

Abstract
Previously we showed L-4F, a novel apolipoprotein A-I (apoA-I) mimetic, improved vasodilation in two dissimilar models of vascular disease; hypercholesterolemic low-density lipoprotein (LDL) receptor null (Ldlr −/−) mice and transgenic sickle cell disease mice. Here we determine the mechanisms by which D-4F improves vasodilation and arterial wall thickness in hypercholesterolemic Ldlr −/− mice and Ldlr −/−/apoA-I null (apoA-I −/−), double knockout mice. Ldlr −/− and Ldlr −/−/apoA-I −/− mice were fed western diet (WD) ± D-4F. Oral D-4F restored endothelium- and eNOS-dependent vasodilation in direct relationship to duration of treatments and reduced wall thickness in as little as 2 weeks in vessels with pre-existing disease in Ldlr −/− mice. D-4F had no effect on total or HDL cholesterol concentrations but reduced proinflammatory HDL levels. D-4F had no effect on plasma myeloperoxidase (MPO) concentrations but reduced MPO association with apoA-I as well as 3-nitrotyrosine in apoA-I. D-4F increased endothelium- and eNOS-dependent vasodilation in Ldlr −/−/apoA-I −/− mice but did not reduce wall thickness as it had in Ldlr −/− mice. Vascular endothelial cells were treated with 22-hydroxycholesterol (22-OHC) ± L-4F. 22-OHC decreased nitric oxide (•NO) and increased superoxide anion (O2 •−) production and increased ABCA-1 and collagen expression. L-4F restored •NO and O2 •− balance, had little effect on ABCA-1 expression but reduced collagen expression. These data demonstrate that although D-4F restores vascular endothelial cell and eNOS function to increase vasodilation, HDL containing apoA-I, or at least some critical concentration of the anti-atherogenic lipoprotein, is required for D-4F to decrease vessel wall thickness.
Keywords: cardiovascular diseases, hypercholesterolemia, lipoproteins, nitric oxide synthase, vasodilation
Introduction
Endothelial cells play an important role in maintaining vascular health. One of the earliest physiologic and most sensitive changes in hypercholesterolemia is loss of endothelium-and endothelial nitric oxide synthase (eNOS)-dependent vasodilation, which appears to occur before structural changes in the vessel wall.1, 2 In a prospective human study, angiography was used to observe impaired endothelium-dependent vasodilation in coronary arteries and was found to be a strong prognostic indicator of vascular pathology.3 Such findings3 as well as those from others4, 5 support the concept that endothelium-dependent vasodilation provides important insights into the atherogenic state of the vessel wall.
We previously reported L-4F, when administered by intraperitoneal injection (ip), increased eNOS–dependent vasodilation of small arteries from hypercholesterolemic Ldlr −/− mice and from transgenic sickle cell mice.6 D-4F is the same as L-4F except it is synthesized from D-amino acids. In the D conformation, 4F is resistant to metabolism compared to 4F synthesized from L-amino acids, which is rapidly degraded after oral administration.8 Oral D-4F reduced lesions in Ldlr −/− mice fed WD by greater than 79% by restoring HDL function without significantly altering cholesterol or HDL-cholesterol levels.7 Interestingly, D-4F also reduced lesions in apoE −/− mice, which were hypercholesterolemic from birth.7 However, to date, no studies have been performed to determine if oral D-4F also restores vasodilation in hypercholesterolemic mice.
D-4F is believed to protect vascular function by binding proinflammatory lipids.8 Proinflammatory oxysterols, such as hydroxycholesterol increase apoptosis of endothelial cells,9, 10 while other proinflammatory lipids have been shown to increase susceptibility of HDL to oxidation.11 Previous studies by Reddy et al.12 revealed 22(R)-hydroxycholesterol (22-OHC), one of several forms of oxysterols found in atherosclerotic lesions,13 increases ATP-binding cassette transporter-1 (ABCA1) expression. Endothelial cells typically maintain an anti-oxidant phenotype until exposed to LDL containing a critical threshold of lipid peroxides, at which point they are converted into a prooxidant phenotype that promotes LDL oxidation.14 Likewise, when endothelial cells are exposed to 22-OHC, they become proinflammatory and promote oxidation of LDL via ABCA1 transportion of oxidized phospholipids from the endothelial cell to the LDL particle.12 As 22-OHC does not contain a hydroperoxide for initiating or propagating oxidation, the mechanism by which 22-OHC-treated endothelial cells take on a proinflammatory phenotype is likely due to altered cell signaling more than lipid peroxidation.
Recently, it was reported myeloperoxidase (MPO) binds to HDL and subsequently increases nitrosation of apoA-I which correlates with a decrease in the ability of HDL to promote cholesterol efflux from cholesterol-loaded macrophages.15 Although MPO’s role in atherosclerosis in mice is controversial,16,17 in murine studies where neutrophils are clearly involved, MPO has been implicated as a mechanism of lipid peroxidation and vascular endothelial cell dysfunction18 in ways that are consistent with how MPO is believed to promote atherosclerosis in humans.19,20
The objectives here were to determine 1) mechanisms by which D-4F improves vasodilation in murine models of hypercholesterolemia; 2) if increases in vasodilation correlate with changes in vessel wall architecture; 3) mechanisms by which D-4F protects HDL against oxidative stress; and, 4) mechanisms by which D-4F reduces vessel wall thickness. Our findings indicate D-4F improves vasodilation in hypercholesterolemic Ldlr −/− mice by increasing eNOS function but, reduces vessel wall thickness in hypercholesterolemic Ldlr −/− mice by an HDL-dependent mechanism.
Materials and Methods
Mice
Male Ldlr −/− mice (6–8 weeks) on a C57BL/6 background were from Jackson Laboratory (Bar Harbor, MA) or from a colony established at the Medical College of Wisconsin. Ldlr −/− mice were maintained on WD, a high-fat, cholesterol diet (No. 88137) from Teklad (Madison, WI) and given D-4F in drinking water (50 μg/mL, ~ 2.5 mL/d/mouse) or by ip (1 mg/Kg/d). Feeding and D-4F schedules for time-dependent studies are shown in Figure 1. Male C57BL/6 mice from Jax Laboratory were maintained on lab chow. Male Ldlr −/− /apoA-I −/− double knockout mice were from a colony maintained at Wake-Forest University School of Medicine.21
Figure 1.
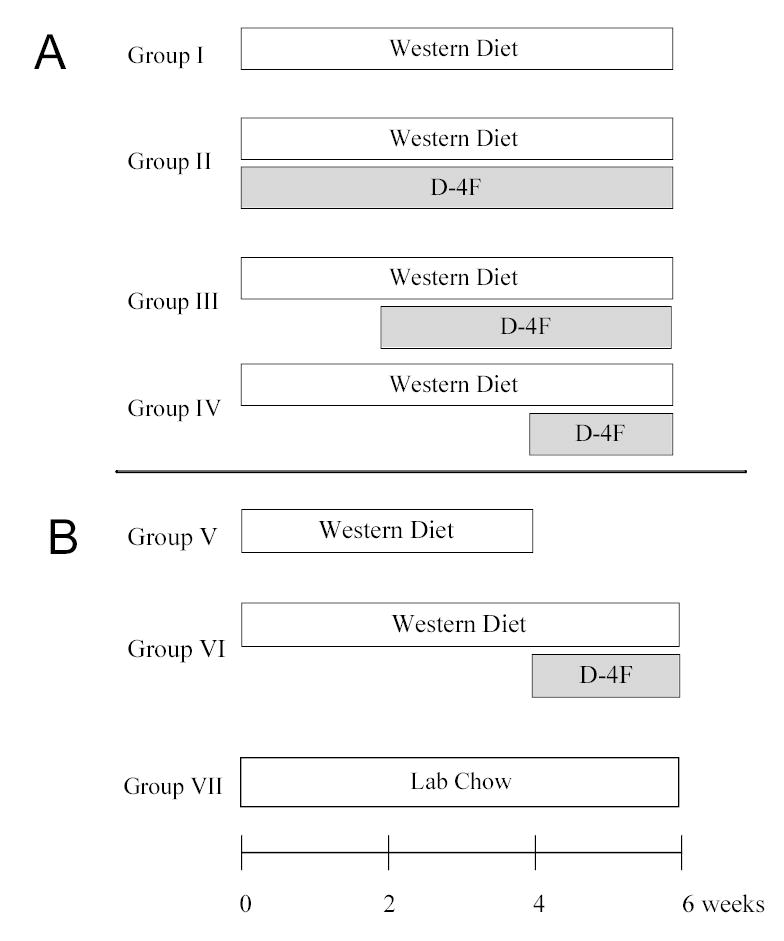
Feeding and D-4F Treatment Protocol. (A) Time-dependent study. Ldlr −/− mice were divided into 4 groups. Group I received WD alone for 6 weeks. In addition to WD, groups II, III and IV received D-4F in their drinking water for the weeks indicated. (B) Pre-existing disease study. Ldlr −/− mice were divided into 3 groups, Group V received WD alone for 4 weeks while group VI was fed WD for 6 weeks with D-4F in their drinking water after 4 weeks on WD. Group VII was fed lab chow diet for 6 weeks.
4F
D-4F and L-4F (Ac-DWFKAFYDKVAEKFKEAFNH2) were synthesized as described.6,7
Vasodilation
Vasodilation of pressurized facialis arteries (180 to 280 microns) was determined as before.6
Wall Thickness
At the end of incubation with papaverine (10−4 M, 4 min),6 an index of wall thickness was calculated using the following formula:
Plasma Cholesterol, HDL Cholesterol and Proinflammatory HDL
Plasma cholesterol and HDL cholesterol were determined by cholesterol oxidase/esterase assays using Cholesterol E and HDL Cholesterol E kits, respectively from Wako Chemicals USA, Inc. (Richmond, VA). After quantification of HDL, proinflammatory HDL was determined.
The proinflammatory HDL assay is based on the observation relative rates of dichlorofluoresceine (DCF) fluorescence are proportional to the levels of seeding molecules of lipid hydroperoxides in HDL.11 Briefly, 1 μg of HDL cholesterol was incubated with CuCl2 (5 μM, final concentration) for 1 hour using a 384-well microtiter plate from MJ Research, Inc. (Waltham, MA). After incubation, 1 μL of DCF solution (2 mg/mL) was added to the HDL-Cu2+ mixture in a total volume of 30 μL. Rates of fluorescence (Ex 485nm; Em 530 nm) were determined on a LJL Biosystem AnalystTM HT from Molecular Devices Corp. (Sunnyvale, CA) over the next 2 hours at 30 minutes intervals.
Effects of 22-OHC on Vascular Endothelial Cell •NO and O2 •− Balance and Expression of ABCA-1 and Collagen
Confluent bovine aortic endothelial cells (EC) were incubated with 22-OHC (25 μM, Sigma, H9384) ± L-4F (10 μg/mL) overnight, and then prepared for 1) A23187 stimulated •NO and O2 •− production as described,22,23 2) Western analysis of ABCA-1 as described,12 or 3) measurements of collagen synthesis based on collagenase-sensitive [3H]proline incorporation as described by Siwik et al.24
MPO/apoA-I and 3-Nitrotyrosine(3NT)/apoA-I
MPO association with apo A-1 was determined by Western blot analysis of apoA-I immunoprecipitates (goat anti-mouse apoA-I, K59166G, Biodesign, Saco, Maine) using a rabbit anti-mouse MPO antibody (07-009, Upstate Biomedical, Charlottesville, VA). 3NT in apoA-I was determined by Western blot analysis of apoA-I immunoprecipitates using mouse anti-nitrotyrosine (05–233, Upstate Biomedical) and anti-apoA-I (K59166G, Biodesign) as primary antibodies and the appropriate HRP conjugated secondary antibodies as described.15 In both cases apoA-I was detected by Western blot analysis using a rabbit anti-mouse apoA-I antibody (K23001R, Biodesign).
Statistical Analysis
Comparisons between two groups were by the student’s t-test. Comparisons between multiple groups were by one-way ANOVA with Boniferroni correction for multiple groups. Comparisons between vasodilation curves were by two-way ANOVA. Minimum levels of significance were set at p<0.05. Statistical analysis was performed using Prism 3 from GraphPad Sotware, Inc.
Results
After 6 weeks on WD, vasodilation of facialis arteries from WD-fed Ldlr −/− mice was reduced compared to vasodilation in C57BL/6 mice and Ldlr −/− mice fed standard lab chow (Figure 2A). Time-dependent studies revealed D-4F improved vasodilation in Ldlr −/− mice fed WD in direct relation to duration of D-4F treatments (Figure 2B). The longer WD-fed Ldlr −/− mice were treated with D-4F, the more vasodilation was increased. Vasodilation in WD-fed Ldlr −/− mice treated with D-4F for 6 weeks (Group II) was increased even beyond that observed in chow-fed Ldlr −/− mice but not in C57BL/6 mice (Figure 2A). As before, L-nitroargininemethylester (L-NAME) essentially ablated acetylcholine (ACh)-induced vasodilation in the pressurized vessels from the experimental groups (I-IV) and papaverine dilated these vessels to ~98% of their maximal diameter, confirming that the loss in eNOS-dependent vasodilation was due to endothelial cell dysfunction, not vascular smooth muscle cell dysfunction (data not shown).6
Figure 2.
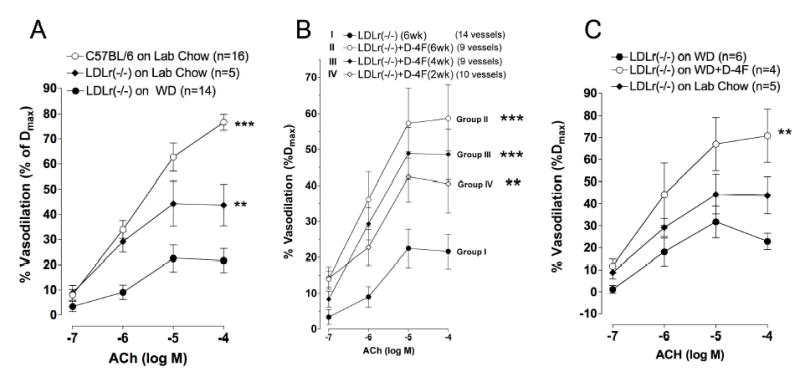
Effects of D-4F on vasodilation. (A) Line graph showing vasodilation from C57BL/6 mice, Ldlr −/− mice fed lab chow and Ldlr −/− mice fed WD for 6 weeks. vasodilation for C57BL/6 mice and Ldlr −/− mice fed lab chow are significantly different from vasodilation for Ldlr −/− mice fed WD. (B) Line graph showing time-dependent effects of D-4F on vasodilation in hypercholesterolemic Ldlr −/− mice. Vasodilation for Ldlr −/− mice fed WD is restored and improved in direct relation to the duration of D-4F treatments. (C) Line graph showing effects of D-4F on vasodilation in hypercholesterolemic mice with pre-existing disease. Two weeks of D-4F treatments increases vasodilation in vessels with pre-existing disease. (** = p<0.02, *** = p<0.01).
To confirm vessels in Ldlr −/− mice from Group IV had pre-existing disease, prior to D-4F treatments, we fed another set of Ldlr −/− mice WD. Vasodilation was impaired in Group V mice after 4 weeks of WD compared to vasodilation in Group VI and Group VII (Figure 2C). With just 2 weeks of oral D-4F treatments, concurrently with WD, vasodilation in Group VI was improved beyond levels in Ldlr −/− mice that were fed lab chow (Group VII) (Figure 2C).
As a non-biased measure of arterial structure, we simply determined the effects of D-4F on arterial wall thickening induced by WD by measuring inside and outside diameters of arteries after incubation with papaverine to eliminate any contribution of endothelial cell function/dysfunction to vessel diameter. Wall thickness in Ldlr −/− mice fed WD was markedly increased compared to the wall thickness in C57BL/6 or Ldlr −/− mice fed lab chow diet (Figure 3A). At all time points tested, oral D-4F reduced wall thickness in Ldlr −/− mice fed WD regardless of duration of oral D-4F treatments (Figure 3B). Wall thickness data confirmed that the vessels in Ldlr −/− mice from Group IV had pre-existing disease at 4 weeks of WD, prior to D-4F treatments (Figure 3C). More importantly, treating WD-fed Ldlr −/− mice with D-4F for only 2 weeks, concurrently with WD, reduced wall thickness to essentially the same thickness as Ldlr −/− mice fed lab chow (Figure 3C).
Figure 3.
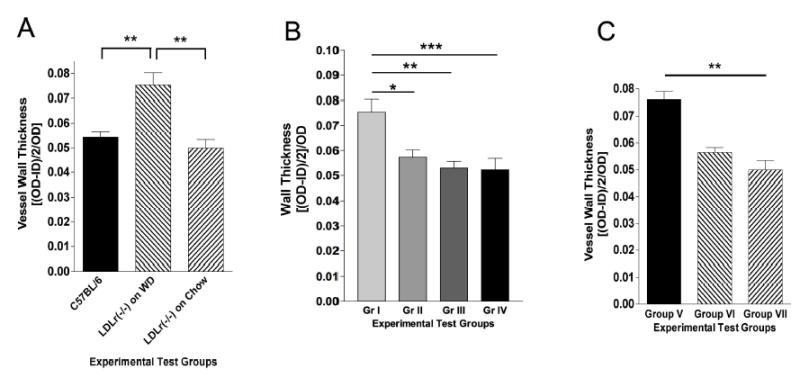
Effects of D-4F on vessel wall thickness. (A) Bar graph showing wall thickness in vessels used for physiological studies in Figure 2A. vessels wall thickness in Ldlr −/− mice fed WD is significantly thicker than in C57BL/6 mice and Ldlr −/− mice fed lab chow. (B) Bar graph showing effects of D-4F on wall thickness in the vessels used for physiological studies in Figure 2B. D-4F decreases wall thickness in as little as 2 weeks of treatments. (C) Bar graph showing wall thickness in the vessels used for physiological studies in Figure 2C. Two weeks of D-4F treatments decrease wall thickness in vessels with pre-exisiting disease. (*=p<0.05, ** = p<0.02, *** = p<0.01).
D-4F had no effect on total cholesterol or HDL cholesterol in Ldlr −/− mice fed WD (Table 1). Although there was a tendency for HDL levels to be higher in Ldlr −/− mice fed WD and treated with D-4F, statistical significance was not achieved. In contrast to D-4F effects on cholesterol, proinflammatory HDL levels were significantly decreased.
Table 1.
Effect of D-4F on plasma total cholesterol, HDL cholesterol and proinflammatory HDL in Ldlr −/− mice fed WD. This table shows the mean ± SEM for total cholesterol, HDL cholesterol and proinflammatory HDL in plasma from WD-fed Ldlr −/− mice treated with D-4F (ip) for 6 weeks. D-4F significantly reduces proinflammatory HDL levels while having little effect on total or HDL cholesterol in the plasma of these mice.
| Ldlr−/− + PBS (n=5) | Ldlr−/− + D-4F (n=6) | p-value | |
|---|---|---|---|
| Total Cholesterol (mg/dL) | 1012±154 | 1014±129 | ns |
| HDL Cholesterol (mg/dL) | 112±11 | 89 ±13 | ns |
| Proinflammatory HDL* | 3.95 x 106 ± 0.34 x106 | 2.43 x 106 ± 0.33 x106 | 0.01 |
Arbitrary Units of Fluorescence per 1 μg HDL cholesterol per minute
To determine mechanisms by which proinflammatory lipids impair endothelial cell function, we treated endothelial cell cultures with 22-OHC ± L-4F. 22-OHC decreased stimulated •NO production (Figure 4A) and increased stimulated O2 •− generation (Figure 4B). L-4F restored stimulated •NO production (Figure 4A) while decreasing stimulated O2 •− production in the cultures treated with 22-OHC (Figure 4B). Western blot analysis confirms 22-OHC increased ABCA1 expression by a liver X receptor-dependent mechanism as was shown earlier by Reddy et al.,12 which was unaltered by L-4F. Next, we examined effects of 22-OHC and L-4F on collagen production as a means of determining whether or not this oxysterol and apo A-I mimetic modulated endothelial cell matrix production. 22-OHC increased collagen synthesis, which L-4F reduced to control levels (Figure 4D).
Figure 4.
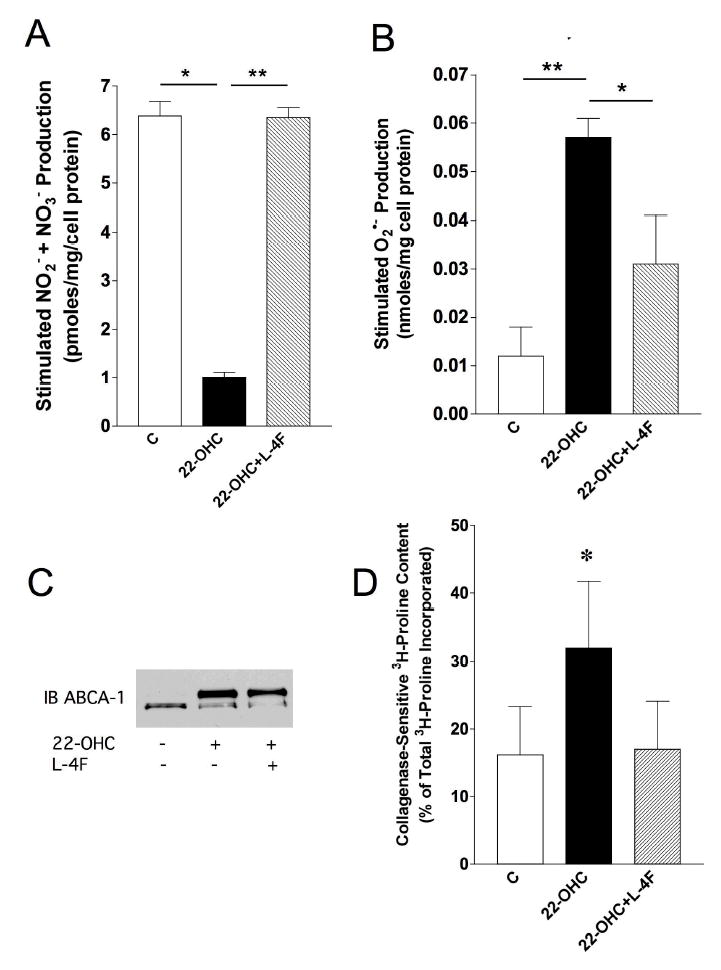
Effects of 4F on EC function. EC cultures, treated with 22-OHC and L-4F were examined for changes in stimulated (A23187, 5 μM, 30 minutes) •NO and O2 •− production as well as expression of ABCA-1 and collagen systhesis. (A) Bar graph showing 22-OHC impairs stimulated •NO production and that L-4F restores •NO in the 22-OHC-treated cultures to control levels. (n=7). (B) Bar graph showing 22-OHC increases stimulated O2 •− production which L-4F significantly reduce. (n=5). (C) Autoradiogram of western blot analysis demonstrating 22-OHC increases ABCA-1 expression, which is unaltered by L-4F. (n=6). (D) Bar graph showing 22-OHC increases collagen synthesis and L-4F decreases synthesis to the levels of control cultures. (n=5). (*=p<0.05, ** = p<0.02).
Plasma meaurements of MPO revealed D-4F had no effect on total MPO concentrations in Ldlr −/− mice fed WD (Figure 5A). However, immunoblotting for MPO and 3NT in apoA-I of apoA-I immunoprecipitates from these mice revealed D-4F decreased both MPO association with and 3NT in apoA-I (Figure 5B and 5C). Immunoblots of apo A-I immunoprecipitates from C57BL/6 plasma that was treated with D-4F in vitro revealed D-4F had no direct effect on MPO association with apoA-I (Figure 5D).
Figure 5.
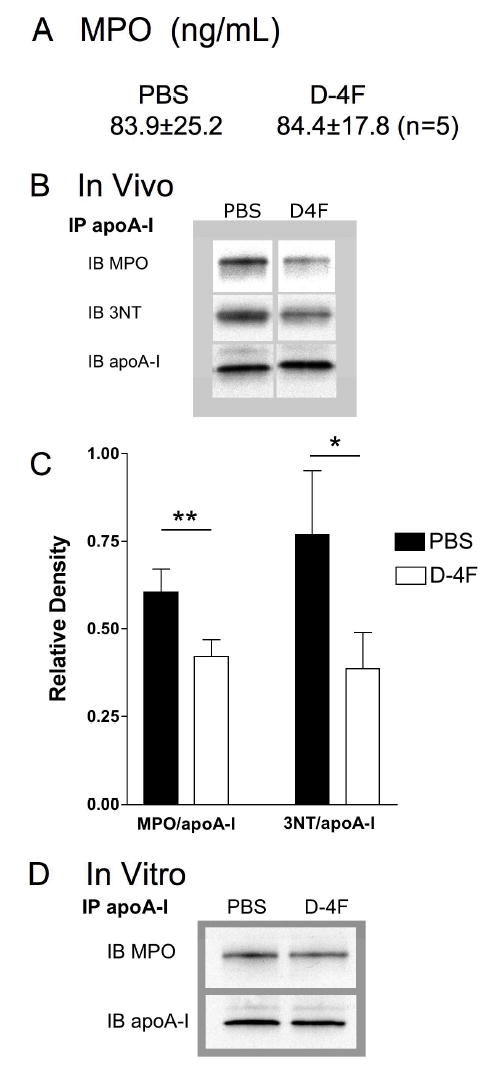
Effects of D-4F Treatments on Plasma MPO Concentrations, MPO association with apoA-I and 3NT formation in apoA-I in Plasma of Hypercholesterolemic Ldlr −/− mice. (A) Plasma MPO concentrations in WD-fed Ldlr −/− mice ± D-4F (ip) were measured by ELISA (HK210, Cell Sciences, Canton, MA). D-4F has no effect on total MPO concentrations in Ldlr −/− mice fed WD. (B) Autoradiograms of western blot analysis for MPO association with and 3NT formation in apoA-I that was immunoprecipitiated from hypercholesterolemia Ldlr −/− mice treated with PBS or D-4F in PBS by ip. Autoradiograms showing D-4F decreases MPO association with apoA-I, with a concurrent decrease in 3NT formation in apoA-I in the plasma of the hypercholesterolemic Ldlr −/− mice. (C) Bar graph showing means ± SEMs of relative density of MPO association and 3NT formation in the WD-fed Ldlr −/− mice ± D-4F treatments (n=5). D-4F decreases MPO association with apoA-I and 3NT residues in apoA-I in WD-fed Ldlr −/− mice. (D) Autoradiograms of western blots for MPO and 3NT in apoA-I of apoA-I immunoprecipitates from plasma from C57BL/6 mice. Plasma was spiked with D-4F (10 μg/mL, final concentration) and incubated overnight. ApoA-I was immunoprecipitated as described in Methods. Western blot analysis for MPO and apoA-I was as above. (*=p<0.05, ** = p<0.02).
Next, we fed Ldlr −/− /apoA-I −/− mice WD and treated them ± D-4F to determine the extent to which D-4F required HDL to improve vasodilation and/or reduce vessel wall thickness. Although ACh promoted a moderate increase in vasodilation in vessels from Ldlr −/− /apoA-I −/− mice fed WD, the upward slope of the ACh dose response curves in the presence of L-NAME revealed only a small portion of endothelium-dependent vasodilation in these mice was actually mediated by eNOS (Figure 6A). Comparing hatched regions in Figure 6A and 6B reveals D-4F increased eNOS-dependent vasodilation in the hypercholesterolemic Ldlr −/− /apoA-I −/− mice by nearly 2-fold. However, in hypercholesterolemic double knock-out mice, D-4F failed to decrease vessel wall thickness (Figure 6C). Analysis of plasma lipids reveals HDL cholesterol in the Ldlr −/− /apoA-I −/− mice (Table 2) was decreased compared to the concentrations in Ldlr −/− mice (Table 1). D-4F had little effect on cholesterol in the hypercholesterolemic Ldlr −/− /apoA-I −/− mice but did increase HDL cholesterol (Table 2). More importantly, D-4F failed to protect HDL against oxidative modification in the double knock out mice (Table 2), in contrast to its ability to protect HDL against oxidation in hypercholesterolemic Ldlr −/− mice (Table 1).
Figure 6.
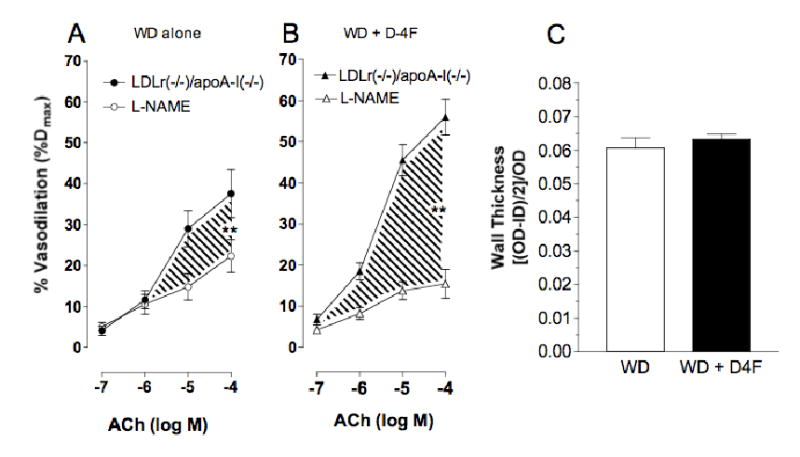
Effects of D-4F on Vasodilation and Wall Thickness in Hypercholesterolemic Ldlr −/− /apoA-I −/− mice. (A) Line graph showing effects of D-4F on vasodilation in Ldlr −/− /apoA-I −/− mice fed WD and treated with D-4F (1mg/kg/d) for 6–8 weeks. Vasodilation for Ldlr −/− /apoA-I −/− mice fed WD is increased in mice treated with D-4F (n=7–8, ** = p<0.02). (B) Bar graph showing D-4F has no effect on wall thickness in the vessels in the Ldlr −/− /apoA-I −/− mice (n=7–8).
Table 2.
Effect of D-4F on plasma total cholesterol, HDL cholesterol and proinflammatory HDL in Ldlr −/− /apoA-I −/− mice fed WD. This table shows the mean ± SEM for total cholesterol, HDL cholesterol and proinflammatory HDL in plasma from WD-fed Ldlr −/− /apoA-I −/− mice treated with D-4F (ip) for 6 weeks. D-4F does not alter total cholesterol concentrations but does increase HDL concentrations in these mice. D-4F has no effect on proinflammatory HDL levels in Ldlr−/−/apoA-I−/− mice.
| Ldlr−/−/ApoA-I−/− + PBS (n=4–5) | Ldlr−/−/ApoA-I−/− + D-4F (n=4) | p-value | |
|---|---|---|---|
| Total Cholesterol (mg/dL) | 1084 ± 135 | 1361 ± 249 | ns |
| HDL Cholesterol (mg/dL) | 18 ± 3.6 | 28 ± 2.5 | 0.04 |
| Proinflammatory HDL* | 5.49 x 108 ± 1.98 x 108 | 6.19 x 108 ± 2.28 x 108 | ns |
Arbitrary Units of Fluorescence per 1 μg HDL cholesterol per minute
Discussion
In recent years, much attention has focused on HDL as a therapeutic target for preventing vascular disease. Here time-dependent studies reveal D-4F increases vasodilation in direct relation to duration of treatments and interrupts or halts atherogenic mechanisms in vivo induced by feeding WD at all time points tested. Even short-term D-4F treatments reduced wall thickness in vessels from hypercholesterolemic Ldlr −/− mice before vasodilation was fully restored. These findings indicate improvements in vessel architecture precede improvements in vascular physiology. It is important to note D-4F decreased wall thickness even in vessels with pre-existing disease in mice with HDL that contains apo A-I, the major atheroprotective apolipoprotein of HDL, but not in mice whose HDL was apo A-I-deficient. Although D-4F still improved vasodilation in mice lacking apo A-I, we think its inability to reduce wall thickness is linked its inability to protect the apo A-I-deficient HDL against oxidation. As HDL function is impaired by oxidation,25,26 such findings strongly support the concept D-4F helps HDL maintain low levels of proinflammatory lipids, which should, in turn, improve vasodilation and reduce vessel wall thickness. Taken together, these findings underscore the importance of D-4F interacting with apo A-I on HDL to increase atheroprotection.
In the studies here, we assessed vessel wall architecture in the same vessel used to measure vasodilation. Therefore we could not perform histological analysis on these vessels. Instead, we relied on an unbiased assessment of wall thickness and explored potential mechanisms in endothelial cell cultures. Additional studies will be required to determine the precise cause of thickening of the facialis arteries in the future. In mechanistic studies, we showed that the proinflammatory oxysterol, 22-OHC, shifts •NO and O2 •− balance and increases collagen synthesis, both common features in vessels with early lesions.27 More importantly, 4F’s ability to restore •NO and O2 •− balance and reduce collagen production in 22-OHC-treated endothelial cell cultures is consistent with the marked increases in endothelium- and eNOS-dependent vasodilation and reductions in vessel wall thickness that we observed in hypercholesterolemic Ldlr −/− mice treated with D-4F.
Mechanistically, many of our findings are consistent with the notion that D-4F binds8 and removes oxidized lipids to protect vascular endothelial cell function.28 As eNOS function (coupled and uncoupled activity) in endothelial cell cultures is dramatically altered by native LDL (n-LDL) and oxidized LDL (ox-LDL),23, 29, 30 logically, anything limiting vascular endothelial cell exposure to proinflammatory lipids should improve eNOS-dependent vasodilation. The fact 4F did not alter 22-OHC-induced increases in ABCA1 expression, suggests endothelial cells exposed to 22-OHC are under a constant state of oxidative stress even in the presence of 4F. Accordingly, 4F’s ability to restore •NO balance and reduce collagen expression in 22-OHC-treated endothelial cells is likely related more to this apo A-I mimetic’s ability to bind oxidized phospholipids than oxysterols. If this is the case, then one can envision 22-OHC increasing endothelial cell production of oxidized phospholipids that in turn, shift •NO balance and increase collagen production. Further, 4F, via binding oxidized phospholipids, should limit this autocrine-type exposure to the very proinflammatory lipids inducing this prooxidant and inflammatory phenotype (i.e., ↑O2 •−, ↓ •NO and increased collagen). Evidence supporting this hypothesis is the fact that 4F restored •NO and O2 •− balance without altering expression of ABCA1. The fact that 4F did not alter ABCA1 expression is also important by itself, because ABCA1 provides a means for endothelial cells subjected to oxidative stress to more easily rid themselves of oxidized phospholipids.12 As ABCA1 activity can be impaired via oxidation,12 our findings demonstrate 4F represents a therapeutic agent that helps to preserve ABCA1’s critical function within the endothelium.
Probing mechanisms for D-4F protecting HDL against oxidative modification, we observed D-4F had no effect on total MPO concentrations in the plasma of hypercholesterolemic Ldlr −/− mice, but did reduce MPO association with apo A-I and subsequent 3NT formation in apo A-I (an index of oxidative modification supporting our proinflammatory HDL findings). Such findings indicate that parallels do exist between the MPO-mediated mechanisms in mice and those in humans who have documented increased risk of heart disease.15 The fact that D-4F decreased MPO association with apo A-I in vivo but not in vitro suggests that D-4F’s ability to reduce MPO association with HDL in vivo is not due simply to displacement. It is possible that D-4F may also protect HDL indirectly by restoring •NO and O2 •− balance to the endothelium,6, 31 which should in turn, decrease O2 •− and subsequently, the H2O2 with which MPO nitrates tyrosine residues in apoA-I.15 Another possibility is 4F increased extracellular superoxide dismutase, which should in turn, decrease O2 •− as shown in diabetic rats by Kruger et al.32
Our studies using the Ldlr −/−/apoA-I −/− mice and D-4F revealed some remarkable insight into HDL’s atheroprotective mechanisms. First, HDL deficient in apoA-I is more susceptible to oxidation than apoA-I containing HDL by a mechanism that can not be counteracted by D-4F treatments. Second, expression of apoA-I deficient HDL can be increased by D-4F, suggesting D-4F may up-regulate expression of other apolipoprotein A proteins as it has been shown for apoA-I.33 Third, the marked increase in proinflammatory HDL in Ldlr −/−/apoA-I −/− mice directly correlates with an increase in wall thickness in these animals which is in direct contrast to D-4F’s ability to improve vasodilation. This disconnect between D-4F’s ability to improve vasodilation and reduce vessel wall thickness was unexpected. One explanation for D-4F’s inability to protect HDL against oxidative modification in the Ldlr −/−/apoA-I −/− mice is the relative rates of DCF fluorescence, an index of seeding molecules of lipid hydroperoxides in HDL,11 were 100 times greater in HDL from Ldlr −/− /apoA-I −/− mice than it was in HDL from Ldlr −/− mice. Such large increases in proinflammatory HDL levels suggest two distinct possibilities; either apoA-I is essential for protecting HDL against oxidation and D-4F protects HDL against oxidation via interacting with apoA-I or the absolute amount of HDL plays a critical role in the mechanisms by which D-4F protects HDL against oxidation to reduce vessel wall thickness. Such observations are consistent with recent reports indicating cardiovascular events are mediated more by proinflammatory HDL than by total HDL,11, 34 a report showing high concentrations of HDL are essential for reducing plaque growth in patients with pre-existing carotid artery disease,35 and our report showing 4F afforded greater protection in vivo than in vitro when compared to vessels pre-incubated with LDL.6
Perhaps D-4F was unable to interact with apoA-I-deficient HDL to bind and remove proinflammatory lipids that increase HDL susceptibility to oxidation.36 Alternatively, D-4F may interact directly with vascular endothelium to protect endothelial cell function as was recently shown by Gupta et al., where L-4F displaced lipopolysaccaride (LPS) from vascular endothelium to preserve endothelial cell function.37 Another possibility is D-4F may act preferentially with vascular endothelial cells rather than vascular smooth muscle cells, which upon exposure to oxidized LDL express less matrix metalloproteases which is considered to increase matrix deposition.38 Such questions, although important, must remain unanswered until more mechanistic studies can be performed.
In conclusion, D-4F reduces proinflammatory HDL levels in hypercholesterolemic Ldlr −/− mice but not Ldlr −/−/apoA-I −/− fed WD. The anti-atherogenic properties of D-4F appear to go hand-in-hand with the apoA-I mimetic’s ability to reduce vessel wall thickness early on and then, with longer treatments, increase vasodilation to almost control levels in Ldlr −/− mice. Our studies suggest the mechanisms mediating impaired vasodilation in this early model of vascular disease may be different from those mediating vessel wall thickness. If this is the case then, HDL with either the proper apolipoprotein composition and/or a critical concentration of HDL may be required for D-4F to be able to both reduce wall thickness and improve vasodilation. Finally, our findings suggest D-4F may be useful for treatment of arteries with pre-existing disease.
Acknowledgments
This study was supported in part by the Marie Z. Uihlein Endowed Chair Award, the Children’s Hospital Foundation (Milwaukee, WI) (K.T.O.), (HL 61417 and HL 71214), to K.A.P. and (HL 064163) to M.S-T., AHA 0325546Z to J.O.; AHA 0520103Z to H.X., The Scientific Research Foundation for the Returned Overseas Chinese Scholars, Guangzhou Government and State Education Ministry of China (J.O.), The Administration of Public Health of Guangdong, China (A2005302, J.O.), Guangzhou Bureau of Education, China (1036 in 2004, J.O.) and Guangzhou Medical College, China (00-Q-06 and 03-G-06, J.O. and 03-G-07, Z.O.).
Footnotes
Results have been presented in part at the American Heart Association Scientific Sessions, Orlando, Florida, November 2003 and New Orland, Louisiana, November 2004.
Conflicts of interest
None
References
- 1.Candipan RC, Wang BY, Buitrago R, Tsao PS, Cooke JP. Regression or progression. Dependency on vascular nitric oxide. Arterioscler Thromb Vasc Biol. 1996;16:44–50. doi: 10.1161/01.atv.16.1.44. [DOI] [PubMed] [Google Scholar]
- 2.Cohen RA, Zitnay KM, Haudenschild CC, Cunningham LD. Loss of selective endothelial cell vasoactive functions caused by hypercholesterolemia in pig coronary arteries. Circ Res. 1988;63:903–910. doi: 10.1161/01.res.63.5.903. [DOI] [PubMed] [Google Scholar]
- 3.Schachinger V, Britten MB, Zeiher AM. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation. 2000;101:1899–1906. doi: 10.1161/01.cir.101.16.1899. [DOI] [PubMed] [Google Scholar]
- 4.Nishimura RA, Lerman A, Chesebro JH, Ilstrup DM, Hodge DO, Higano ST, Holmes DR, Jr, Tajik AJ. Epicardial vasomotor responses to acetylcholine are not predicted by coronary atherosclerosis as assessed by intracoronary ultrasound. J Am Coll Cardiol. 1995;26:41–49. doi: 10.1016/0735-1097(95)00142-m. [DOI] [PubMed] [Google Scholar]
- 5.Widlansky ME, Gokce N, Keaney JF, Jr, Vita JA. The clinical implications of endothelial dysfunction. J Am Coll Cardiol. 2003;42:1149–1160. doi: 10.1016/s0735-1097(03)00994-x. [DOI] [PubMed] [Google Scholar]
- 6.Ou J, Ou Z, Jones DW, Holzhauer S, Hatoum OA, Ackerman AW, Weihrauch DW, Gutterman DD, Guice K, Oldham KT, Hillery CA, Pritchard KA., Jr L-4F, an apolipoprotein A-1 mimetic, dramatically improves vasodilation in hypercholesterolemia and sickle cell disease. Circulation. 2003;107:2337–2341. doi: 10.1161/01.CIR.0000070589.61860.A9. [DOI] [PubMed] [Google Scholar]
- 7.Navab M, Anantharamaiah GM, Hama S, Garber DW, Chaddha M, Hough G, Lallone R, Fogelman AM. Oral administration of an Apo A-I mimetic Peptide synthesized from D-amino acids dramatically reduces atherosclerosis in mice independent of plasma cholesterol. Circulation. 2002;105:290–292. doi: 10.1161/hc0302.103711. [DOI] [PubMed] [Google Scholar]
- 8.Navab M, Anantharamaiah GM, Reddy ST, Hama S, Hough G, Grijalva VR, Yu N, Ansell BJ, Datta G, Garber DW, Fogelman AM. Apolipoprotein A-I mimetic peptides. Arterioscler Thromb Vasc Biol. 2005;25:1325–1331. doi: 10.1161/01.ATV.0000165694.39518.95. [DOI] [PubMed] [Google Scholar]
- 9.Harada-Shiba M, Kinoshita M, Kamido H, Shimokado K. Oxidized low density lipoprotein induces apoptosis in cultured human umbilical vein endothelial cells by common and unique mechanisms. J Biol Chem. 1998;273:9681–9687. doi: 10.1074/jbc.273.16.9681. [DOI] [PubMed] [Google Scholar]
- 10.Lizard G, Deckert V, Dubrez L, Moisant M, Gambert P, Lagrost L. Induction of apoptosis in endothelial cells treated with cholesterol oxides. Am J Pathol. 1996;148:1625–1638. [PMC free article] [PubMed] [Google Scholar]
- 11.Navab M, Hama SY, Hough GP, Subbanagounder G, Reddy ST, Fogelman AM. A cell-free assay for detecting HDL that is dysfunctional in preventing the formation of or inactivating oxidized phospholipids. J Lipid Res. 2001;42:1308–1317. [PubMed] [Google Scholar]
- 12.Reddy ST, Hama S, Ng C, Grijalva V, Navab M, Fogelman AM. ATP-binding cassette transporter 1 participates in LDL oxidation by artery wall cells. Arterioscler Thromb Vasc Biol. 2002;22:1877–1883. doi: 10.1161/01.atv.0000035700.82829.2a. [DOI] [PubMed] [Google Scholar]
- 13.Carpenter KL, Taylor SE, van der Veen C, Williamson BK, Ballantine JA, Mitchinson MJ. Lipids and oxidised lipids in human atherosclerotic lesions at different stages of development. Biochim Biophys Acta. 1995;1256:141–150. doi: 10.1016/0005-2760(94)00247-v. [DOI] [PubMed] [Google Scholar]
- 14.Smalley DM, Hogg N, Kalyanaraman B, Pritchard KA., Jr Endothelial cells prevent accumulation of lipid hydroperoxides in low-density lipoprotein. Arteriosclerosis, Thrombosis and Vascular Biology. 1997;17:3469–3474. doi: 10.1161/01.atv.17.12.3469. [DOI] [PubMed] [Google Scholar]
- 15.Zheng L, Nukuna B, Brennan ML, Sun M, Goormastic M, Settle M, Schmitt D, Fu X, Thomson L, Fox PL, Ischiropoulos H, Smith JD, Kinter M, Hazen SL. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J Clin Invest. 2004;114:529–541. doi: 10.1172/JCI21109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nauseef WM. The proper study of mankind. J Clin Invest. 2001;107:401–403. doi: 10.1172/JCI11713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brennan ML, Anderson MM, Shih DM, Qu XD, Wang X, Mehta AC, Lim LL, Shi W, Hazen SL, Jacob JS, Crowley JR, Heinecke JW, Lusis AJ. Increased atherosclerosis in myeloperoxidase-deficient mice. J Clin Invest. 2001;107:419–430. doi: 10.1172/JCI8797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nicholls SJ, Hazen SL. Myeloperoxidase and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2005;25:1102–1111. doi: 10.1161/01.ATV.0000163262.83456.6d. [DOI] [PubMed] [Google Scholar]
- 19.Malle E, Waeg G, Schreiber R, Grone EF, Sattler W, Grone HJ. Immunohistochemical evidence for the myeloperoxidase/H2O2/halide system in human atherosclerotic lesions: colocalization of myeloperoxidase and hypochlorite-modified proteins. Eur J Biochem. 2000;267:4495–4503. doi: 10.1046/j.1432-1327.2000.01498.x. [DOI] [PubMed] [Google Scholar]
- 20.Podrez EA, Abu-Soud HM, Hazen SL. Myeloperoxidase-generated oxidants and atherosclerosis. Free Radic Biol Med. 2000;28:1717–1725. doi: 10.1016/s0891-5849(00)00229-x. [DOI] [PubMed] [Google Scholar]
- 21.Zabalawi M, Bhat S, Loughlin T, Thomas MJ, Alexander E, Cline M, Bullock B, Willingham M, Sorci-Thomas MG. Induction of fatal inflammation in LDL receptor and ApoA-I double-knockout mice fed dietary fat and cholesterol. Am J Pathol. 2003;163:1201–1213. doi: 10.1016/S0002-9440(10)63480-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ou J, Fontana JT, Ou Z, Jones DW, Ackerman AW, Oldham KT, Yu J, Sessa WC, Pritchard KA., Jr Heat shock protein 90 and tyrosine kinase regulate eNOS •NO generation but not •NO bioactivity. Am J Physiol Heart Circ Physiol. 2004;286:H561–H569. doi: 10.1152/ajpheart.00736.2003. [DOI] [PubMed] [Google Scholar]
- 23.Ou Z, Ou J, Ackerman AW, Oldham KT, Pritchard KA., Jr L-4F, an apolipoprotein A-1 mimetic, restores nitric oxide and superoxide anion balance in low-density lipoprotein-treated endothelial cells. Circulation. 2003;107:1520–1524. doi: 10.1161/01.cir.0000061949.17174.b6. [DOI] [PubMed] [Google Scholar]
- 24.Siwik DA, Pagano PJ, Colucci WS. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am J Physiol Cell Physiol. 2001;280:C53–60. doi: 10.1152/ajpcell.2001.280.1.C53. [DOI] [PubMed] [Google Scholar]
- 25.Van Lenten BJ, Hama SY, de Beer FC, Stafforini DM, McIntyre TM, Prescott SM, La Du BN, Fogelman AM, Navab M. Anti-inflammatory HDL becomes pro-inflammatory during the acute phase response. Loss of protective effect of HDL against LDL oxidation in aortic wall cell cocultures. J Clin Invest. 1995;96:2758–2767. doi: 10.1172/JCI118345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feingold KR, Memon RA, Moser AH, Grunfeld C. Paraoxonase activity in the serum and hepatic mRNA levels decrease during the acute phase response. Atherosclerosis. 1998;139:307–315. doi: 10.1016/s0021-9150(98)00084-7. [DOI] [PubMed] [Google Scholar]
- 27.Stary HC. The Evolution of Human Atherosclerotic Lesions. West Point, PA: Merck; 1993.
- 28.Navab M, Van Lenten BJ, Reddy ST, Fogelman AM. High-density lipoprotein and the dynamics of atherosclerotic lesions. Circulation. 2001;104:2386–2387. [PubMed] [Google Scholar]
- 29.Stepp DW, Ou J, Ackerman AW, Welak S, Klick D, Pritchard KA., Jr Native LDL and minimally oxidized LDL differentially regulate superoxide anion in vascular endothelium In Situ. Am. J. Physiology Heart and Circulation. 2002;283:H750–H759. doi: 10.1152/ajpheart.00029.2002. [DOI] [PubMed] [Google Scholar]
- 30.Vergnani L, Hatrik S, Ricci F, Passaro A, Manzoli N, Zuliani G, Brovkovych V, Fellin R, Malinski T. Effect of native and oxidized low-density lipoprotein on endothelial nitric oxide and superoxide production: key role of L-arginine availability. Circulation. 2000;101:1261–1266. doi: 10.1161/01.cir.101.11.1261. [DOI] [PubMed] [Google Scholar]
- 31.Ou Z, Ou J, Ackerman AW, Oldham KT, Pritchard KA., Jr L-4F, an apolipoprotein A-1 mimetic, restores nitric oxide and superoxide anion balance in low-density lipoprotein treated endothelial cells. Circulation. 2003;107:1520–1524. doi: 10.1161/01.cir.0000061949.17174.b6. [DOI] [PubMed] [Google Scholar]
- 32.Kruger AL, Peterson S, Turkseven S, Kaminski PM, Zhang FF, Quan S, Wolin MS, Abraham NG. D-4F induces heme oxygenase-1 and extracellular superoxide dismutase, decreases endothelial cell sloughing, and improves vascular reactivity in rat model of diabetes. Circulation. 2005;111:3126–3134. doi: 10.1161/CIRCULATIONAHA.104.517102. [DOI] [PubMed] [Google Scholar]
- 33.Navab M, Anantharamaiah GM, Hama S, Hough G, Reddy ST, Frank JS, Garber DW, Handattu S, Fogelman AM. D-4F and statins synergize to render HDL antiinflammatory in mice and monkeys and cause lesion regression in old apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2005;25:1426–1432. doi: 10.1161/01.ATV.0000167412.98221.1a. [DOI] [PubMed] [Google Scholar]
- 34.Ansell BJ, Navab M, Hama S, Kamranpour N, Fonarow G, Hough G, Rahmani S, Mottahedeh R, Dave R, Reddy ST, Fogelman AM. Inflammatory/antiinflammatory properties of high-density lipoprotein distinguish patients from control subjects better than high-density lipoprotein cholesterol levels and are favorably affected by simvastatin treatment. Circulation. 2003;108:2751–2756. doi: 10.1161/01.CIR.0000103624.14436.4B. [DOI] [PubMed] [Google Scholar]
- 35.Johnsen SH, Mathiesen EB, Fosse E, Joakimsen O, Stensland-Bugge E, Njolstad I, Arnesen E. Elevated high-density lipoprotein cholesterol levels are protective against plaque progression: a follow-up study of 1952 persons with carotid atherosclerosis the Tromso study. Circulation. 2005;112:498–504. doi: 10.1161/CIRCULATIONAHA.104.522706. [DOI] [PubMed] [Google Scholar]
- 36.Bielicki JK, Oda MN. Apolipoprotein A-I(Milano) and apolipoprotein A-I(Paris) exhibit an antioxidant activity distinct from that of wild-type apolipoprotein A-I. Biochemistry. 2002;41:2089–2096. doi: 10.1021/bi011716p. [DOI] [PubMed] [Google Scholar]
- 37.Gupta H, Dai L, Datta G, Garber DW, Grenett H, Li Y, Mishra V, Palgunachari MN, Handattu S, Gianturco SH, Bradley WA, Anantharamaiah GM, White CR. Inhibition of lipopolysaccharide-induced inflammatory responses by an apolipoprotein AI mimetic peptide. Circ Res. 2005;97:236–243. doi: 10.1161/01.RES.0000176530.66400.48. [DOI] [PubMed] [Google Scholar]
- 38.Wilson D, Massaeli H, Pierce GN, Zahradka P. Native and minimally oxidized low density lipoprotein depress smooth muscle matrix metalloproteinase levels. Mol Cell Biochem. 2003;249:141–149. [PubMed] [Google Scholar]