimer’s disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol. 1998;8:65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Markesbery WR. The role of oxidative stress in Alzheimer disease. Arch Neurol. 1999;56:1449–1452. doi: 10.1001/archneur.56.12.1449. [DOI] [PubMed] [Google Scholar]
- 17.Selkoe DJ. Molecular pathology of Alzheimer’s disease: the role of amyloid. In: Growdon JH, Rossor MN, eds. Blue Books of Practical Neurology: The Dementias London, England: Butterworth-Heinemann, Inc; 1998:257–283.
- 18.Selkoe DJ. Alzheimer’s disease: genotypes, phenotypes, and treatments. Science. 1997;275:630–631. doi: 10.1126/science.275.5300.630. [DOI] [PubMed] [Google Scholar]
- 19.Glenner GG, Wong CW. Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;122:1131–1135. doi: 10.1016/0006-291x(84)91209-9. [DOI] [PubMed] [Google Scholar]
- 20.Goldgaber D, Lerman MI, McBride OW, Saffriotti U, Gajdusek DC. Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer’s disease. Science. 1987;235:877–880. doi: 10.1126/science.3810169. [DOI] [PubMed] [Google Scholar]
- 21.Tanzi RE, Gusella JF, Watkins PC, Bruns GA, St George-Hyslop P, Van Keuren ML, Patterson D, Pagan S, Kurnit DM, Neve RL. Amyloid beta protein gene: cDNA MRNA distribution, and genetic linkage near the Alzheimer locus. Science. 1987;235:880–884. doi: 10.1126/science.2949367. [DOI] [PubMed] [Google Scholar]
- 22.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kowal NW, Beal MF, Busciglio J, Duffy LK, Yankner BA. An in-vivo model for the neurodegenerative effects of βamyloid and protection by substance P. Proc Natl Acad Sci U S A. 1991;88:7247–7251. doi: 10.1073/pnas.88.16.7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geula C, Wu CK, Saroff D, Lorenzo A, Yuan M, Yankner BA. Aging renders the brain vulnerable to amyloid β-protein neurotoxicity. Nature Med. 1998;4:827–831. doi: 10.1038/nm0798-827. [DOI] [PubMed] [Google Scholar]
- 25.Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, Song YQ, Rogaeva E, Chen F, Kawarai T, Supala A, Levesque L, Yu H, Yang DS, Holmes E, Milman P, Liang Y, Zhang DM, Xu DH, Sato C, Rogaev E, Smith M, Janus C, Zhang Y, Aebersold R, Farrer LS, Sorbi S, Bruni A, Fraser P, St George-Hyslop P. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature. 2000;407:48–54. doi: 10.1038/35024009. [DOI] [PubMed] [Google Scholar]
- 26.Tanzi RE, Kovacs DM, Kim TW, Moir RD, Guenette SY, Wasco W. The gene defects responsible for familial Alzheimer’s disease. Neurobiol Dis. 1996;3:159–168. doi: 10.1006/nbdi.1996.0016. [DOI] [PubMed] [Google Scholar]
- 27.Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- 28.Baskin F, Rosenberg R, Davis R. Morphological differentiation and proteoglycan synthesis regulate Alzheimer amyloid precursor protein processing in PC12 and human astrocyte cultures. J Neurosci Res. 1992;32:274–279. doi: 10.1002/jnr.490320217. [DOI] [PubMed] [Google Scholar]
- 29.Miller BC, Eckman EA, Sambamurti K, Dobbs N, Chow KM, Eckman CB, Hersh LB, Thiele DL. Amyloid-beta peptide levels in brain are inversely correlated with insulysin activity levels in vivo. Proc Natl Acad Sci U S A. 2003;100:6221–6226. doi: 10.1073/pnas.1031520100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosenberg RN. Genomic neurology: a new beginning. Arch Neurol. 2001;58:1739–1741. doi: 10.1001/archneur.58.11.1739. [DOI] [PubMed] [Google Scholar]
- 31.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 32.Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, McKeel D, Wozniak D, Paul SM. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2000;97:2892–2897. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosenberg RN, Baskin F, Fosmire JA, Risser R, Adams P, Svetlik D, Honig LS, Cullum CM, Weiner MF. Altered amyloid protein processing in platelets of patients with Alzheimer disease. Arch Neurol. 1997;54:139–144. doi: 10.1001/archneur.1997.00550140019007. [DOI] [PubMed] [Google Scholar]
- 34.Baskin F, Rosenberg RN, Fang X, Hynan LS, Moore CB, Weiner M, Vega GL. Correlation of statin-increased platelet APP ratios and reduced blood lipids in AD patients. Neurology. 2003;60:2006–2007. doi: 10.1212/01.wnl.0000068029.56740.96. [DOI] [PubMed] [Google Scholar]
- 35.Weiner MF, Rosenberg RN, Svetlik D, Hynan LS, Womack KB, White C, III, Good S, Fuller C, Wharton D, Richter R. Comparison of Alzheimer’s disease in Native Americans and Whites. Int Psychogeriatr. 2003;15:367–375. doi: 10.1017/s104161020300961x. [DOI] [PubMed] [Google Scholar]
- 36.Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J. Reeler/disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell. 1999;97:689–701. doi: 10.1016/s0092-8674(00)80782-5. [DOI] [PubMed] [Google Scholar]
- 37.Beffert U, Arguin C, Poirier J. The polymorphism in exon 3 of the low-density lipoprotein receptor-related protein gene is weakly associated with Alzheimer’s disease. Neurosci Lett. 1999;259:29–32. doi: 10.1016/s0304-3940(98)00888-x. [DOI] [PubMed] [Google Scholar]
- 38.Cummings JL. Alzheimer’s disease . N Engl J Med. 2004;351:56–67. doi: 10.1056/NEJMra040223. [DOI] [PubMed] [Google Scholar]
- 39.Courtney C, Farrell D, Gray R, Hills R, Lynch L, Sellwood E, Edwards S, Hardyman W, Raftery J, Crome P, Lendon C, Shaw H, Bentham P AD2000 Collaborative Group. Long-term donepezil treatment in 565 patients with Alzheimer’s disease (AD2000): randomised double-blind trial. Lancet. 2004;363:2105–2115. doi: 10.1016/S0140-6736(04)16499-4. [DOI] [PubMed] [Google Scholar]
- 40.Petersen R, Grundman M, Thomas R, Thal L. Donepezil and vitamin E as treatments for mild cognitive impairment [abstract]. Presented at: Ninth International Conference on Alzheimer’s Disease and Related Disorders; July 17–22, 2004; Philadelphia, Pa.
- 41.Holmes C, Wilkinson D, Dean C, Vethanayagam S, Olivieri S, Langley A, Pandita-Gunawardena N, Hogg F, Clare C, Damms J. The efficacy of donepezil in the treatment of neuropsychiatric symptoms in Alzheimer disease. Neurology. 2004;63:214–219. doi: 10.1212/01.wnl.0000129990.32253.7b. [DOI] [PubMed] [Google Scholar]
- 42.Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ. Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med. 2003;348:1333–1341. doi: 10.1056/NEJMoa013128. [DOI] [PubMed] [Google Scholar]
- 43.Tariot PN, Farlow MR, Grossberg GT, Graham SM, McDonald S, Gergel I. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291:317–324. doi: 10.1001/jama.291.3.317. [DOI] [PubMed] [Google Scholar]
- 44.Shumaker SA, Legault C, Rapp SR, Thal L, Wallace RB, Ockene JK, Hendrix SL, Jones BN, III, Assaf AR, Jackson RD, Kotchen JM, Wassertheil-Smoller S, Wactawski-Wende J. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: the Women’s Health Initiative Memory Study: a randomized controlled trial. JAMA. 2003;289:2651–2662. doi: 10.1001/jama.289.20.2651. [DOI] [PubMed] [Google Scholar]
- 45.Shumaker SA, Legault C, Kuller L, Rapp SR, Thal L, Lane DS, Fillit H, Stefanick ML, Hendrix SL, Lewis CE, Masaki K, Coker LH. Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women’s Health Initiative Memory Study. JAMA. 2004;291:2947–2958. doi: 10.1001/jama.291.24.2947. [DOI] [PubMed] [Google Scholar]
- 46.Espeland MA, Rapp SR, Shumaker SA, Brunner R, Manson JE, Sherwin BB, Hsia J, Margolis KL, Hogan PE, Wallace R, Dailey M, Freeman R, Hays J. Conjugated equine estrogens and global cognitive function in postmenopausal women: Women’s Health Initiative Memory Study. JAMA. 2004;291:2959–2968. doi: 10.1001/jama.291.24.2959. [DOI] [PubMed] [Google Scholar]
- 47.Sano M, Ernesto C, Thomas RG, Klauber MR, Schafer K, Grundman M, Woodbury P, Growdon J, Cotman CW, Pfeiffer E, Schneider LS, Thal LJ. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease: the Alzheimer’s Disease Cooperative Study. N Engl J Med. 1997;336:1216–1222. doi: 10.1056/NEJM199704243361704. [DOI] [PubMed] [Google Scholar]
- 48.Tabet N, Birks J, Grimley Evans J. Vitamin E for Alzheimer’s disease. Cochrane Database Syst Rev. 2000;(4):CD002854. doi: 10.1002/14651858.CD002854. [DOI] [PubMed] [Google Scholar]
- 49.Zandi PP, Anthony JC, Khachaturian AS, Stone SV, Gustafson D, Tschanz JT, Norton MC, Welsh-Bohmer KA, Breitner JC Cache County Study Group. Reduced risk of Alzheimer disease in users of antioxidant vitamin supplements. Arch Neurol. 2004;61:82–88. doi: 10.1001/archneur.61.1.82. [DOI] [PubMed] [Google Scholar]
- 50.Aisen PS, Schafer KA, Grundman M, Pfeiffer E, Sano M, Davis KL, Farlow MR, Jin S, Thomas RJ, Thal LJ Alzheimer’s Disease Cooperative Study. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled study. JAMA. 2003;289:2819–2826. doi: 10.1001/jama.289.21.2819. [DOI] [PubMed] [Google Scholar]
- 51.Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol. 2000;57:1439–1443. doi: 10.1001/archneur.57.10.1439. [DOI] [PubMed] [Google Scholar]
- 52.Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA. Statins and the risk of dementia. Lancet. 2000;356:1627–1631. doi: 10.1016/s0140-6736(00)03155-x. [DOI] [PubMed] [Google Scholar]
- 53.Hoglund K, Wiklund O, Vanderstichele H, Eikenberg O, Vanmechelen E, Blennow K. Plasma levels of beta-amyloid (1–40), beta-amyloid (1–42), and total beta-amyloid remain unaffected in adult patients with hypercholesterolemia after treatment with statins. Arch Neurol. 2004;61:333–337. doi: 10.1001/archneur.61.3.333. [DOI] [PubMed] [Google Scholar]
- 54.Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,506 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360:7–22. [Google Scholar]
- 55.Shepherd J, Blauw GJ, Murphy MB, Bollen EL, Buckley BM, Cobbe SM, Ford I, Gaw A, Hyland M, Jukema JW, Kamper AM, Macfarlane PW, Meinders AE, Norrie J, Packard CJ, Perry IJ, Stott DJ, Sweeney BJ, Twomey C, Westendorp RG PROSPER Study Group. Pravastatin in elderly individuals at risk of vascular disease(PROSPER): a randomised controlled trial. Lancet. 2002;360:1623–1630. doi: 10.1016/s0140-6736(02)11600-x. [DOI] [PubMed] [Google Scholar]
- 56.Collins R, Armitage J. High-risk elderly patients PROSPER from cholesterol-lowering therapy. Lancet. 2002;360:1618–1619. doi: 10.1016/S0140-6736(02)11650-3. [DOI] [PubMed] [Google Scholar]
- 57.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 58.Hock C, Konietzko U, Papassotiropoulos A, Wollmer A, Streffer J, von Rotz RC, Davey G, Moritz E, Nitsch RM. Generation of antibodies specific for beta-amyloid by vaccination of patients with Alzheimer disease. Nat Med. 2002;8:1270–1275. doi: 10.1038/nm783. [DOI] [PubMed] [Google Scholar]
- 59.Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Muller-Tillmanns B, Lemke U, Henke K, Moritz E, Garcia E, Wollmer MA, Umbricht D, de Quervain DJ, Hofmann M, Maddalena A, Papassotiropoulos A, Nitsch RM. Antibodies against beta-amyloid slow cognitive decline in Alzheimer’s disease. Neuron. 2003;38:547–554. doi: 10.1016/s0896-6273(03)00294-0. [DOI] [PubMed] [Google Scholar]
- 60.Bayer AJ, Bullock R, Jones RW, Wilkinson D, et al. Evaluation of the safety and immunogenicity of synthetic Aβ 42 (AN1792) in patients with AD. Neurology. 2005;64:94–101. doi: 10.1212/01.WNL.0000148604.77591.67. [DOI] [PubMed] [Google Scholar]
- 61.Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C. Subacute meningoencephalitis in a subset of patients with AD after Aβ 42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- 62.Beckman M. Untangling Alzheimer’s by paring plaques bolsters amyloid theory. Science. 2004;305:762. doi: 10.1126/science.305.5685.762a. [DOI] [PubMed] [Google Scholar]
- 63.Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C, Seubert P, Games D, Kirby L, Schenk D. Aβ vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology. 2005;64:129–131. doi: 10.1212/01.WNL.0000148590.39911.DF. [DOI] [PubMed] [Google Scholar]
- 64.Bennett DA, Holtzman DM. Immunization therapy for Alzheimer disease? Neurology. 2005;64:10–12. doi: 10.1212/01.WNL.0000150527.24596.29. [DOI] [PubMed] [Google Scholar]
- 65.Qu B, Rosenberg RN, Li L, Boyer PJ, Johnston SA. Gene vaccination to bias the immune response to amyloid-β peptide as therapy for Alzheimer disease. Arch Neurol. 2004;61:1859–1864. doi: 10.1001/archneur.61.12.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny R, Li QX, Tammer A, Carrington D, Mavros C, Volitakis I, Xilinas M, Ames D, Davis S, Beyreuther K, Tanzi RE, Masters CL. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Aβ amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol. 2003;60:1685–1691. doi: 10.1001/archneur.60.12.1685. [DOI] [PubMed] [Google Scholar]
- 67.Cacabelos R. The application of functional genomics to Alzheimer’s disease. Pharmacogenomics. 2003;4:597–621. doi: 10.1517/phgs.4.5.597.23795. [DOI] [PubMed] [Google Scholar]
- 68.King HC, Sinha AA. Gene expression profile analysis by DNA microarrays: promise and pitfalls. JAMA. 2001;286:2280–2288. doi: 10.1001/jama.286.18.2280. [DOI] [PubMed] [Google Scholar]
- 69.Subramanian G, Adams MD, Venter JC, Broder S. Implications of the human genome for understanding human biology and medicine. JAMA. 2001;286:2296–2307. doi: 10.1001/jama.286.18.2296. [DOI] [PubMed] [Google Scholar]
- 70.Schonberger SJ, Edgar PF, Kydd R, Faull RL, Cooper GJ. Proteomic analysis of the brain in Alzheimer’s disease: molecular phenotype of a complex disease process. Proteomics. 2001;1:1519–1528. doi: 10.1002/1615-9861(200111)1:12<1519::aid-prot1519>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 71.Liotta LA, Kohn EC, Petriconi EF. Clinical proteomics: personalized molecular medicine. JAMA. 2001;286:2211–2214. doi: 10.1001/jama.286.18.2211. [DOI] [PubMed] [Google Scholar]