

Abstract
Torsades de pointes (TdP) is a potentially lethal arrhythmia that develops as a consequence of amplification of electrical heterogeneities intrinsic to the ventricular myocardium. These heterogeneities exist because of differences in the time course of repolarization of the three predominant cell types that make up the ventricular myocardium, giving rise to transmural voltage gradients and a dispersion of repolarization responsible for inscription of the ECG T wave. Antiarrhythmic agents with class III actions and/or the various mutations and cardiomyopathies associated with the long QT syndrome reduce net repolarizing current and amplify the intrinsic spatial dispersion of repolarization, thus creating the substrate for the development of reentry. The result is prolongation of the QT interval, abnormal T waves, and development of polymorphic reentrant ventricular tachycardia displaying characteristics of TdP. Prolongation of the QT interval apparently is not the sole determinant of a drug's potential to cause TdP. Agents that do not increase transmural dispersion of repolarization have little or no potential to induce TdP despite any ability to prolong the QT interval. In addition, drugs such as amiodarone and sodium pentobarbital can cause large QT prolongations but, by reducing transmural dispersion of repolarization, may reduce the likelihood of TdP. Arterially perfused wedge preparations of canine left ventricle can be used to explore the role of transmural dispersion of repolarization in the genesis of TdP. The purpose of this article is to review recent advances that have improved our understanding of these mechanisms, particularly the role of transmural dispersion of repolarization, in the genesis of drug-induced TdP and to examine how these advances can guide us toward the development of safer and more effective drugs.
Keywords: Long QT syndrome, Torsades de pointes, Transmural dispersion of repolarization, Arterially perfused wedge, Arrhythmia, Arrhythmogenesis, Antiarrhythmia agents
Causes of prolonged QT interval and torsades de pointes
Prolongation of the QT interval (the time elapsed between ventricular depolarization and repolarization) on the surface ECG is caused by an increase in the action potential duration (APD) of ventricular myocytes. It can result from a number of causes. Prolongation of the QT can occur as a consequence of congenital defects that prolong the APD, a reduction in the cardiac slowly activating component (IKS) or the rapidly activating component (IKr) of the delayed rectifier potassium current, a reduction in the inward rectifier potassium current (IK1) or an increase in the late sodium current (INa). The long QT syndrome (LQTS) phenotype is not the consequence of a single defect; it can arise from several possible genetic defects occurring in the genes encoding for the four cardiac ion channels. Since Keating et all confirmed the genetic basis of LQTS in 1991, seven genotypes of LQTS have been identified. The most recently described genotype affects chromosome 17 of the KCNJ2 gene.2 Prolongation of the QT interval also can occur in response to an increasing number of diverse drugs. Many of these agents are IKr blockers, some also block IKS, and some, such as the positive inotropic agent DPI 201-106, increase late INa. Other forms of acquired long QT have been recognized, including the QT prolongation that accompanies hypertrophic cardiomyopathy, dilated cardiomyopathy, or heart failure, and the QT prolongation occurring in the early phases after myocardial infarction. Investigations indicate a coordinated reduction in IKr and IKs is partially responsible for prolongation of the QT interval after myocardial infarction. In some cases, increases in late INa and in the Na+/Ca2+ exchange current (INaCa) occur. Prolongations of the QT interval associated with cardiomyopathy differ from congenital LQTS in that the changes in IKr, IKs, and INaCa occur simultaneously with cardiomyopathy, whereas in the familial form of LQTS individual ion channels are affected in the majority of cases. Prolongation of the QT interval and induction of torsades de pointes (TdP) likely is a consequence of one or more of the aforementioned factors: the presence of one or more drugs, a genetic predisposition, or an underlying heart disease. When these forms of QT prolongation occur concurrently, each can amplify the response to the other.
Uninjured adjacent cells of the same kind likely have the same form and duration of the action potential. However, different kinds of cells express different mixtures of ion channels and thus display different resulting action potentials, in the native state or as affected by drugs. A cross-section of the normal myocardium in humans contains at least three different populations of cells—epicardial, endocardial, and midmyocardial (M)—with different native electrophysiologic properties and different responses to drugs. When the action potentials of adjacent cells have significantly different durations, the scene is set for reentrant arrhythmias, including TdP. Figure 1 illustrates the principal hypothesis of this article. Basically, the article proposes that the intrinsic heterogeneities within the myocardium, principally in the form of transmural dispersion of repolarization (TDR) and transseptal dispersion of repolarization, when amplified by agents with class III actions and/or the various mutations and cardiomyopathies associated with LQTS, lead to a reduction in net repolarizing current. The reduction in net repolarizing current produces prolongation of the APD. However, because APD is preferentially prolonged in the M cells, not only is the QT interval prolonged but TDR increases, creating a vulnerable window across the ventricular wall. The decrease in net repolarizing current predisposes to the development of early afterdepolarization (EAD)-induced triggered activity, which provides the extrasystoles that then capture this vulnerable window and precipitate TdP. Although an EAD-induced extrasystole is believed to be responsible for the premature beat that initiates TdP, maintenance of the arrhythmia is generally thought to be the result of circus movement reentry.3
Figure 1.
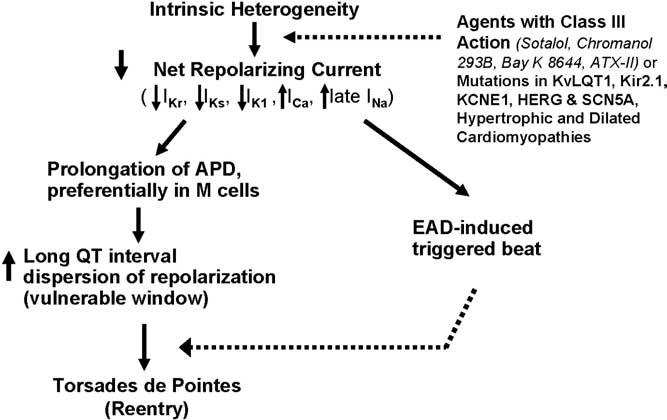
Mechanism by which transmural dispersion of repolarization and transseptal dispersion of repolarization precipitate torsades de pointes. APD = action potential duration; EAD = early afterdepolarization; M cells = midmyocardial cells.
Central role of the M cell
At the heart of this hypothesis is the fact that M cells prolong preferentially. One hallmark of the M cell is the ability of its APD to prolong more than the other cell types in response to a slowing of rate. This feature of the M cell is more prominent in the left ventricle than in the right ventricle (Figure 2).4 Another distinguishing characteristic of the M cell is the ability of its APD to prolong more in response to drugs that normally prolong the APD, that is, agents with class III actions. Figure 3 depicts the action potentials of preparations isolated from the epicardial region, the M region, and from the endocardial region of a canine heart, recorded at progressively slower rates. As shown, low therapeutic concentrations of quinidine produce a preferential prolongation of the APD of the M cells, a prolongation more pronounced than seen in cells from either the epicardium or the endocardium. This effect is most prominent at slower rates. At higher drug concentrations, the dispersion of repolarization that develops is attenuated.5 This result is seen because at lower concentrations quinidine principally blocks IKr, whereas higher drug concentrations also block IKs and INa, producing prolongation of the APD of the M cells more consistent with those of epicardial and endocardial cells. This process is believed to explain why agents such as quinidine produce TdP at low therapeutic concentrations but never at high or toxic concentrations.
Figure 2.
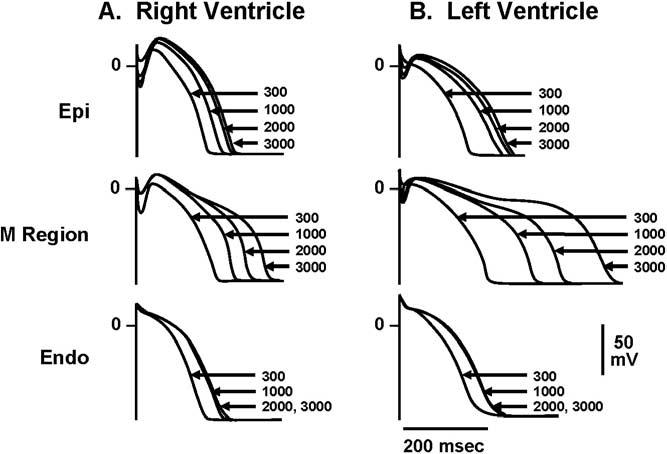
Cells from the midmyocardial region prolong more than other cell types in response to a slowing of rate, an effect more prominent in the left ventricle than the right ventricle. Endo = endocardial region; Epi = epicardial region; M = midmyocardial. (From Antzelevitch C, Sicouri S, Litovsky SH, Lukas A, Krishnan SC, Di Diego JM, Gintant GA, Liu DW. Heterogeneity within the ventricular wall. Electrophysiology and pharmacology of epicardial, endocardial, and M cells. Circ Res 1991;69:1427–1449, with permission.)
Figure 3.
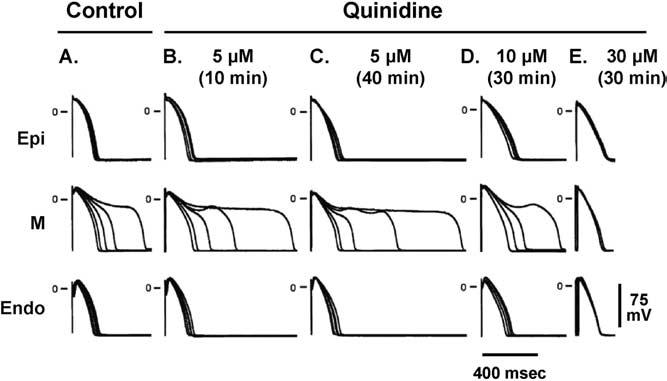
The action potential of M cells is prolonged more than that of epicardial or endocardial cells in the presence agents such as quinidine. Endo = endocardial region; Epi = epicardial region; M = midmyocardial. (From Antzelevitch C, Shimizu W, Yan GX, Sicouri S, Weissenburger J, Nesterenko VV, Burashnikov A, Di Diego J, Saffitz J, Thomas GP. The M cell: its contribution to the ECG and to normal and abnormal electrical function of the heart. J Cardiovasc Electrophysiol 1999;10:1124–1152, with permission from Blackwell Publishing. http://www.blackwell-synergy.com)
M cells exhibit a prominent response to each of the four principal mechanisms responsible for QT prolongation: IKr block, IKs block, activation of INa, and activation of ICa. Endocardial and epicardial cells show only modest prolongation in response to IKr block and activation of the calcium current, whereas Purkinje fibers typically show no response to IKs block. For this reason, we and others have advocated the use of M cells in screening for QT-prolonging drugs.6
The longer APD of the M cell results from the presence of a relatively weak IKs current density and the presence of relatively stronger INaCa and INa current densities compared to endocardial or epicardial cells. These ionic distinctions sensitize the M cells to drugs that prolong the APD. For example, the pure IKr blocker erythromycin produces a dose-dependent prolongation of APD, which is significantly greater in the M cell than in either the endocardium or epicardium, creating a very large TDR. Likewise, another antibiotic, moxifloxacin, produces a dangerous increase in TDR at high concentrations, which once more is a consequence of the preferential prolongation of the APD of the M cell.
Arterially perfused left ventricular wedge preparation
Figure 3 demonstrates the response of the APD to drugs in isolated tissues that have been removed from different regions of the ventricular wall. These cells are not interacting with each other electrotonically. Response to drugs with electrotonic interaction among the various layers of the heart can be observed with a preparation such as that illustrated in Figure 4, the arterially perfused left ventricular wedge preparation. The wedge preparation is a few square centimeters of full-thickness ventricular wall excised from a canine heart. The specimen is immersed in a physiologic bath and perfused through its native vessels. Floating glass microelectrodes record simultaneous action potentials in each layer of tissue from the transmural surface. Electrodes in the bath at some distance from the endocardial and epicardial surfaces simultaneously record the ECG along the same axis, allowing correlation of transmembrane and ECG activity. This preparation is described in detail elsewhere.7
Figure 4.
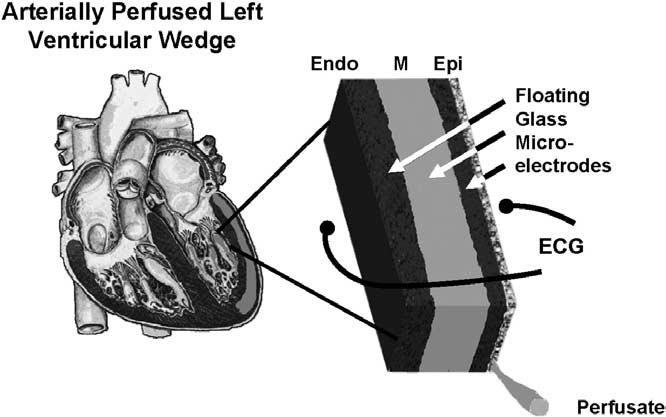
Schematic of arterially perfused canine left ventricular wedge preparation. Transmembrane action potentials can be simultaneously recorded from epicardial, M region, endocardial sites or subendocardial Purkinje fibers using three floating microelectrodes. ECG = electrocardiogram.
Studies using the left ventricular wedge preparation have developed experimental models to assess the contribution of electrical heterogeneity across the ventricular wall to the manifestation of the T wave under conditions of acquired LQTS, mimicking the genetic defects linked to the congenital syndrome. Figure 5 shows a recording of action potentials from endocardium, from the M cell region, and from epicardium, correlated with the ECG. Here, APDs characteristic of the LQT1, LQT2, and LQT3 form of the long QT syndrome have been produced experimentally: LQT1 with an IKs blocker and a β-adrenergic agonist, LQT2 with an IKr blocker and low potassium, and LQT3 with an agent that increases late INa. In all cases, the peak of the T wave in the ECG coincides with the repolarization of the epicardial action potential, whereas the end of the T wave coincides with repolarization of the M cell action potential. Repolarization of the endocardial cell is intermediate between that of the M cell and the epicardial cell. TDR, defined as the difference in repolarization time between M and epicardial cells, is denoted below the ECG traces. The ECG manifestations of the wedge preparation are similar to those observed clinically in LQTS. Isoproterenol in the presence of the experimental agent chromanol 293B produces preferential prolongation of the APD of the M cell, resulting in an accentuated TDR and broad-based T waves as commonly seen in LQT1 patients. D-Sotalol in the presence of low potassium gives rise to low-amplitude T waves with a notched or bifurcated appearance as a result of significant slowing of repolarization, as commonly seen in LQT2 patients. ATX-II, a sea anemone toxin, markedly prolongs the QT interval, widens the T wave, and causes a sharp rise in the TDR. ATX-II also produces a marked delay in onset of the T wave because of relatively large effects of the drug on the APD of epicardium and endocardium, consistent with the late-appearing T-wave pattern observed in LQT3 patients. QT interval prolongs in all cases, but because the M cells are preferentially prolonged by all of these agents, TDR also increases from 42 to 85, 59 to 80, and 53 to 156 in the three models.3 Development of TdP is seen in these wedge preparations only in the presence of large TDR.
Figure 5.
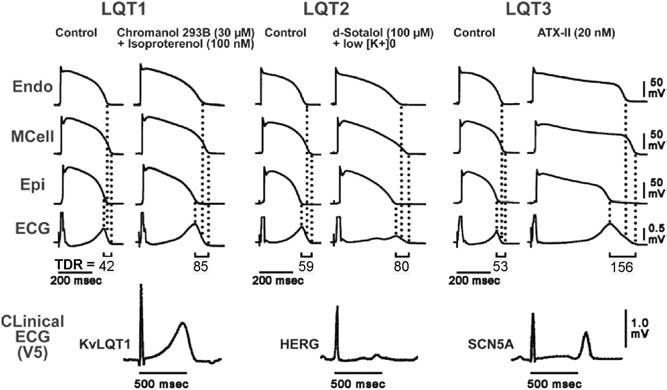
Transmembrane action potentials and transmural electrocardiograms in control and LQT1, LQT2, and LQT3 models of LQTS (arterially perfused canine left ventricular wedge preparations), and clinical ECG (lead V5) of patients with LQT1 (KvLQT1 defect), LQT2 (HERG defect), and LQT3 (SCN5A defect) syndromes. Isoproterenol plus chromanol 293B, an IKs blocker, d-sotalol plus low K+, and ATX-II, an agent that slows inactivation of late INa, are used to mimic the LQT1, LQT2, and LQT3 syndromes, respectively. ECG = electrocardiogram; Endo = endocardial region; Epi = epicardial region; M = midmyocardial. (From Antzelevitch C, Shimizu W. Cellular mechanisms underlying the long QT syndrome: Curr Opin Cardiol 2002;17:43–51, with permission.)
Figure 6 illustrates TdP that developed spontaneously. The first grouping in panel A shows a spontaneous extrasystole that failed to initiate torsade. The second grouping shows a spontaneous premature beat that succeeded. This extrasystole most likely arose from an EAD in subendocardial Purkinje fibers or in an M cell. With lowering of the temperature, this extrasystolic activity and all EAD activity can be eliminated, but the presence of a reentrant substrate in the form of TDR is evidenced by the ability to induce TdP by introducing an extrinsic stimulus to epicardium.
Figure 6.
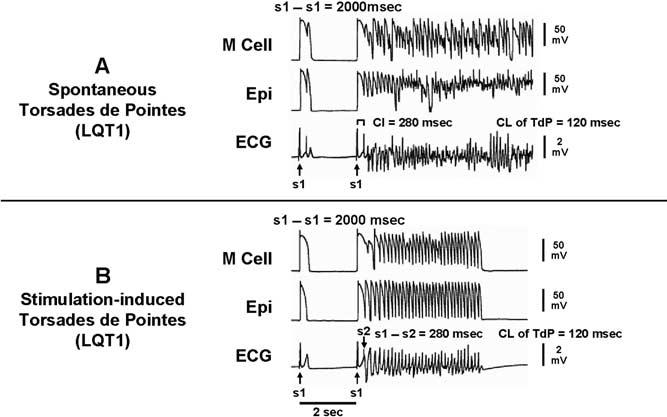
A: Spontaneous torsades de pointes in an LQT1 model of a wedge preparation. Each trace shows action potentials recorded simultaneously from M and epicardial cells, together with a transmural ECG. B: Stimulation induced torsade de pointes. ECG = electrocardiogram; Epi = epicardial region; M = midmyocardial.
Exceptions to the rule
Not all agents that prolong the QT interval increase TDR. Amiodarone, a potent antiarrhythmic agent used in the management of both atrial and ventricular arrhythmias, is rarely associated with TdP. Chronic administration of amiodarone produces greater prolongation of APD in epicardium and endocardium but less of an increase, or even a decrease at slow rates, in the M region, thereby reducing TDR.8 In a dog model of chronic complete AV block and acquired LQTS, administration of amiodarone for 6 weeks produced major QT prolongation without producing TdP. In contrast, after administration of dronedarone for 6 weeks, TdP occurred in four of eight dogs with the highest spatial dispersion of repolarization (105 ± 20 ms).9 Sodium pentobarbital is another agent that prolongs the QT interval but reduces TDR. Figure 7 shows tracings made using arterially perfused canine left ventricular wedge preparations. Pentobarbital produces a dose-dependent prolongation of the QT interval, accompanied by a reduction in TDR from 51 to 27 ms.10 TdP is never seen under these conditions, nor can it be induced with programmed stimulation. Amiodarone and pentobarbital have in common the ability to block IKs, IKr, and late INa. This combination produces preferential prolongation of the APD of epicardium and endocardium so that the QT interval is prolonged, but TDR is reduced and TdP does not occur.
Figure 7.
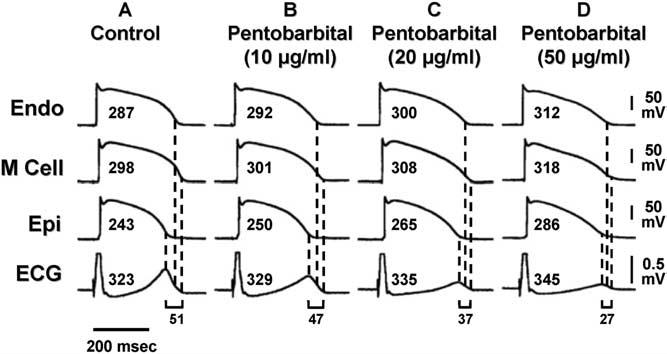
Pentobarbital prolongs the APD of epicardial and endocardial cells and, to a lesser extent, that of the M cell, thus prolonging the QT interval but reducing TDR. The effects of pentobarbital on the QT interval and APD were biphasic. In this study, pentobarbital 10 μg/mL pentobarbital further prolonged APD of epicardial and endocardial cells more than that of the M cell, whereas 20 to 50 μg/mL pentobarbital abbreviated the APD of epicardial and endocardial cells less than that of the M cell, thus abbreviating the QT interval and markedly reducing TDR. Torsades de pointes was never observed, nor could it be induced with programmed electrical stimulation. ECG = electorcardiogram; Endo = endocardial region; Epi = epicardial region; M = midmyocardial; TDR = transmural dispersion of repolarization. (From Shimizu W, McMahon B, Antzelevitch C. Sodium pentobarbital reduces transmural dispersion of repolarization and prevents torsade de pointes in models of acquired and congenital long QT syndrome. J Cardiovasc Electrophysiol 1999;10:154–164, with permission.)
Cisapride is another agent that blocks both inward and outward currents. Cisapride produces a biphasic dose–response curve as a consequence of the multichannel effect. The early effect results from IKr block. The later effect results from the addition of an inward current block. A biphasic dose–response relationship is seen for TDR. Figure 8 depicts a global measure of transmural dispersion in the form of Tpeak-Tend from the ECG recorded across the wedge. Peak dispersion occurred at 0.2 μmol/L. TdP was observed only at this concentration, when TDR was maximally prolonged, and not at peak QT prolongation. At higher cisapride concentrations, QT was further prolonged but TDR was diminished, and TdP could no longer be induced.11 This finding suggests spatial dispersion of repolarization is more significant than prolongation of the QT interval in determining the substrate for TdP.
Figure 8.
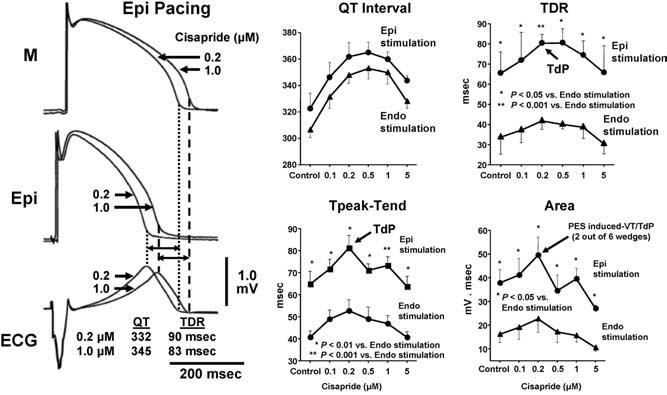
Torsades de pointes (TdP) could be induced with programmed electrical stimulation only at a low concentration of cisapride (0.2 μmol/L), when TDR was maximally prolonged. Moreover, TdP could only be induced during epicardial (but not endocardial) activation of the wedge, which was found to augment TDR. At higher concentrations of cisapride, QT was further prolonged, TDR was diminished, and TdP could no longer be induced. ECG = electrocardiogram; Epi = epicardial region; M = midmyocardial; TdP = torsade de pointes; TDR = transmural dispersion of repolarization. (From Di Diego JM, Belardinelli L, Antzelevitch C. Cisapride-induced transmural dispersion of repolarization and torsades de pointes in the canine left ventricular wedge preparation during epicardial stimulation. Circulation 2003; 108:1027–1033, with permission.)
Sympathetic agonists increase likelihood of TdP
Anesthetics such as pentobarbital can dramatically reduce the ability of QT-prolonging agents to increase TDR. Other agents, such as the sympathetic agonists, can dramatically amplify the effects of such drugs to increase TDR. Chromanol 293B is an agent that blocks the IKs current. Figure 9 shows transmembrane activity recorded simultaneously from endocardial, epicardial, and M regions, together with a transmural ECG in the absence and presence of chromanol 293B 30 μmol/L and in the presence of isoproterenol 100 nmol/L and chromanol 293B. Under control conditions, chromanol 293B produces a transmural dispersion of 42 ms. By blocking the IKs current, chromanol 293B prolonged the APD and the QT interval of the three cell types homogeneously, neither increasing TDR nor widening the T wave. TdP is never seen under these conditions. Chromanol 293B is not the ideal antiarrhythmic drug, however, because β-adrenergic stimulation, as with isoproterenol, abbreviates the APD of epicardial and endocardial cells but not that of the M cell, resulting in an accentuated, almost doubled, TDR.12 TdP develops under these conditions. This finding explains why long QT patients, particularly those with LQT1, are so sensitive to sympathetic influences, and reinforces the previous observation that the risks associated with LQTS result less from prolongation of the QT interval but more from the increase in spatial dispersion that very often, but not always, accompanies prolongation of the QT interval.
Figure 9.
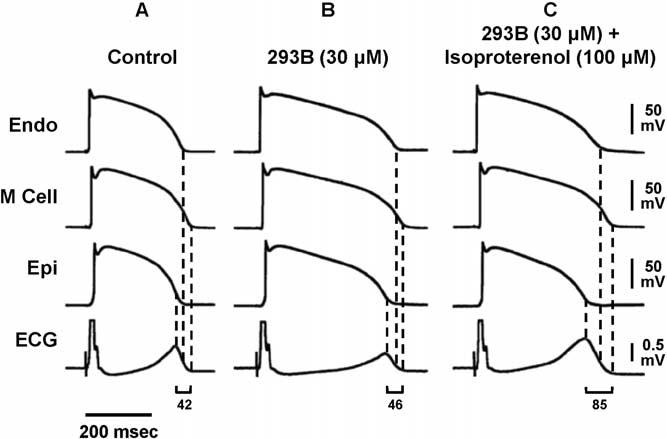
Transmembrane APs and transmural ECG under control conditions (A), after addition of chromanol 293B 30 μmol/L (B), and after further addition of isoproterenol 100 nmol/L (C). Chromanol 293B prolonged APs of three cell types and QT interval but did not increase TDR; 42–46 ms) or widen the T wave. Isoproterenol in the continued presence of chromanol 293B abbreviated AP of epicardial and endocardial cells but not that of M cells, resulting in an accentuated TDR (85 ms) and broad-based T waves as commonly seen in LQT1 patients. ECG = electrocardiogram; Endo = endocardial region; Epi = epicardial region; M = midmyocardial. (From Shimizu W, Antzelevitch C. Cellular basis for the ECG features of the LQT1 form of the long-QT syndrome: effects of beta-adrenergic agonists and antagonists and sodium channel blockers on transmural dispersion of repolarization and torsade de pointes. Circulation 1998;98:2314 –2322, with permission.)
Figure 10 summarizes the effects of the drugs just described on TdP. In the first example, pure IKr blockers such as sotalol, dofetilide, and erythromycin produce a dose-dependent prolongation of the QT interval, which is associated with a dose-dependent prolongation of TDR. When TDR reaches the threshold for reentry, which is approximately 90 ms in the wedge, TdP occurs. With more complex drugs such as quinidine and cisapride, a biphasic dose-response relationship is seen. TDR parallels QT, but the two can peak at different concentrations. TdP occurs when and if TDR reaches the threshold value. Still other drugs produce a dose-dependent prolongation of QT but a much smaller change in TDR, and threshold values for TdP are rarely reached. As shown, chromanol 293B, pentobarbital, amiodarone, and the new antianginal agent ranolazine produce a dose-dependent prolongation of the QT interval that is not associated with an increase in TDP. TdP rarely, if ever, occurs under these circumstances. TdP is never observed with drugs associated with a dose-dependent prolongation of QT but a dose-dependent reduction of TDR or no increase in TDR. Collectively, these examples demonstrate that arrhythmogenesis does not result from prolongation of QT interval but rather from the increase in TDR that often accompanies prolongation of the QT interval. This finding also explains why long QT patients, particularly those with LQT1, are so sensitive to sympathetic influences, as adrenergic stimulation dramatically increases TDR.
Figure 10.
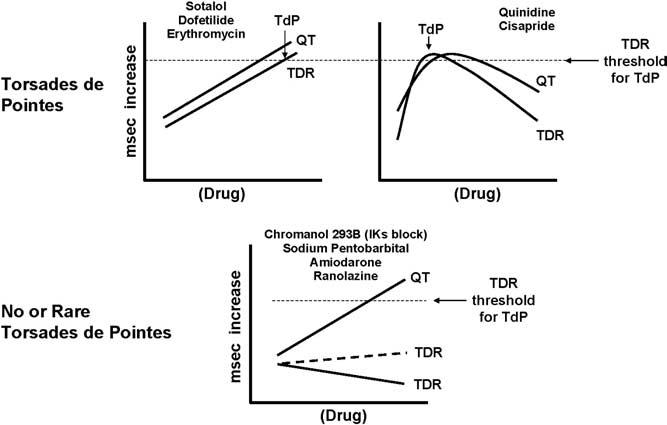
IKr blockers produce a dose-dependent prolongation of QT that is associated with a dose-dependent accentuation of TDR. When TDR reaches the threshold for reentry, TdP develops. More complex drugs, such as quinidine and cisapride, show a biphasic relationship, with a dose-dependent prolongation of QT until increasing concentrations produce inhibition of inward currents, leading to a reduction of QT. TdP occurs when and if TDR reaches the threshold value for reentry. Other drugs, such as chromanol 293B, pentobarbital, and amiodarone, produce a dose-dependent prolongation of QT but a much smaller change in TDR. Here, threshold values for TdP are rarely reached. TdP is never observed with drugs that prolong QT but produce a dose-dependent reduction of TDR. TdP = torsade de pointes; TDR = transmural dispersion of repolarization.
No reliable predictive assay for risk of TdP
Although no single in vitro or in vivo assay can predict with absolute accuracy which drugs will produce TdP in humans, evaluation of the electrophysiologic events underlying TdP, including prolongation of the ventricular APD, generation of EADs, and increased TDR, is useful for identifying drugs having the potential to cause TdP. The arterially perfused canine left ventricular wedge model is particularly useful in this regard. Studies have indicated that drugs that do not increase TDR have little or no potential to induce TdP despite causing prolongation of the QT interval.13 Amiodarone is an exception to this rule. Amiodarone produces TdP in human patients,14 although it does not increase TDR. In fact, amiodarone reduces TDR in the canine model.8
Thus, QT prolongation is only a mediocre predictor of TdP. Some drugs, such as amiodarone and pentobarbital, can cause large QT prolongations but, by reducing TDR, actually reduce the likelihood of TdP. On the other hand, some patients with congenital LQTS have normal QT duration but still have an increased risk for sudden death. TDR, represented by Tpeak-Tend, could provide a more accurate electrophysiologic marker of risk than does the QT interval. Use of this marker will require a great deal of prospective validation, both in in vivo models and in the clinic. Yamaguchi et al15 made an important first step for clinical validation of the use of Tpeak-Tend in predicting TdP in patients with acquired LQTS. These researchers divided a series of 27 patients with acquired LQTS into two groups, those with TdP and those without TdP. They measured QT dispersion, the difference between the maximum and the minimum QT intervals, and the Tpeak-Tend (Tpe) interval divided by the QT interval (Tpe) in lead V5. Logistic regression analysis revealed that Tpe was a reliable predictor for TdP in these patients, whereas QT dispersion was not. A Tpe/QT threshold value of 0.28 ms appeared to discriminate between patients who developed TdP and those who did not. The authors concluded that Tpe may be the best predictor for TdP in patients with acquired LQTS. Although this finding represents only the first step in the long process of validation, clearly prolongation of the ventricular action potential, and presumably the QT interval, is not the sole determinant of a drug's potential to cause TdP. Other electrophysiologic markers must be developed in order to more accurately assign clinical risk.
References
- 1.Keating M, Atkinson D, Dunn C, Timothy K, Vincent GM, Leppert M. Linkage of a cardiac arrhythmia, the long-QT syndrome, and the Harvey ras-1 gene. Science. 1991;252:704–706. doi: 10.1126/science.1673802. [DOI] [PubMed] [Google Scholar]
- 2.Ricardo Perez Riera A, Ferreira C, Dubner SJ, Schapachnik E. Andersen syndrome: the newest variant of the hereditary-familial long QT syndrome. Ann Noninvas Electrocardiol. 2004;9:175–179. doi: 10.1111/j.1542-474X.2004.92552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antzelevitch C, Shimizu W. Cellular mechanisms underlying the long QT syndrome. Curr Opin Cardiol. 2002;17:43–51. doi: 10.1097/00001573-200201000-00007. [DOI] [PubMed] [Google Scholar]
- 4.Antzelevitch C, Sicouri S, Litovsky SH, Lukas A, Krishnan SC, Di Diego JM, Gintant GA, Liu DW. Heterogeneity within the ventricular wall. Electrophysiology and pharmacology of epicardial, endocardial, and M cells. Circ Res. 1991;69:1427–1449. doi: 10.1161/01.res.69.6.1427. [DOI] [PubMed] [Google Scholar]
- 5.Antzelevitch C, Shimizu W, Yan GX, Sicouri S, Weissenburger J, Nesterenko VV, Burashnikov A, Di Diego J, Saffitz J, Thomas GP. The M cell: its contribution to the ECG and to normal and abnormal electrical function of the heart. J Cardiovasc Electrophysiol. 1999;10:1124–1152. doi: 10.1111/j.1540-8167.1999.tb00287.x. [DOI] [PubMed] [Google Scholar]
- 6.Fenichel RR, Malik M, Antzelevitch C, Sanguinetti M, Roden DM, Priori SG, Ruskin JN, Lipicky RJ, Cantilena LR. Independent Academic Task Force. Drug-induced torsade de pointes and implications for drug development. J Cardiovasc Electrophysiol. 2004;15:475–495. doi: 10.1046/j.1540-8167.2004.03534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan GX, Shimizu W, Antzelevitch C. Characteristics and distribution of M cells in arterially perfused canine left ventricular wedge preparations. Circulation. 1998;98:1921–1927. doi: 10.1161/01.cir.98.18.1921. [DOI] [PubMed] [Google Scholar]
- 8.Sicouri S, Moro S, Litovsky S, Elizari MV, Antzelevitch C. Chronic amiodarone reduces transmural dispersion of repolarization in the canine heart. J Cardiovasc Electrophysiol. 1997;8:1269–1279. doi: 10.1111/j.1540-8167.1997.tb01018.x. [DOI] [PubMed] [Google Scholar]
- 9.van Opstal JM, Schoenmakers M, Verduyn SC, de Groot SH, Leunissen JD, van Der Hulst FF, Molenschot MM, Wellens HJ, Vos MA. Chronic amiodarone evokes no torsade de pointes arrhythmias despite QT lengthening in an animal model of acquired long-QT syndrome. Circulation. 2001;104:2722–2727. doi: 10.1161/hc4701.099579. [DOI] [PubMed] [Google Scholar]
- 10.Shimizu W, McMahon B, Antzelevitch C. Sodium pentobarbital reduces transmural dispersion of repolarization and prevents torsade de pointes in models of acquired and congenital long QT syndrome. J Cardiovasc Electrophysiol. 1999;10:154–164. doi: 10.1111/j.1540-8167.1999.tb00656.x. [DOI] [PubMed] [Google Scholar]
- 11.Di Diego JM, Belardinelli L, Antzelevitch C. Cisapride-induced transmural dispersion of repolarization and torsade de pointes in the canine left ventricular wedge preparation during epicardial stimulation. Circulation. 2003;108:1027–1033. doi: 10.1161/01.CIR.0000085066.05180.40. [DOI] [PubMed] [Google Scholar]
- 12.Shimizu W, Antzelevitch C. Cellular basis for the ECG features of the LQT1 form of the long-QT syndrome: effects of beta-adrenergic agonists and antagonists and sodium channel blockers on transmural dispersion of repolarization and torsade de pointes. Circulation. 1998;98:2314–2322. doi: 10.1161/01.cir.98.21.2314. [DOI] [PubMed] [Google Scholar]
- 13.Belardinelli L, Antzelevitch C, Vos MA. Assessing predictors of drug-induced torsade de pointes. Trends Pharmacol Sci. 2003;24:619–625. doi: 10.1016/j.tips.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 14.Hohnloser SH, Klingenheben T, Singh BN. Amiodarone-associated proarrhythmic effects: a review with special reference to torsade de pointes tachycardia. Ann Intern Med. 1994;121:529–535. doi: 10.7326/0003-4819-121-7-199410010-00009. [DOI] [PubMed] [Google Scholar]
- 15.Yamaguchi M, Shimizu M, Ino H, Terai H, Uchiyama K, Oe K, Mabuchi T, Konno T, Kaneda T, Mabuchi H. T wave peak-to-end interval and QT dispersion in acquired long QT syndrome: a new index for arrhythmogenicity. Clin Sci (Lond) 2003;105:671–676. doi: 10.1042/CS20030010. [DOI] [PubMed] [Google Scholar]