

Abstract
Sequencing of full-insert clones from full-length cDNA libraries from both Xenopus laevis and Xenopus tropicalis has been ongoing as part of the Xenopus Gene Collection Initiative. Here we present 10,967 full ORF verified cDNA clones (8049 from X. laevis and 2918 from X. tropicalis) as a community resource. Because the genome of X. laevis, but not X. tropicalis, has undergone allotetraploidization, comparison of coding sequences from these two clawed (pipid) frogs provides a unique angle for exploring the molecular evolution of duplicate genes. Within our clone set, we have identified 445 gene trios, each comprised of an allotetraploidization-derived X. laevis gene pair and their shared X. tropicalis ortholog. Pairwise dN/dS, comparisons within trios show strong evidence for purifying selection acting on all three members. However, dN/dS ratios between X. laevis gene pairs are elevated relative to their X. tropicalis ortholog. This difference is highly significant and indicates an overall relaxation of selective pressures on duplicated gene pairs. We have found that the paralogs that have been lost since the tetraploidization event are enriched for several molecular functions, but have found no such enrichment in the extant paralogs. Approximately 14% of the paralogous pairs analyzed here also show differential expression indicative of subfunctionalization.
Xenopus laevis (the African claw-toed frog) has long been a preferred model organism among developmental biologists. Features such as ease of maintenance, oocyte size and number, and an easily manipulated reproductive system make it an ideal organism for the study of early embryonic development (De Sa and Hillis 1990). Studies of embryonic development in Xenopus have provided insights into many salient aspects of vertebrate development that would be difficult to study in other vertebrate systems (Gilchrist et al. 2004). However, the study of Xenopus genetic material is difficult because of an allotetraploidization event in the Xenopus lineage ∼30 million years ago (Mya) (Bisbee et al. 1977; Evans et al. 2004) that generated a more complex genome in all extant Xenopus species except for Xenopus tropicalis, whose genome remains diploid (Graf and Kobel 1991; Hirsch et al. 2002). With a less complex genome as well as a shorter generation time, X. tropicalis is more amenable to genetic manipulation and has become the preferred Xenopus species for genetic analyses (Hirsch et al. 2002).
Several groups have performed large-scale EST studies on libraries from various tissues from both X. laevis and X. tropicalis (Klein et al. 2002; Blackshear et al. 2001; Gilchrist et al. 2004). However, for analysis of transcripts and gene structures, the quality of data and coverage provided by EST reads can be limiting. Sequence-verified full-length cDNA clones are more informative and have a higher sequence quality standard. Here we report the full open reading frame (ORF) sequencing and coding DNA segment (CDS) analysis of 10,967 Xenopus full-length cDNA clones (8049 from X. laevis and 2918 from X. tropicalis). These clones are from libraries that were constructed using RNA from numerous tissues and whole animals in various developmental stages. We expect that these clones and their verified full ORF sequences will be a valuable resource for the community. Furthermore, the availability of full ORF sequences for a large set of Xenopus clones provides a unique opportunity to study molecular evolution in the context of allotetraploidization. The putative X. laevis ancestral allotetraploidization event created a full set of paralogs, each from one of the parent species involved in the mating (Graf and Kobel 1991). The redundancy of the resultant tetraploid X. laevis genome has, in theory, afforded this species greater freedom to accumulate mutations that may otherwise be deleterious in a diploid genome, such as that of X. tropicalis. In the present study, carefully defining gene trios (gene sets comprised of the two allotetraploidization-derived X. laevis paralogs and their shared X. tropicalis ortholog) has allowed us to distinguish paralogs arising from genome duplication from paralogs arising by ordinary within-species gene family expansion. We focus our initial analysis, presented here, on detecting signatures of purifying and positive selection, and on exploring the evolution of tissue-specific gene expression.
Results
X. laevis and X. tropicalis cDNA libraries (Supplemental Tables S1 and S2) were end-sequenced by the National Intramural Sequencing Center, Washington University Genome Sequencing Center, and Agencourt Bioscience Corporation (Gerhard et al. 2004). Candidate full ORF clones were selected as previously described (Klein et al. 2002; Gerhard et al. 2004). Each candidate full ORF clone was fully sequenced to a consensus phred score of no less than 30 (Ewing and Green 1998) at each consensus position by either transposon insertion or primer walking as previously described (Wilson and Mardis 1997; Gordon et al. 1998; Butterfield et al. 2002; Strausberg et al. 2002; Yang et al. 2005). Coding DNA segment (CDS) annotation of the full insert sequences was performed as previously described (Gerhard et al. 2004). Distinct from previous MGC projects, however, we took a second approach to clone selection in an attempt to identify clones that might encode either amphibian-specific proteins or proteins too weakly conserved at the N terminus to be identified by comparison with proteins from other organisms. The technique we used (see Methods) assumes that a stronger conservation will be observed in the CDS than in the 5′-untranslated region (UTR) of paralogous genes. Most of the clones identified by this technique (∼80%) were also identified by comparison with proteins from other species. The full-length sequences identified by this method are listed in Table 1. While there are seven proteins with no significant hits (E < 10−10) in any mammalian protein, there are also six proteins with alignment scores more than two standard deviations below the mean value to the most closely related human protein (mean 73%, 13.8% standard deviation). The small number of novel proteins identified as well as the overlap with proteins identified from protein or mRNA comparison suggest that there are few proteins present in this cDNA collection that are structurally distinct from previously identified proteins.
Table 1.
X. laevis genes identified without protein comparison
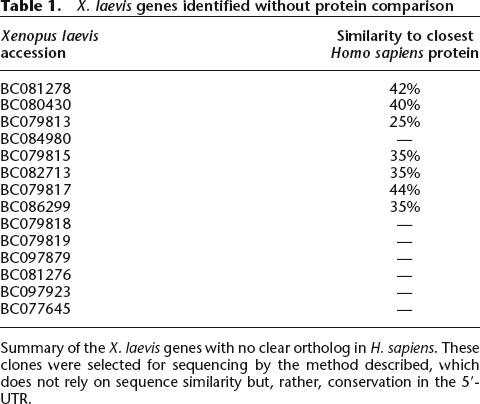
Summary of the X. laevis genes with no clear ortholog in H. sapiens. These clones were selected for sequencing by the method described, which does not rely on sequence similarity but, rather, conservation in the 5′-UTR.
Following full insert sequencing of candidate clones, only those clones with verified complete ORFs were given an XGC (Xenopus Gene Collection) identifier. These 10,967 clones (8049 from X. laevis and 2918 from X. tropicalis) are considered the core group of XGC clones and are the basis of the analysis presented here. All clone sequences have been submitted to GenBank, and the physical clones are available through the IMAGE distribution network.
From the set of 10,967 clones, we identified 445 distinct gene trios for analysis (see Methods). Again, a trio is a gene set comprised of the two allotetraploidization-derived X. laevis paralogs and their shared X. tropicalis ortholog. To explore the signature of selection between X. laevis and X. tropicalis, we applied the dN/dS test using the maximum likelihood method of Yang and Nielsen available as the codeml component of the PAML software package (PAML software release 3.14) (Yang et al. 1997). This method allows inference of evolutionary selection for mutations using the ratio of nonsynonymous (dN) to synonymous (dS) mutations in the coding DNA sequence of a phylogeny of homologous genes. In general, a dN/dS ratio (ω) >1 is evidence for positive selection acting to modify the function of a gene (Thornton and Long 2002; Zhang et al. 2002), whereas an ω significantly <1 suggests negative or purifying selection where functional constraint on the gene product has restricted the amount of nonsynonymous mutation. None of the pairwise comparisons between X. laevis paralogs or between each X. laevis gene and its X. tropicalis ortholog resulted in an ω significantly >1 (Fig. Fig. 1), suggesting that purifying selection has continued to act on X. laevis genes duplicated by allotetraploidization, and that in general both copies have retained function. Interestingly, taken together, ωs from pairwise comparisons between X. laevis paralogs are significantly larger (Kruskal-Wallis rank sum test, P = 2.184 × 10−7) than either of the ωs from pairwise comparisons between each X. laevis gene and X. tropicalis paralog. These observations suggest that overall there has been relaxation of selective pressures on X. laevis duplicated gene pairs, allowing them more freedom to accumulate nonsynonymous substitutions.
Figure 1.

Frequencies of dN/dS ratio (ω) for pairwise comparisons between X. laevis and X. tropicalis genes. The distribution of dN/dS from pairwise comparisons of genes within gene trios is shown. ωs from X. laevis paralog pairs (shown in red) indicate a weaker selective constraint than the ω obtained from the comparisons of X. laevis paralogs with their X. tropicalis ortholog (shown in gray and blue). The ωs from both paralog–ortholog pairs follow a similar distribution with a lower median than the ω obtained from the paralogs in each trio (P = 2.184 × 10−7).
Pairwise dS estimates provide a relative measure of time since divergence of homologous genes and give an independent estimate of the topology of our defined trios. As expected, in all of the gene trios, the dS between the X. laevis paralogs is lower than both dSs between each X. laevis paralog and its X. tropicalis ortholog. The trend (Fig. Fig. 2) shows a peak representing the divergence between the two species (blue and gray curves, median = 0.2915) and a peak representing the tetraploidization within the X. laevis genome (red curve, median = 0.2039). We calculated the effective number of codons (ENC) for all clones in this study using the codonW program (http://codonw.sourceforge.net) and did not see any evidence for an effect of codon bias on synonymous substitution rates.
Figure 2.

Distribution of dS for pairwise comparisons between paralogs and orthologs. Distribution of dS from pairwise comparison between X. laevis paralogs (red) and from pairwise comparisons between each X. laevis paralog from a trio with its X. tropicalis ortholog (blue and gray). The small number (eight in total) of dS values that were >1 were eliminated to provide an appropriate scale.
To identify the function of genes for which duplicate copies are preferentially retained or lost after tetraploidization, we explored the Gene Ontology (GO) representation of X. laevis genes with and without paralogs. We used the High-Throughput GoMiner tool (http://discover.nci.nih.gov/gominer/htgm.jsp) (Zeeberg et al. 2005) to search for GO categories overrepresented in the clones with either active paralogs in X. laevis or no evidence for extant paralogs (see Methods). Several categories are enriched in the set of genes with no evidence for an extant paralog (Table 2; Fig. Fig. 3; Supplemental Fig. S6), but we saw no significant enrichment of GO categories in the set of genes with retained paralogs. GO categories enriched in the single-copy genes, which have lost the tetraploidization-derived paralog, can be grouped into three main clusters related to respiration (cluster 1), nucleic acid processing (cluster 2), and nucleoside metabolism (cluster 3) (Table 2; Supplemental Fig. S5).
Table 2.
GO Categories enriched in genes with no paralogs in X. laevis

All nonredundant Gene Ontology (Ashburner et al. 2000) terms enriched in the set of clones suspected to have no expressed paralogs in X. laevis. Categories with a false discovery rate (FDR) ≤0.10 are shown. Several very large generic categories were removed for clarity. No statistically significant categories were found for genes for which a tetraploidization-derived paralog was found.
Figure 3.

Clustered Image Map of genes with no paralog versus GO categories for categories with significant enrichment. Thumbnail clustered image map (CIM) of genes (top) versus categories (right) for categories with a false discovery rate (FDR) ≤0.10. Very large generic categories have been removed to improve visualization. Clustering was performed with the Genesis Client (Sturn et al. 2002; http://genome.tugraz.at/Software/GenesisCenter.html). Three major clusters can be seen. Processes involved in general metabolism (far left) include “cofactor catabolism,” “acetyl-CoA catabolism,” “aerobic respiration,” “cellular respiration,” and “tricarboxylic acid cycle.” Processes involved in nucleic acid processing (bottom right) include “RNA metabolism,” “transcription,” “nucleobase metabolism,” “DNA replication,” and “DNA metabolism.” The third cluster contains GO categories involved in nucleoside metabolism such as “nucleobase metabolism,” “pyrimidine base biosynthesis,” and “nucleobase biosynthesis.” The full-size CIM in which all genes are displayed is available as Supplemental Figure S6.
Next, we explored whether paralogous genes in X. laevis have begun to subfunctionalize at the level of gene expression. Subfunctionalized genes may retain similar or identical CDS while obtaining tissue-specific functions through mutations that alter their expression (Force et al. 1999). The UniGene project contains ∼27,000 expressed sequence tags each from X. laevis and X. tropicalis that are derived exclusively from tissue-specific libraries (builds 63 and 24 respectively) (Pontius et al. 2003). Taking these tissue-specific ESTs, we matched each to its corresponding X. laevis and X. tropicalis gene where matches could be unambiguously assigned (BLASTN) (Altschul et al. 1997). For the 1039 X. laevis allotetraploidization-derived paralogs in our gene set (see Methods), 842 had EST matches in tissue-specific libraries for both clones. Of these, 118 (14.0%) showed significant (P < 0.05, corrected for multiple testing) differential tissue expression (Table 3; Supplemental Table S3) consistent with subfunctionalization. Next, we ranked X. laevis paralogs by the number of ESTs from any tissue in which each member of the pair is expressed. This approach gave a Spearman’s rank correlation coefficient of 0.49. A correlation of 1 would indicate that all paralogous gene pairs were similarly expressed, whereas a correlation near 0 would indicate that all gene pairs were expressed independently and randomly; the observed correlation of aggregate expression is intermediate to these extremes, which again is consistent with a degree of subfunctionalization in X. laevis. To assess how often paralogous genes have differential aggregate expression, we examined the highest and lowest deciles of aggregate expression. In the highest expression decile, 68% of genes have a paralog that is also in the highest expression decile, and in the lowest expression decile, 38% of genes have a paralog also in the lowest expression decile. This observation suggests that the function of both copies of highly expressed genes in X. laevis is more often conserved than is the function of sparsely expressed gene pairs.
Table 3.
Differential tissue expression for X. laevis paralogs from EST information (top 20 genes)

A summary of the top 20 paralog pairs with evidence for subfunctionalization (sorted with most significant P-value at the top). The EST count for gene 1 and gene 2 of each paralog pair in the tissue showing differential expression is shown. The Gene Ontology (GO) category (Ashburner et al. 2000) that best describes the putative function of each gene pair is included in addition to the name of the best Homo sapiens BLASTP hit. All 118 cases of potential subfunctionalization are supplied in Supplemental Table S3.
Discussion
We report 10,967 full insert verified clones and sequences from X. laevis and X. tropicalis that have been generated by the XGC project. This clone set provides the community with a useful resource for functional genomic studies in these important model organisms. Furthermore, we have gained insight into the evolution of protein-coding sequences in a genome duplicated by species hybridization. It is well established that new genes typically arise through duplication of existing genes (Ohno 1970; Force et al. 1999). It is also known that whole genome duplications are common among the pipid frogs (Evans et al. 2004), and it is thought that these events are the major method of speciation within this genus (Kobel 1996; Evans et al. 2004). Because one member of a new duplicate gene pair is initially redundant, it is likely to quickly become a pseudogene after fixation of a null mutation within the population (Ohno 1970). The present study focuses on cDNA sequences; thus only actively transcribed genes (and potentially, rarely transcribed pseudogenes) contribute to our data set. For these expressed genes, the results of the present study indicate that many of the redundant copies have remained active in X. laevis over tens of millions of years. This result is consistent with previous observations in Xenopus, fish, and various plants (Hughes and Hughes 1993; Taylor et al. 2001; Adams and Wendel 2005).
Given that we observe 1039 paralog pairs after sampling only 8049 genes and using a conservative paralog assignment method, it is possible that retention of both copies within X. laevis has benefited the species. It has previously been estimated that X. laevis has retained about half of its duplicate genes (Bisbee et al. 1977). While there is no evidence for selective retention by functional category, we do provide evidence consistent with a model of selective loss of certain duplicate genes after genome duplication. Interestingly, the functional groups selectively lost in Xenopus, namely, respiration, nucleic acid processing, and nucleoside metabolism (Table 2; Supplemental Fig. S5), are included in those known to have been selectively lost after whole genome duplication in Arabidopsis thaliana (Maere et al. 2005). Genes in these categories, therefore, appear more likely than other genes to confer dosage sensitivity.
We searched for evidence of selection on the remaining tetraploidization-derived paralogs in X. laevis. Using the Yang and Nielsen maximum likelihood method (Yang 1997, 1998), we calculated pairwise dN/dS ratios between tetraploidization-derived X. laevis paralogs and between each of these X. laevis paralogs and its X. tropicalis ortholog. While none of the paralog pairs observed in this study provides direct evidence of positive Darwinian selection, it must be noted that the evolutionary distance between X. laevis and X. tropicalis is approaching the practical limit of detecting positive selection by the dN/dS test (Hughes et al. 2000). Also, all dN/dS ratios are averages across the length of each gene, and may mask situations in which positive selection has only occurred in a site-specific manner. Interestingly, overall, dN/dS ratios between tetraploidization-derived X. laevis paralogs were significantly elevated relative to dN/dS ratios between X. laevis/X. tropicalis orthologs (Fig. Fig. 1). This result is consistent with the notion that gene duplication has, to a small but measurable degree, freed expressed X. laevis sequence from functional constraint.
A subset of X. laevis paralogs also shows strong evidence for subfunctionalization, as indicated by the differential expression observed between ∼14% of the paralog pairs analyzed. The paralogs that show differential expression at the highest significance levels are overrepresented in a few molecular functions (Table 3). In the top 20 subfunctionalized pairs, five are involved in protein translation, and five are potentially involved in either iron or calcium transport. With the application of Spearman’s rank to paralog pairs, we have detected a global trend of many of the paralog pairs toward subfunctionalization. The observation of numerous examples in which two allotetraploidization-derived gene copies show differential expression lends support to the theory of regulatory evolution (King and Wilson 1975), which holds that evolution is mediated by modification of gene expression patterns as well as by coding sequence changes.
Methods
The analyses described here were performed on the 10,967 Xenopus sequences available from the XGC Web site as of September 9, 2005 (XGC homepage, http://xgc.nci.nih.gov/).
Selection of clones for sequencing
Candidate clones for full insert sequencing were selected as previously described (Gerhard et al. 2004). Identification of additional putatively novel Xenopus clones was as follows. Pairs of orthologous (in the reciprocal best-match sense) X. tropicalis and X. laevis sequences were aligned, and the 5′-most ATG in the sequence was used to divide the alignment. Then 674 pairs of characterized genes were examined to determine mean conservation in CDS and 5′-UTR (92.9% and 89.7%, respectively), as well as to compute the variance of the conservation difference between CDS and UTRs. Sequences showing any increase in conservation (corresponding to a sequence conservation of 1.6 standard deviations below the mean) and with a 5′-UTR of at least 25 nt were selected for sequencing.
Paralog and ortholog assignment
For each species, an all-by-all BLASTN search was executed, and any sequences with >98% nucleic acid identity over 90% of their length were removed as redundant copies of the same clone, allelic copies, or recent gene duplicates. Next, unmatched sequences from the X. laevis reciprocal BLASTN and those with more than one significant (e < 10−20) match were excluded, leaving 1354 putative allotetraploidization-derived X. laevis paralogs with 93.1% ± 2.72% nucleic acid identity (mean ± SD). Finally, a reciprocal BLASTP search of each X. laevis paralogous against the set of X. tropicalis predicted nonredundant proteins (EnsEMBL homepage, http://www.ensembl.org/Xenopus_ tropicalis) identified 1039 paralog pairs for which both clones had a common best X. tropicalis hit. Note that the above approach automatically prevents us from considering gene families with more than 1:1 paralog mapping and prevents assignment of tetraploidization-derived paralogs to those that formed prior to the speciation between X. laevis and X. tropicalis. Also, by considering the best nonredundant match of each X. laevis clone sequence, we assigned the most recent paralog pairs, assuming that few paralog pairs have formed since the tetraploidization event. Of the 1039 gene trios, 445 have an ortholog that is represented in the set of 2918 X. tropicalis full ORF cDNAs. Our dN/dS analysis is based on these 445 trios, and we included the additional paralog pairs with no X. tropicalis XGC ortholog in the retained paralog and subfunctionalization analyses.
Multiple sequence alignments
We performed all protein alignments using CLUSTALW with default parameters (Thompson et al. 1994). We used the RevTrans program (Wernersson and Pedersen 2003) to produce codon-aware alignments for more accurate predictions of substitution rates. Files were formatted from RevTrans output (FASTA) to codeml input format with a Perl script.
dN/dS estimation
We performed pairwise dN,dS and ω calculations using the yn00 component of the PAML package. We also calculated dN/dS with the maximum likelihood method of the codeml program (Yang et al. 1997) in order to perform likelihood ratio tests. Each dN/dS calculation was performed twice. In one run, the dN/dS was fixed at 1, and in the other run it was free to vary. Twice the difference between the two log likelihood values was compared to the χ2 distribution with 1 degree of freedom for rejection of the hypothesis that dN/dS = 1 (P < 0.05).
Gene Ontology analysis of retained paralogs
The aim of this analysis was to determine whether genes of specific functions have been selectively retained in multiple copies within the X. laevis genome or whether redundant copies have been selectively lost after duplication. Clones with no expressed paralog were defined by first assigning a UniGene cluster to each clone as described above. We limited our analysis to clones for which this corresponding UniGene cluster contained at least 20 ESTs to minimize the potential of assigning genes with low expression as single-copy genes. Single-copy genes were then assigned by the absence of a second significant (e < 10−30) BLASTN hit in UniGene. We searched for GO categories enriched in either the 1527 single-copy genes or the 1039 dual-copy genes using High-Throughput GoMiner (http://discover.nci.nih.gov/gominer/htgm.jsp) (Zeeberg et al. 2005) and an in-house customized version of the GO Consortium (Ashburner et al. 2000) database. This database was populated with all GO annotations assigned by Interproscan (Apweller et al. 2001) to the X. laevis genes. The statistical significance of enrichment was based on a false discover rate (FDR) threshold value of 0.10, using High-Throughput GoMiner’s correction for multiple testing.
Tissue expression comparisons
We matched every clone to its best UniGene cluster by a BLASTN of each clone against a representative EST from each UniGene cluster in the files Xl.seq.uniq and Str.seq.uniq downloaded from NCBI (UniGene Download page, ftp://ftp.ncbi.nih.gov/repository/UniGene). We then performed a second BLASTN search of each clone against all ESTs in its representative cluster and only considered matches with alignment lengths >200 bp and percent identities >90%. As some (∼20) of the genes shared a common UniGene cluster, we only used ESTs from those clusters if they could be unambiguously assigned to one copy by differential percent identity. In all other cases, all reads from the best UniGene cluster for each clone were used. Using the library name for each EST, we determined the source tissue of the EST as defined in the files Xl.lib.info and Str.lib.info. A Perl script counted the total number of ESTs of each gene found in each tissue. Using a previously described Bayesian method (Audic and Claverie 1997), we assigned P-values to each gene pair showing a significant difference in expression (P < 0.05). As the variability in expression patterns was strongest in tissues where the genes are highly expressed, we chose to only compare the expression of the two paralogs in the tissue in which they showed to be most highly expressed (highest EST count). A Bonferroni correction was used to compensate for multiple hypothesis testing where applicable, although a maximum of two tests were done for each paralog pair.
Acknowledgments
This project has been funded in part with Federal funds from the National Human Genome Research Institute, National Institutes of Health, under contract No. U01 HG002155-06S1. This research was supported (in part) by the Intramural Research Program of the National Cancer Institute (NCI) of the National Institutes of Health (NIH). This work would also not have been possible without the cDNA libraries and EST sequences provided by various groups and we specifically thank Igor Dawid and Thomas Sargent (NICHD), Donald Brown (Carnegie Institute), Bruce Blumberg (UC at Irvine), and Robert Grainger (UVA). Work by C.P.P. was performed under the auspices of the U.S. Department of Energy by the University of California, Lawrence Livermore National Laboratory under contract No. W-7405-Eng-48. We thank Diana Palmquist and Elizabeth Chun, who assisted with finishing some of the clones. R.A.H., S.J.M.J., and M.A.M. are Michael Smith Foundation for Health Research Scholars.
Footnotes
[Supplemental material is available online at www.genome.org. The sequence data from this study have been submitted to GenBank under accession nos. BC040971–BC100665 (not exclusively). Keyword MGC and organism Xenopus will be required to get only the XGC sequences in the range.]
The content of this publication does not necessarily reflect the views or policies of the U.S. Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the U.S. Government.
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.4871006
References
- Adams K.L., Wendel J.F., Wendel J.F. Polyploidy and genome evolution in plants. Curr. Opin. Plant Biol. 2005;8:135–141. doi: 10.1016/j.pbi.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D., Zhang J., Zhang Z., Miller W., Lipman D., Zhang Z., Miller W., Lipman D., Miller W., Lipman D., Lipman D. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apweller R., Attwood T.K., Bairoch A., Bateman A., Birney E., Biswas M., Bucher P., Cerutti L., Corpet F., Croning M.D.R., Attwood T.K., Bairoch A., Bateman A., Birney E., Biswas M., Bucher P., Cerutti L., Corpet F., Croning M.D.R., Bairoch A., Bateman A., Birney E., Biswas M., Bucher P., Cerutti L., Corpet F., Croning M.D.R., Bateman A., Birney E., Biswas M., Bucher P., Cerutti L., Corpet F., Croning M.D.R., Birney E., Biswas M., Bucher P., Cerutti L., Corpet F., Croning M.D.R., Biswas M., Bucher P., Cerutti L., Corpet F., Croning M.D.R., Bucher P., Cerutti L., Corpet F., Croning M.D.R., Cerutti L., Corpet F., Croning M.D.R., Corpet F., Croning M.D.R., Croning M.D.R., et al. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res. 2001;29:37–40. doi: 10.1093/nar/29.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al The Gene Ontology Consortium , Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al The Gene Ontology Consortium , Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al The Gene Ontology Consortium , Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al The Gene Ontology Consortium , Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al The Gene Ontology Consortium , Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al The Gene Ontology Consortium , Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al The Gene Ontology Consortium , Dolinski K., Dwight S.S., Eppig J.T., et al The Gene Ontology Consortium , Dwight S.S., Eppig J.T., et al The Gene Ontology Consortium , Eppig J.T., et al The Gene Ontology Consortium , et al The Gene Ontology Consortium Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audic S., Claverie J.M., Claverie J.M. The significance of digital gene expression profiles. Genome Res. 1997;7:986–995. doi: 10.1101/gr.7.10.986. [DOI] [PubMed] [Google Scholar]
- Bisbee C.A., Baker M.A., Wilson A.C., Irandokht H.A., Fischberg M., Baker M.A., Wilson A.C., Irandokht H.A., Fischberg M., Wilson A.C., Irandokht H.A., Fischberg M., Irandokht H.A., Fischberg M., Fischberg M. Albumin phylogeny for clawed frogs. Science. 1977;195:785–787. doi: 10.1126/science.65013. [DOI] [PubMed] [Google Scholar]
- Blackshear P.J., Lai W.S., Thorn J.M., Kennington E.A., Staffa N.G., Moore D.T., Bouffard G.G., Beckstrom-Sterberg S.M., Touchman J.W., de Fatima Bonaldo M., Lai W.S., Thorn J.M., Kennington E.A., Staffa N.G., Moore D.T., Bouffard G.G., Beckstrom-Sterberg S.M., Touchman J.W., de Fatima Bonaldo M., Thorn J.M., Kennington E.A., Staffa N.G., Moore D.T., Bouffard G.G., Beckstrom-Sterberg S.M., Touchman J.W., de Fatima Bonaldo M., Kennington E.A., Staffa N.G., Moore D.T., Bouffard G.G., Beckstrom-Sterberg S.M., Touchman J.W., de Fatima Bonaldo M., Staffa N.G., Moore D.T., Bouffard G.G., Beckstrom-Sterberg S.M., Touchman J.W., de Fatima Bonaldo M., Moore D.T., Bouffard G.G., Beckstrom-Sterberg S.M., Touchman J.W., de Fatima Bonaldo M., Bouffard G.G., Beckstrom-Sterberg S.M., Touchman J.W., de Fatima Bonaldo M., Beckstrom-Sterberg S.M., Touchman J.W., de Fatima Bonaldo M., Touchman J.W., de Fatima Bonaldo M., de Fatima Bonaldo M., et al. The NIEHS Xenopus maternal EST project: Interim analysis of the first 13,879 ESTs from unfertilized eggs. Gene. 2001;267:71–87. doi: 10.1016/s0378-1119(01)00383-3. [DOI] [PubMed] [Google Scholar]
- Butterfield Y.S., Marra M.A., Asano J.K., Chan S.Y., Guin R., Krzywinski M.I., Lee S.S., MacDonald K.W., Mathewson C.A., Olson T.E., Marra M.A., Asano J.K., Chan S.Y., Guin R., Krzywinski M.I., Lee S.S., MacDonald K.W., Mathewson C.A., Olson T.E., Asano J.K., Chan S.Y., Guin R., Krzywinski M.I., Lee S.S., MacDonald K.W., Mathewson C.A., Olson T.E., Chan S.Y., Guin R., Krzywinski M.I., Lee S.S., MacDonald K.W., Mathewson C.A., Olson T.E., Guin R., Krzywinski M.I., Lee S.S., MacDonald K.W., Mathewson C.A., Olson T.E., Krzywinski M.I., Lee S.S., MacDonald K.W., Mathewson C.A., Olson T.E., Lee S.S., MacDonald K.W., Mathewson C.A., Olson T.E., MacDonald K.W., Mathewson C.A., Olson T.E., Mathewson C.A., Olson T.E., Olson T.E., et al. An efficient strategy for large-scale high-throughput transposon-mediated sequencing of cDNA clones. Nucleic Acids Res. 2002;30:2460–2468. doi: 10.1093/nar/30.11.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sa R.O., Hillis D.M., Hillis D.M. Phylogenetic relationships of the pipid frogs Xenopus and Silurana: An integration of ribosomal DNA morphology. Mol. Biol. Evol. 1990;7:365–376. doi: 10.1093/oxfordjournals.molbev.a040612. [DOI] [PubMed] [Google Scholar]
- Evans B.J., Kelley D.B., Tinsley R.C., Melnick D.J., Cannatella D.C., Kelley D.B., Tinsley R.C., Melnick D.J., Cannatella D.C., Tinsley R.C., Melnick D.J., Cannatella D.C., Melnick D.J., Cannatella D.C., Cannatella D.C. A mitochondrial DNA phylogeny of African clawed frogs: Phylogeography and implications for polyploid evolution. Mol. Phylogenet. Evol. 2004;33:197–213. doi: 10.1016/j.ympev.2004.04.018. [DOI] [PubMed] [Google Scholar]
- Ewing B., Green P., Green P. Base-calling of automated sequencer traces using Phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- Force A., Lynch M., Pickett F.B., Amores A., Yan Y., Postlethwait J., Lynch M., Pickett F.B., Amores A., Yan Y., Postlethwait J., Pickett F.B., Amores A., Yan Y., Postlethwait J., Amores A., Yan Y., Postlethwait J., Yan Y., Postlethwait J., Postlethwait J. Preservation of duplicate genes by complementary, degenerative mutations. Genetics. 1999;151:1531–1545. doi: 10.1093/genetics/151.4.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard D.S., Wagner L., Feingold E.A., Shenmen C.M., Grouse L.H., Schuler G., Klein S.L., Old S., Rasooly R., Good P., Wagner L., Feingold E.A., Shenmen C.M., Grouse L.H., Schuler G., Klein S.L., Old S., Rasooly R., Good P., Feingold E.A., Shenmen C.M., Grouse L.H., Schuler G., Klein S.L., Old S., Rasooly R., Good P., Shenmen C.M., Grouse L.H., Schuler G., Klein S.L., Old S., Rasooly R., Good P., Grouse L.H., Schuler G., Klein S.L., Old S., Rasooly R., Good P., Schuler G., Klein S.L., Old S., Rasooly R., Good P., Klein S.L., Old S., Rasooly R., Good P., Old S., Rasooly R., Good P., Rasooly R., Good P., Good P., et al. The status, quality, and expansion of the NIH Full-Length cDNA Project: The Mammalian Gene Collection (MGC). Genome Res. 2004;14:2121–2127. doi: 10.1101/gr.2596504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist M.J., Zorn A.M., Voigt J., Smith J.C., Papalopulu N., Amaya E., Zorn A.M., Voigt J., Smith J.C., Papalopulu N., Amaya E., Voigt J., Smith J.C., Papalopulu N., Amaya E., Smith J.C., Papalopulu N., Amaya E., Papalopulu N., Amaya E., Amaya E. Defining a large set of full-length clones from a Xenopus tropicalis EST project. Dev. Biol. 2004;271:498–516. doi: 10.1016/j.ydbio.2004.04.023. [DOI] [PubMed] [Google Scholar]
- Gordon D., Abajian C., Green P., Abajian C., Green P., Green P. Consed: A graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- Graf J., Kobel H., Kobel H. Genetics of Xenopus laevis. Methods Cell Biol. 1991;36:663–669. doi: 10.1016/s0091-679x(08)60270-8. [DOI] [PubMed] [Google Scholar]
- Hirsch N., Zimmerman L., Grainger R., Zimmerman L., Grainger R., Grainger R. Xenopus, the next generation: X. tropicalis genetics and genomics. Dev. Dyn. 2002;225:422–433. doi: 10.1002/dvdy.10178. [DOI] [PubMed] [Google Scholar]
- Hughes M.K., Hughes A.L., Hughes A.L. Evolution of duplicate genes in a tetraploid animal, Xenopus laevis. Mol. Biol. Evol. 1993;10:1360–1369. doi: 10.1093/oxfordjournals.molbev.a040080. [DOI] [PubMed] [Google Scholar]
- Hughes M.K., Green J.A., Garbayo J.M., Roberts R.M., Green J.A., Garbayo J.M., Roberts R.M., Garbayo J.M., Roberts R.M., Roberts R.M. Adaptive diversification within a large family of recently duplicated, placentally expressed genes. Proc. Natl. Acad. Sci. 2000;97:3319–3323. doi: 10.1073/pnas.050002797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King M.C., Wilson A.C., Wilson A.C. Evolution at two levels in humans and chimpanzees. Science. 1975;188:107–116. doi: 10.1126/science.1090005. [DOI] [PubMed] [Google Scholar]
- Klein S.L., Strausberg R.L., Wagner L., Pontius J., Clifton S.W., Richardson P., Strausberg R.L., Wagner L., Pontius J., Clifton S.W., Richardson P., Wagner L., Pontius J., Clifton S.W., Richardson P., Pontius J., Clifton S.W., Richardson P., Clifton S.W., Richardson P., Richardson P. Genetic and genomic tools for Xenopus research: The NIH Xenopus Initiative. Dev. Dyn. 2002;225:384–391. doi: 10.1002/dvdy.10174. [DOI] [PubMed] [Google Scholar]
- Kobel H.R.1996. Allopolyploid speciation. In The biology of Xenopus (eds. R.C. Tinsley and H.R. Kobel) pp. 391–401. Clarendon Press; Oxford [Google Scholar]
- Maere S., De Bodt S., Raes J., Casneuf T., Van Montagu M., Kuiper M., de Van Peer Y., De Bodt S., Raes J., Casneuf T., Van Montagu M., Kuiper M., de Van Peer Y., Raes J., Casneuf T., Van Montagu M., Kuiper M., de Van Peer Y., Casneuf T., Van Montagu M., Kuiper M., de Van Peer Y., Van Montagu M., Kuiper M., de Van Peer Y., Kuiper M., de Van Peer Y., de Van Peer Y. Modeling gene and genome duplication in eukaryotes. Proc. Natl. Acad. Sci. 2005;102:5454–5459. doi: 10.1073/pnas.0501102102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S. Evolution by gene duplication. Springer-Verlag; New York.: 1970. [Google Scholar]
- Pontius J.U., Wagner L., Schuler G.D., Wagner L., Schuler G.D., Schuler G.D.2003. UniGene: A unified view of the transcriptome. In The NCBI Handbook, pp. 857–868. National Center for Biotechnology Information; Bethesda, MD [Google Scholar]
- Strausberg R.L., Feingold E.A., Grouse L.H., Derge J.G., Klausner R.D., Collins F.S., Wagner L., Shenmen C.M., Schuler G.D., Altschul S.F., Feingold E.A., Grouse L.H., Derge J.G., Klausner R.D., Collins F.S., Wagner L., Shenmen C.M., Schuler G.D., Altschul S.F., Grouse L.H., Derge J.G., Klausner R.D., Collins F.S., Wagner L., Shenmen C.M., Schuler G.D., Altschul S.F., Derge J.G., Klausner R.D., Collins F.S., Wagner L., Shenmen C.M., Schuler G.D., Altschul S.F., Klausner R.D., Collins F.S., Wagner L., Shenmen C.M., Schuler G.D., Altschul S.F., Collins F.S., Wagner L., Shenmen C.M., Schuler G.D., Altschul S.F., Wagner L., Shenmen C.M., Schuler G.D., Altschul S.F., Shenmen C.M., Schuler G.D., Altschul S.F., Schuler G.D., Altschul S.F., Altschul S.F. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc. Natl. Acad. Sci. 2002;99:16899–16903. doi: 10.1073/pnas.242603899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturn A., Quackenbush J., Trajanoski Z., Quackenbush J., Trajanoski Z., Trajanoski Z. Genesis: Cluster analysis of microarray data. Bioinformatics. 2002;18:207–208. doi: 10.1093/bioinformatics/18.1.207. [DOI] [PubMed] [Google Scholar]
- Taylor J.S., de Van Peer Y., Braasch I., Myer A., de Van Peer Y., Braasch I., Myer A., Braasch I., Myer A., Myer A. Comparative genomics provides evidence for an ancient genome duplication event in fish. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2001;356:1661–1679. doi: 10.1098/rstb.2001.0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.D., Higgins D.G., Gibson T.J., Higgins D.G., Gibson T.J., Gibson T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton K., Long M., Long M. Rapid divergence of gene duplicates on the Drosophila melanogaster X chromosome. Mol. Biol. Evol. 2002;19:918–925. doi: 10.1093/oxfordjournals.molbev.a004149. [DOI] [PubMed] [Google Scholar]
- Wernersson R., Pedersen A.G., Pedersen A.G. RevTrans: Multiple alignment of coding DNA from aligned amino acid sequences. Nucleic Acids Res. 2003;31:3537–3539. doi: 10.1093/nar/gkg609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R.K., Mardis E.R., Mardis E.R.1997. Fluorescence-based DNA sequencing. In Genome analysis: A laboratory manual: Analyzing DNA (eds. B. Birren et al.), Vol. 1, pp. 301–395. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY [Google Scholar]
- Yang Z. PAML, a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- Yang Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol. Biol. Evol. 1998;15:568–573. doi: 10.1093/oxfordjournals.molbev.a025957. [DOI] [PubMed] [Google Scholar]
- Yang G., Stott J.M., Smailus D.M., Barber S.A., Balasundaram M., Marra M.A., Holt R.A., Stott J.M., Smailus D.M., Barber S.A., Balasundaram M., Marra M.A., Holt R.A., Smailus D.M., Barber S.A., Balasundaram M., Marra M.A., Holt R.A., Barber S.A., Balasundaram M., Marra M.A., Holt R.A., Balasundaram M., Marra M.A., Holt R.A., Marra M.A., Holt R.A., Holt R.A. High-throughput sequencing: A failure mode analysis. BMC Genomics. 2005;6:2. doi: 10.1186/1471-2164-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeeberg B.R., Qin H., Narasimhan S., Sunshine M., Cao H., Kane D.W., Reimers M., Stephens R.M., Bryant D., Burt S.K., Qin H., Narasimhan S., Sunshine M., Cao H., Kane D.W., Reimers M., Stephens R.M., Bryant D., Burt S.K., Narasimhan S., Sunshine M., Cao H., Kane D.W., Reimers M., Stephens R.M., Bryant D., Burt S.K., Sunshine M., Cao H., Kane D.W., Reimers M., Stephens R.M., Bryant D., Burt S.K., Cao H., Kane D.W., Reimers M., Stephens R.M., Bryant D., Burt S.K., Kane D.W., Reimers M., Stephens R.M., Bryant D., Burt S.K., Reimers M., Stephens R.M., Bryant D., Burt S.K., Stephens R.M., Bryant D., Burt S.K., Bryant D., Burt S.K., Burt S.K., et al. High-Throughput GoMiner, an ‘industrial-strength’ integrative gene ontology tool for interpretation of multiple-microarray experiments, with application to studies of Common Variable Immune Deficiency (CVID). BMC Bioinformatics. 2005;6:168. doi: 10.1186/1471-2105-6-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Vision T., Gaut B., Vision T., Gaut B., Gaut B. Patterns of nucleotide substitution among simultaneously duplicated gene pairs in Arabidopsis thaliana. Mol. Biol. Evol. 2002;19:1464–1473. doi: 10.1093/oxfordjournals.molbev.a004209. [DOI] [PubMed] [Google Scholar]