

Abstract
Pretreatment of animals with butyrylcholinesterase (EC 3.1.1.8 BChE) provides complete protection from the acute effects of organophosphorus nerve agents. Butyrylcholinesterase has also been shown to protect from cocaine toxicity. Large amounts of highly purified butyrylcholinesterase are needed to test the effectiveness of this new therapeutic agent in monkeys. Only a minimum amount of endotoxin can be present in a therapeutic intended for injection into monkeys. Our goal was to develop a large scale purification method for human BChE from human plasma with precautions to minimize endotoxin content. A protocol was developed that processed up to 100 L of human plasma at a time. Dialysis in pH 4.0 buffer, ion exchange chromatography at pH 4, affinity chromatography on procainamide-Sepharose, and HPLC ion exchange at pH 7.4 yielded highly purified human BChE containing a low endotoxin level of about 800 EU/ml. The purified BChE produced by this method had a mean residence time of 56 h in mice and 93 h in monkeys, and caused no toxic effects. The absence of a toxic effect in monkeys demonstrates that the endotoxin level of 800 EU/ml was well tolerated by monkeys.
INTRODUCTION
Butyrylcholinesterase (EC 3.1.1.8) protects against cocaine toxicity (Hoffman et al., 1996; Lynch et al., 1997; Carmona et al., 1998; Duysen et al., 2002; Sun et al., 2002) and protects from the toxicity of nerve agents (Broomfield et al., 1991; Brandeis et al., 1993; Raveh et al., 1993; Raveh et al., 1997; Allon et al., 1998). Large amounts of highly purified BChE are needed to conduct studies in monkeys. Substances injected into monkeys must contain minimum amounts of endotoxin to avoid toxic shock. This paper describes how these goals were achieved in a university laboratory, without industry-scale equipment. The starting material was outdated human plasma. The key steps in the protocol were ion exchange chromatography at pH 4.0, affinity chromatography on procainamide-Sepharose, and HPLC ion exchange at pH 7.4. The scale up to processing 50-100 liters of plasma at a time, and the necessity to minimize endotoxin, required us to modify procedures we had previously published (Lockridge and La Du, 1978; Lockridge, 1990).
Our protocol uses no large-scale centrifugation. Therefore we did not need a continuous flow centrifuge as in the purification procedure by Grünwald et al. (Grunwald et al., 1997). This paper describes in detail the procedures we developed while processing a total of 1,700 L of human plasma.
BChE in plasma is a tetramer of 340,000 Da. Each 85,000 Da subunit contains 574 amino acids and 9 asparagine linked carbohydrate chains (Lockridge et al., 1987b). The carbohydrates, which account for 24% of the total weight, are located on the surface of the globular protein molecule and serve to protect BChE from proteolysis. Each subunit contains 3 internal disulfide bonds (Lockridge et al., 1987a; Nicolet et al., 2003). A 40 residue tetramerization domain at the C-terminus of each subunit functions to assemble the four subunits by folding into an alpha helix. The tetramer assembly is stabilized by interchain disulfide bonds at Cys571.
MATERIALS AND METHODS
Materials. Two 55 gallon (200 L) Nalgene cylindrical tanks with cover and spigot (11102-0055, Fisher Scientific) were used for dialysis. The widest dialysis tubing available had a flat width of 120 mm, Spectra/Por MWCO 6-8000 manufactured by Spectrum labs (part no. #132675) and sold by Fisher Scientific. The top of each dialysis bag was closed with Spectra/Por closures, gripping width 150 mm (142 250 Spectrum Labs, Fisher Scientific). The bottom of each dialysis bag was tied in three knots. Dialysis bags were recycled and reused. Used bags were washed with water and stored in 25% ethanol.
The first ion exchange step used a 10 × 90 cm glass chromatography column, maximum capacity 7.1 L (Part No. 123965 Spectrum Chromatography, Houston TX). This column was built into a table on wheels by Edward Rao in the University of Nebraska instrument shop. Ion exchange chromatography at pH 4 used 4 to 5 Liters of Q Sepharose Fast Flow (cat. no. 17-0510-04 Amersham Biosciences, Piscataway, NJ). The flow rate was 1 L in 18 min.
The affinity gel was custom synthesized by Dr. Yacov Ashani, Institute of Biotechnology, Ness-Ziona, Israel, in 1 L quantities. He attached procainamide to Sepharose 4B through a 6 carbon spacer arm (Grunwald et al., 1997). The density of covalently bound procainamide was 34 micromoles procainamide per ml beads. This affinity gel was introduced for purification of BChE in 1978 (Lockridge and La Du, 1978); it can be synthesized in a small scale from Sepharose beads already derivatized with a 6 carbon spacer arm. The affinity gel was packed in a Pharmacia column C26/40, maximum capacity 212 ml.
A plastic 20 L carboy was used to deliver pH 4.0 buffer to the ion exchange column. A plastic 2 L carboy was used to deliver pH 7.0 buffer to the affinity column.
Recycling and equilibrating Q-Sepharose Fast Flow. The Q-Sepharose Fast Flow (QFF) column was unpacked after each use, and the QFF was recycled in a 22 L plastic bucket. It was washed twice with 10 L of 0.5 M sodium hydroxide, and then 3 times with endotoxin-free water. The QFF was resuspended in water and the pH was adjusted to 3.9 with glacial acetic acid. The QFF was washed once more with endotoxin-free water to bring the conductivity to 0.29 mS. The washed QFF was stored in 25% ethanol by adding 2 L of ethanol to a 6 L slurry. The bucket was covered with a plastic lid and stored at 4°C. Immediately before use, the ethanol was poured off and the QFF was washed twice with endotoxin-free water. Each use of QFF resulted in some loss of gel. The starting 5 L of gel had diminished to 4 L after 10 rounds of use and recycling.
Recycling and equilibrating affinity gel. Used affinity gel was stored in 20% ethanol or in 0.1% sodium azide. Just before use the 150 ml affinity gel was washed in a coarse scinterred glass funnel with 1 L of 0.5 M acetic acid in endotoxin-free water, followed by 2 L of endotoxin-free water, and 1 L of 20 mM potassium phosphate, 1 mM EDTA pH 7.0. The washed affinity gel was packed into Pharmacia column C26/40 and equilibrated with 2 L of 20 mM potassium phosphate, 1 mM EDTA pH 7.0 at 4°C.
Amicon stirred cell. BChE was concentrated and dialyzed in an Amicon stirred cell Model 8200 with a PM10 membrane, cut off 10,000 kDa (13132 MEM 5436C Diaflo Ultrafiltration Membrane, Amicon).
HPLC. Waters 625 LC system, with a Waters 486 tunable absorbance detector. Only highly purified BChE was applied to the Protein-Pak anion exchange column, DEAE 8HR 1000Å 8 microm, 10 × 100 mm, Part No. 35650 (Waters Chromatography Division/Millipore Corporation, Milford, MA). The loading loop was an empty glass column with a 25 ml capacity; HR10/30 column, code no. 18-1470-01 (Amersham Pharmacia Biotech, Piscataway, NJ). The maximum pressure tolerated by this glass loading loop was 900 psi. Protein samples were filtered through a 0.2 micron syringe filter before being placed into the glass loading loop. The precolumn was a 2 microm titanium frit installed in a holder (A-340 analytical filter with A-108 frit, Upchurch Scientific, Oak Harbor, WA). The standard operating pressure was about 400 psi. When the pressure reached about 800 psi at a flow rate of 1 ml per min, the frit in the precolumn was replaced. Frits were cleaned by immersion in nitric acid for 24 hours.
The HPLC column was operated at room temperature at a flow rate of 1 ml per min. All buffers were made in endotoxin-free glass bottles (2 L glacial acetic acid bottles) with endotoxin-free water, and autoclaved. The HPLC column was cleaned with 0.1 M NaOH and 0.5 M NaCl to remove contaminating proteins between runs.
A 1 M solution of dibasic potassium phosphate (Acros Organics 215470010) had an absorbance at 280 nm of 0.040, whereas a 1 M solution of monobasic potassium phosphate (Sigma P-5379) had an absorbance at 280 nm of 0.003. These low absorbance values verify that the reagents were greater than 99% pure. The HPLC buffers contained 11 mM potassium phosphate pH 7.4. A contaminant from the phosphate buffer bound to the ion exchange column and eluted with 0.5 M NaCl. The contaminant in the phosphate buffer accumulated on the ion exchanger during the BChE loading step, when 600 ml of buffer were pumped through the loading loop and Protein-Pak column. This artifact from the phosphate buffer was first noticed when a BChE preparation that looked 100% pure on SDS gel, nevertheless showed a contaminant peak of absorbance at 280nm when the ion exchange column was eluted with 0.5 M NaCl. No such artifact was found when the buffer was 20 mM TrisCl pH 7.5.
Human plasma. Outdated human plasma was a gift from the University of Nebraska Hospital Blood Bank. Bags of plasma were stored at 4°C until 50-100 L had accumulated, a period of about 3 to 5 months. Plasma that was visibly turbid with lipids was not used because the lipids clogged the ion exchange gel, reducing the flow rate to almost zero. Clots of fibrin were not a problem, however, because they settled to the bottom of the dialysis bag.
Platelet depleted plasma gave normal yields of purified BChE activity. However, aphoresis plasma often yielded only half of the expected amount of BChE and was therefore not a preferred starting material.
Endotoxin-free water. Buffers were made with double distilled water taken directly from the glass distillation tank. This water was free of endotoxin as measured with the chromogenic kinetic assay. In contrast, Nanopure water, which is water filtered through Millipore filters, contained significant amounts of endotoxin (1200 EU/ml) and was not used. Buffers that were not used immediately were autoclaved to prevent bacterial growth.
Endotoxin-free plasticware and glassware. Plastic containers were soaked in 1 M sodium hydroxide for 24 hours or longer to destroy endotoxin (Sharma, 1986). Then they were rinsed with endotoxin-free water. Glass beakers were heated in a muffle oven at 1000°F (750°C) for 16 hours. Glass bottles that had previously contained 2 L of glacial acetic acid made good endotoxin-free bottles for buffers. They were rinsed with endotoxin-free water before use. Fractions from QFF and affinity chromatography were collected into endotoxin-free bottles (500 ml and 1 L serum-free culture-medium bottles). Volume was determined by weight.
Kinetic chromogenic assay for measuring endotoxin. The Limulus Amebocyte Lysate Endochrome-K kit was used for measuring endotoxin levels (LVR17000, Charles River Laboratories, Charleston, SC). The kit contains lyophilized amebocyte lysate, buffer, and the chromogenic substrate p-nitroaniline. Immediately before use, the contents of a vial were rehydrated with 3.4 ml of LAL reagent water (Cat. No. W50-500, BioWhittaker, Walkersville, MD). A multichannel pipettor was used to deliver 100 microliters of the reagent to 100 microliters of test sample in a 96-well plate. Absorbance increase at 405 nm was recorded in a Molecular Devices Spectra Max 190 plate reader for up to 60 minutes, at 25°C. The time to reach an absorbance of 0.20 was determined. A standard curve was prepared from endotoxin standard (catalog 210-SE, Sigma).
100X buffer for dialysis. 70 L of plasma was dialyzed in 120 L of 20 mM sodium acetate, 1 mM EDTA pH 4.0. The 120 L of buffer was prepared by diluting 1.2 L of 100x buffer to 120 L with distilled water. The 100x buffer was made in a glass bottle and contained 152 g EDTA tetrasodium salt (Aldrich E2,629-0, 98% pure) plus 456 ml glacial acetic acid, and 18 g sodium hydroxide in a total volume of 4 L. After 1:100 dilution this buffer had a conductivity of 0.36 mS and a pH of 4.0. Neither the 100X stock buffer nor the 120 L of 1X buffer was autoclaved.
50X buffer for affinity chromatography. The buffer for affinity chromatography was 20 mM potassium phosphate, 1mM EDTA pH 7.0. This buffer was prepared by diluting 40 ml of a 50X stock buffer to 2 L with endotoxin-free water. The 50X stock buffer contained 108.8 g K2HPO4 plus 51.0 g KH2PO4 plus 19.0 g Na4EDTA in 1 L total volume. The stock buffer solution was autoclaved.
Activity assay. BChE activity was measured with 1 mM butyrylthiocholine as substrate in the presence of 0.5 mM dithiobisnitrobenzoic acid, in 0.1 M potassium phosphate buffer pH 7.0 at 25°C in 1 cm cuvettes. A recording Gilford spectrophotometer connected to a Macintosh computer via a MacLab interface was used to record the change in absorbance at 412 nm. The extinction coefficient for the product was 13,600 M−1 cm−1 (Ellman et al., 1961). Units of activity were defined as micromoles butyrylthiocholine hydrolyzed per minute per ml.
Nondenaturing gel electrophoresis. Migration of BChE on nondenaturing gel stained for activity revealed the relative amounts of tetramers, dimers, and monomers. In addition this method was useful for identifying BChE samples that had been desialylated by bacterial contaminants. A 4-30% gradient polyacrylamide gel was prepared in a Hoefer SE600 gel apparatus (Hoefer, San Francisco, CA; presently part of Pharmacia Inc.). Electrophoresis was at 200 volts constant voltage for 24 hours at 4°C. Gels were stained for BChE activity in the presence of 2 mM butyrylthiocholine iodide by the method of Karnovsky and Roots (Karnovsky and Roots, 1964).
SDS gel electrophoresis. 4-30% gradient gels were run at 120 volts constant voltage for 15 hours at 4°C. Gels were stained with Coomassie Blue R-250.
Protein concentration. The protein concentration was calculated from absorbance at 280 nm where a 1 mg/ml solution had an absorbance of 1.8 in a 1 cm quartz cuvette. Percent purity was calculated by using a value of 720 units/mg as the specific activity of 100% pure BChE.
Animals. Animal studies were carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted by the U.S. National Institutes of Health. Mice were strain 129Sv from Taconic (n =5). Rhesus monkeys (Macaca mulata) (n=2) were maintained at the University of Michigan. The monkeys weighed 12.6 and 14.2 kg. Monkeys were anesthetized with ketamine before being injected with purified human BChE (3 mg/ml) at a dose of 4 mg/kg.
Pharmacokinetic calculations. The data for elimination of BChE from the circulation were fitted to the double exponential equation in Sigma Plot (Jandel Scientific), where A is the fraction of material removed from the circulation in the fast phase, B is the fraction of material removed in the slow phase, k1 is the first order rate constant for the fast phase, k2 is the first order rate constant for the slow phase, and Ct is the concentration of BChE in the blood at time t in units/ml. This equation has been used by Kronman et al. (Kronman et al., 2000) as well as by Saxena et al. (Saxena et al., 1998). Another way to describe these rate constants is to say that k1 is the first-order rate constant of enzyme distribution while k2 is the first-order rate constant of enzyme elimination from the blood stream. The half-time calculated from the slowest process, k2, is called the biological half-life.
The mean residence time (MRT) was calculated according to Gabrielson and Weiner (Gabrielson and Weiner, 1994) using the following equations. AUC = A/k1 + B/k2 ; AUMC = A/ (k2)2 + B /(k2)2 ; MRT = AUMC/AUC.
The half-life was calculated from the relationship, t1/2 = 0.693/k, where 0.693 is ln2, and k is the first order rate constant.
RESULTS
Purification method
Dialysis. Dialysis in cellulose membrane bags was chosen as the first step in the purification because it avoided centrifugation. A heavy precipitate settled to the bottom of the dialysis bags, removing about 9% of the total protein without loss of BChE activity. In addition, this step reduced the salt concentration to 0.02 M and the pH to 4.3, to a level that enabled selective binding of BChE to the ion exchange gel (Das and Liddell, 1970). Most plasma proteins do not bind to ion exchange gel at pH 4.3.
A typical starting volume of plasma was 70 L. The plasma was poured into 26 dialysis bags, the bags were clamped shut and placed into 120 L of precooled 20 mM sodium acetate 1 mM EDTA pH 4.0 in a 200 L tank. Fresh buffer was prepared in the second tank and allowed to cool overnight to 4°C before the bags were transferred to the fresh buffer. The buffer was changed every day until the pH of the tank buffer at the end of 24 hours was 4.0. The number of days required for dialysis depended on the volume of plasma: 12 days for 100 L, 9 days for 70 L, 8 days for 47 L.
The pH of the dialyzed plasma at the end of the dialysis period was 4.3 to 4.4.
Binding BChE to ion exchanger. Dialyzed plasma was poured into a 22 L plastic cylinder containing 4 L of Q-Sepharose Fast Flow (QFF) that had been equilibrated to pH 3.8 and low ionic strength (conductivity 0.3 mS/cm) Only the clear plasma was poured out of the dialysis bag, while the heavy precipitate was left undisturbed in the bottom of the bag. The plasma and QFF were stirred with a plastic rod, allowed to settle for 30 minutes, stirred again, and allowed to settle for another 30 min. The supernatant was tested for activity. When 80-90 % of the starting BChE activity had disappeared from the plasma by binding to QFF, the yellow supernatant was discarded. The cylinder was refilled with more dialyzed plasma to allow BChE to bind. This was repeated 5 to 6 times until all the dialysis bags had been emptied.
pH 4 chromatography. The QFF in the bucket was washed with 10 L of cold 20 mM sodium acetate, 1 mM EDTA pH 4.0, then resuspended to make a slurry of less than 7 L, and poured into the 7.1 L column. The column was washed with 200 L of pH 4.0 buffer at 4°C to elute contaminating proteins. Buffer was made with double-distilled water because double-distilled water, taken directly from the still, was free of endotoxin. The column was washed with 200 L of buffer until the eluant had an absorbance of 0.04 at 280 nm. It took about 7 days to wash the column with 200 L of buffer, depending on the availability of endotoxin-free water in the still. The 200 L of buffer was not autoclaved.
BChE was eluted with 0.05 M NaCl in 20 mM sodium acetate, 1 mM EDTA pH 4.0. The first 2 L contained no BChE activity and no protein. The next 1 L contained 18% of the BChE, which was 8.3% pure. The next 0.6 L contained 60% of the BChE and was 4.1% pure. The next 0.5 L contained 17% of the BChE and was 0.9% pure. See Figure 1. The final two fractions, containing about 5% of the BChE activity, were discarded. The QFF column purified the BChE about 800 fold. Figure 2 shows the proteins in plasma and in the QFF fractions on SDS gel stained with Coomassie blue. The most prominent protein in plasma is the 66 kDa albumin. The QFF fractions have almost no albumin but have a prominent band at 45 kDa. Fractions rich in the 45 kDa protein are blue-green in color.
Figure 1.
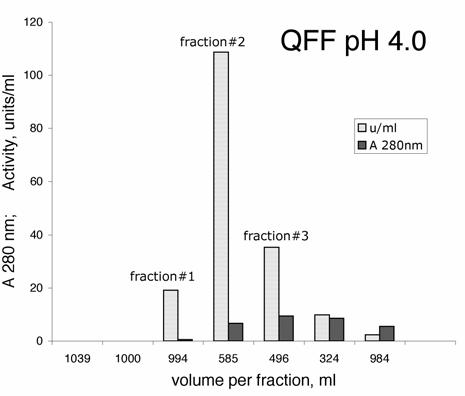
Purification of BChE by ion exchange at pH 4.0 The BChE in 62 L of dialyzed human plasma was bound to 4 L of Q-Sepharose Fast Flow (QFF) at pH 4.0 in a batch process. The QFF was packed into a 7 L glass column and washed with 200 L of endotoxin-free 20 mM sodium acetate, 1 mM EDTA pH 4.0, at 4°C. The graph shows elution of BChE from QFF with 0.05 M NaCl in pH 4.0 buffer at a flow rate of 1.7 L per hour. The BChE in fraction #1 was purified 1200-fold.
Figure 2.
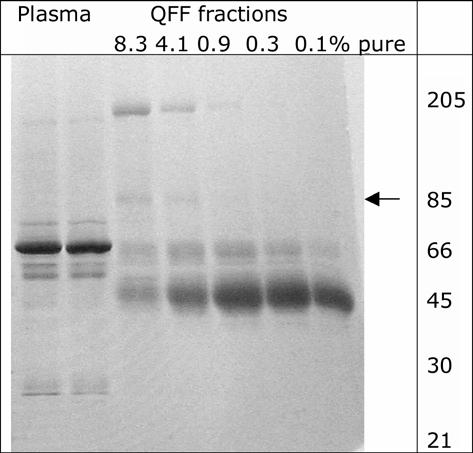
SDS gel stained with Coomassie blue Plasma before and after dialysis and fractions from QFF were applied at 5 micrograms protein per lane. The BChE band at 85 kDa is indicated by an arrow. BChE is a faint band in the 8.3 and 4.1% pure fractions. The 66 kDa albumin band is intense in plasma but not in the QFF fractions, indicating that most of the albumin has been removed by ion exchange at pH 4.0.
The pH of protein-containing fractions eluted from QFF was about 4.5.
Affinity chromatography. There was no need to adjust the pH or ionic strength before loading the BChE on the affinity column. The BChE that had eluted from the ion exchange column was immediately loaded onto a column packed with 150 ml of recycled procainamide-Sepharose. The affinity gel had been equilibrated with 20 mM potassium phosphate, 1 mM EDTA pH 7. 99% of the activity bound to the affinity gel, whereas contaminating proteins eluted during loading. The gel was washed with 2 L of 20 mM potassium phosphate, 1mM EDTA pH 7.0 and then with 2 L of 0.2 M NaCl in buffer to remove contaminating proteins. Finally the BChE was eluted with 1 M NaCl in 20 mM potassium phosphate, 1mM EDTA pH 7.0. The BChE eluted in the first 300 ml. See Figure 3. The affinity column increased the purity about 10 fold.
Figure 3.
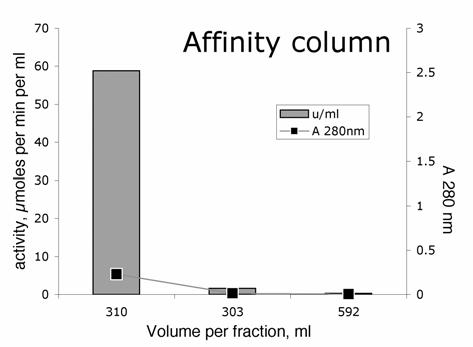
Affinity column purification of BChE The 1200-fold purified BChE from QFF was loaded onto a 150 ml affinity column at a flow rate of 350 ml per hour. The column was washed with 2 L of 20 mM potassium phosphate, 1mM EDTA pH 7.0 followed by 2 L of 0.2 M NaCl in buffer to elute contaminants. The graph shows elution of BChE from procainamide-Sepharose with 1 M NaCl in pH 7 buffer.
Sodium chloride was used to elute BChE because sodium chloride had no absorbance at 280 nm, was not inhibitory, and was inexpensive. Elution with 0.2 M procainamide, or 0.2 M procaine hydrochloride, or 0.2 M succinylcholine chloride, or 0.2 M acetyl-beta-methylcholine, or 0.2 M decamethonium also worked but the purification was no better. The disadvantage of using these inhibitors for elution was that an accurate specific activity could not be determined until after the inhibitors had been removed. The inhibitors procainamide and procaine not only inhibited activity but they also interfered with measurement of absorbance at 280 nm. Another disadvantage of using an inhibitor for elution is that inhibitors remain in the enzyme active site gorge even after extensive dialysis. The presence of decamethonium in highly purified acetylcholinesterase was seen in the x-ray structure of Torpedo c. acetylcholinesterase (Axelsen et al., 1994), and also detected using capillary electrophoresis and differential scanning calorimetry (Rochu et al., 2002).
The BChE could also be eluted with substrate, for example with 0.2 M acetylcholine, but the pH of the BChE solution rapidly dropped to 3.6 and inactivated the enzyme. Other compounds that worked for elution were 0.1 M trimethylammonium bromide in 0.3 M NaCl, or 0.2 M choline chloride.
Desalting. The BChE was dialyzed in an Amicon stirred cell to reduce the salt concentration below 0.03 M NaCl and to reduce the volume to about 20 ml.
HPLC. The BChE was centrifuged in microfuge tubes to remove solids and then filtered through a 0.2 microm syringe filter before it was loaded into the 25 ml glass loading loop of the HPLC. Autoclaved buffer, 11 mM potassium phosphate pH 7.4 prepared in endotoxin-free water, was pumped through the column and glass loading loop to load the BChE onto the HPLC ion exchange column. It took 500-600 ml to wash the 25 ml of BChE out of the loading loop. Loading was usually accomplished overnight. The BChE was eluted from the HPLC column with autoclaved 0.15 M NaCl in 11 mM potassium phosphate pH 7.4, a buffer equivalent to phosphate buffered saline. Figure 4 shows the elution profile detected as absorbance at 280 nm versus time in minutes. Step elution with 0.15 M NaCl in buffer eluted contaminating proteins at the front and tail end of the peak. The purest BChE eluted in the center of the peak. Additional contaminating proteins and a yellow color eluted with 0.5 M NaCl in buffer. A Coomassie blue stained gel in Figure 5 shows that fraction 3 was the purest. To minimize the possibility of bacterial growth, the BChE recovered from HPLC was immediately filter sterilized and frozen at −70°C.
Figure 4.
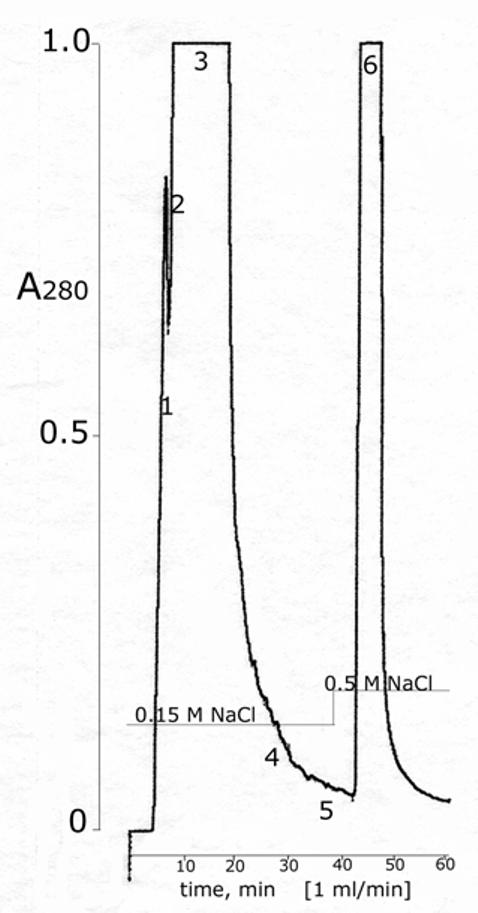
HPLC purification of BChE The 64% pure BChE recovered from the affinity column was desalted to about 0.04 M NaCl, reduced in volume, and filtered into a 25 ml sample loop in preparation for loading onto the Protein-Pak DEAE column. About 600 ml of 11 mM potassium phosphate pH 7.4 were pumped through the sample loop to load all of the BChE onto the HPLC column. The BChE was eluted with 0.15 M NaCl in 11 mM potassium phosphate pH 7.4 at a flow rate of 1 ml per min, at ambient temperature, at 400 psi. Fractions 1 and 2 at the leading edge of the peak, between 4.6-7.0 min, were 25% and 34% pure and contained 180 units out of the 18,200 units applied. Fraction 3 between 7.0-18.5 min was 80% pure and contained 16,100 units. Fractions 4 and 5 at the tail end of the peak between 18.5 and 38 min were 74% and 43% pure and contained a total of 970 units. Fraction 6, which contained additional contaminating proteins as well as 190 units of 5% pure BChE, eluted with 0.5 M NaCl.
Figure 5.
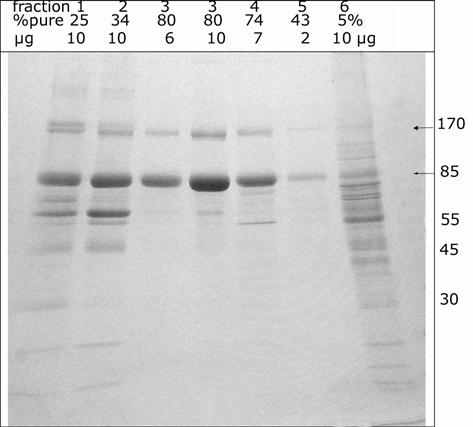
HPLC fractions on SDS gel stained with Coomassie blue Fractions 1-5 were eluted with 0.15 M NaCl. Fraction 6 was eluted with 0.5 M NaCl. The % purity of the BChE in each fraction is indicated. Up to 10 micrograms protein were loaded per lane. The BChE monomer and dimer are 85 and 170 kDa.
Table 1 summarizes the purification of BChE from human plasma. The data for the purification of each fraction from the QFF column are shown in groups (QFF/Affinity/HPLC). To reach a specific activity of 720 units/mg, the BChE must be purified 14,000 fold. This level of purity can be achieved by additional HPLC runs where only the cleanest fractions are pooled.
Notes on HPLC purification.
The binding capacity of the 7.8 ml HPLC column (Protein-Pak DEAE, 10×100 mm, Millipore) is very high. When the BChE was 55% pure, this column bound a maximum of 76,000 units or 105 mg BChE. The binding capacity decreased when the BChE was less pure. For example, it bound a maximum of 15 mg BChE (11,000 units) when the BChE was 11% pure. Step elution gave a more concentrated BChE than gradient elution.
An alternative buffer that gave very good results was 20 mM TrisCl pH 7.5. When Tris buffer was used, contaminating proteins could be eluted with 0.1 M NaCl in buffer, provided the BChE was at least 36% pure when it was loaded. If the BChE was 5-20% pure, then some of the BChE eluted during washing with 0.1 M NaCl in 20 mM TrisCl pH 7.5. Elution of BChE in a small volume was achieved with 0.19 M NaCl in 20 mM TrisCl pH 7.5 buffer. Use of a lower salt concentration spread the BChE into a larger volume. TrisCl buffer was used for BChE samples intended for non-animal experiments.
Purification of side-fraction. Approximately 20,000 units out of 100,000 units in the starting plasma, eluted late in the first chromatography step on QFF at pH 4. This fraction was about 0.9% pure, was green in color, and had a major protein band at 45 kDa (Figure 2). This side-fraction was carried through the purification steps separately and was not combined with the cleaner BChE because this fraction was difficult to purify. Table 1 shows that after affinity column chromatography this side-fraction became 30% pure, and after HPLC it became 58% pure, values significantly lower than the best BChE fractions.
Additional steps that helped to further purify this side-fraction included ion exchange at pH 4.5, affinity chromatography on a 10 ml affinity column, and ion exchange at pH 7.5.
Characteristics of purified BChE
Purified BChE consists of tetramers
The BChE in human plasma consists of 95% tetramers, and 5% dimers and monomers. The minor forms are cleared from the BChE during the purification. The final HPLC purified BChE consists predominantly of tetramers. These forms are visualized on a nondenaturing gel stained for BChE activity (Karnovsky and Roots, 1964) in Figure 6 . Monomers are the result of proteolysis which removes the tetramerization domain at the C-terminus (Lockridge and La Du, 1982; Saxena et al., 2003).
Figure 6.
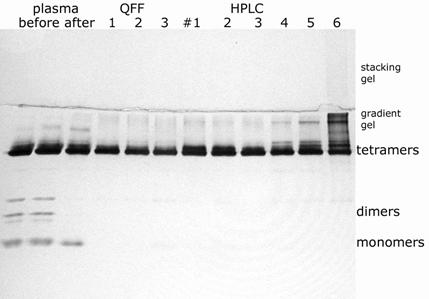
Nondenaturing 4-30% gradient gel stained for BChE activity The samples are plasma before (2 lanes) and after (1 lane) dialysis in pH 4 buffer, 3 fractions from QFF (see Figure 2), and 6 fractions from HPLC (see Figure 3). Each lane contains the amount of BChE activity found in 5 microliters of plasma, 0.015 units. After electrophoresis at 200 volts constant voltage for 24 hours at 4°C, the gel was stained for BChE activity.
Endotoxin content. The endotoxin content of purified human BChE was generally in the range 300 to 1000 EU/ml, similar to the endotoxin level found in purified horse BChE purchased from Sigma. Table 2.
The endotoxin level in various sources of water varied widely (Table 2). Double distilled water prepared at UNMC and taken directly from the glass still was as pure as LAL reagent water purchased from a commercial vendor. Buffers for washing the QFF column, the affinity column, and the HPLC column were made with this water. When this water was stored in a plastic carboy at room temperature, the endotoxin level rose significantly. Nanopure water had a high level of endotoxin even after a new filter was installed. Surprisingly, city tap water had less endotoxin than Nanopure water.
N-terminal amino acid sequence. The goal here was to determine whether the purified BChE contained a second peptide that might represent a tetramer organizing peptide. Recombinant AChE requires a proline-rich tetramer organizing peptide in a ratio of 1:4 before AChE assembles into tetramers (Simon et al., 1998). Proline-rich peptides also promote tetramerization of BChE from cell culture (Altamirano and Lockridge, 1999). The amount of BChE sequenced (370 pmoles) would have allowed detection of a second peptide if the second peptide constituted 25% of the N-termini. Only one amino acid sequence was found and that sequence was EDDIIIATKN, a result in agreement with the published amino acid sequence of human BChE (Lockridge et al., 1987b). No evidence for a second peptide sequence was found. This result does not rule out the possibility that a proline-rich tetramer organizing peptide may be present in the tetramer organizing domain because the N-terminus could be blocked. Alternatively the peptide could promote tetramerization in catalytic quantities.
The finding of only one N-terminal sequence confirms the purity of the BChE preparation.
Storage. BChE solutions containing at least 1 mg/ml protein (720 units/ml) could be stored frozen, in the absence of any cryoprotectant (i.e. glycerol, sucrose, or polyethylene glycol), without loss of activity. Dilute solutions required a cryoprotectant such as 30% glycerol. Purified BChE could also be stored at 4°C indefinitely provided it was either filter sterilized, or it contained sodium azide (0.02%= 3 mM) to prevent bacterial and fungal growth. Ten to twenty years of storage in aqueous solution caused no loss of activity in sterile BChE, as measured with butyrylthiocholine or benzoylcholine, but did have an effect on the carbohydrate content. The BChE became desialylated as observed on nondenaturing gel stained for BChE activity.
After frozen BChE containing 1-4 mg/ml in phosphate buffered saline was thawed, a white pellet was observed at the bottom of the tube. This pellet was easily dissolved. There was no adverse affect on BChE activity.
Bacterial contamination of BChE. Bacterial contamination of a BChE preparation could be recognized in 4 ways. 1) A heavy contamination was accompanied by a distinct odor. 2) A heavily contaminated preparation had an endotoxin level in millions of units per ml. 3) Bacterial contamination changed the migration pattern of BChE on nondenaturing gel because bacterial enzymes desialylated the BChE. Partially desialylated BChE tetramers migrated more slowly than native tetramers. Figure 7 shows that completely desialylated BChE tetramers barely entered the separating gel. In addition, a broad smear of BChE activity was found in the 4% stacking gel. The effects of bacterial contamination were reproduced by incubating BChE with viral neuraminidase, confirming that the shift in migration was caused by desialylation. Native human BChE tetramers contain approximately 72 sialic acids on 36 N-linked carbohydrate chains. 4) As a result of desialylation, bacterially contaminated BChE had a short residence time of less than 1 h in the circulation of rats and mice. The asialo receptors in liver rapidly cleared BChE from the circulation.
Figure 7.
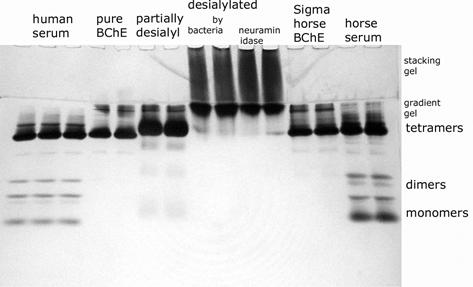
Detection of bacterial contamination BChE contaminated with bacteria migrates more slowly on a nondenaturing gel stained for BChE activity. Lanes 1-3, human serum; lanes 4-5, highly purified human BChE, 800 EU/ml; lanes 6-7, partially desialylated BChE; lanes 8-9, completely desialylated human BChE by bacterial contamination 30,000,000 EU/ml; lanes 10-11, completely desialylated human BChE , by treatment with viral neuraminidase; lanes 12-13, purified horse BChE from Sigma, 800 EU/ml; lanes 14-15, horse serum. Each lane contains the amount of BChE enzyme activity in 5 microliters of human serum, 0.015 units.
Bacterial contamination of purified BChE had no apparent effect on BChE activity as measured with butyrylthiocholine.
Stability of BChE at low pH. BChE in plasma was stable during dialysis and chromatography in pH 4 buffer for at least one month at 4°C. It was noticed that though the pH of the buffer was 4.0, the pH of the plasma never dropped below 4.3. After partial purification, the BChE was no longer stable to dialysis and chromatography in pH 4.0 buffer. A 20% pure BChE preparation lost 22 % activity after 66 hours in pH 4.1 buffer. Therefore, ion exchange chromatography at pH 4.0 could not be repeated. However, partially purified BChE was stable at pH 4.5. No activity was lost after 66 hours in 20 mM NaAcetate pH 4.5.
Half-life in mouse. The BChE purified by this protocol had a half-life of 43 h and a mean residence time of 56 hours in the circulation of mice. Figures 8 shows that 168 h (7 days) after injection of human BChE, the mouse circulation still had 8.9 u/ml of BChE, a value 5 fold above endogenous levels.
Figure 8.
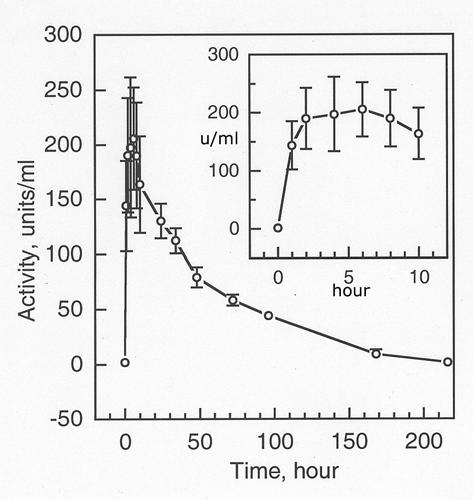
Residence time of purified human BChE in mice 0.2 ml of purified native human BChE containing 438 units was injected i.p. into 5 mice, at a dose of 27 mg/kg. Blood samples were collected at 0, 1, 2, 4, 6, 8, 10, 24, 34, 48, 72, 96, 168, and 216 hours and assayed for BChE activity. At time zero the BChE activity in mouse blood was 1.79±0.16 u/ml. The mean residence time was 56.6 hours.
Half-life in monkey. The BChE purified by this protocol was injected iv into rhesus monkeys at a dose of 4 mg/kg. Blood samples were drawn for 14 days and tested for BChE activity. Figure 9 shows the biphasic disappearance of human BChE from the circulation of two monkeys. In the first few minutes after injection, the BChE activity increased 30-fold, from an endogenous value of about 4 u/ml to 120 u/ml. After 11 days, the BChE activity was still slightly elevated, at about 6 u/ml. After 13 days, the BChE activity had returned to the starting activity of 4 u/ml. The data in Figure 9 were fit to a double exponential equation using the curve fitting program in Sigma Plot. The double exponential equation gave the following values. The fast phase had a first order rate constant of 3.02±1.49 days, which calculates to t1/2 = 5.5 h; 28% of the BChE activity disappeared in the fast phase. The slow phase had a first order rate constant of 0.250±0.024 days, which calculates to t1/2 = 66.4 h; 72% of the BChE activity disappeared in the slow phase. The mean residence time of our purified human BChE in monkeys was 93.2 h.
Figure 9.

Residence time of purified human BChE in monkeys Two monkeys were injected with 16.8 ml and 19.5 ml of purified human BChE iv to give a dose of 4 mg/kg. The BChE solution had a concentration of 3 mg/ml. Blood was withdrawn at 10, 30, 60, 120 min, 6 h, 12 h, 24 h, 2 days, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, and 14 days and assayed in triplicate for BChE activity. The symbols are the data, the line is the fit to the double exponential equation Ct = Ae(−k1t) + Be(−k2t). The half-life, calculated from the first order rate constant was 5.5 h for the fast phase and 66.4 h for the slow phase. The mean residence time was 93.2 h.
DISCUSSION
What is new in this purification protocol.
Methods for purifying BChE from human plasma have been described previously (Surgenor and Ellis, 1954; Malmstrom et al., 1956; Das and Liddell, 1970; Gaffney, 1970; Lockridge and La Du, 1978; Ralston et al., 1983; Lockridge, 1990; Lynch, 1993; Grunwald et al., 1997; Lynch et al., 1997). Our previous reports have been brief outlines. This report is the first detailed description of the methods we use, developed during 30 years experience of purifying human BChE.
The new features of our purification protocol are the scale-up to processing large volumes of plasma and the use of HPLC. We simplified the protocol by using a batch method to bind BChE to ion exchanger, and by using step elution in place of gradient elution for each chromatography column. We introduced the use of an HPLC chromatography step. The ion exchange gels for HPLC have a higher binding capacity than the gravity flow ion exchangers and give better separations. Finally, we showed how to prepare human BChE so that the purified BChE contained a minimum amount of endotoxin.
Procainamide-Sepharose affinity gel.
The affinity gel we introduced in 1978 (Lockridge and La Du, 1978), is widely used today for purification of both butyrylcholinesterase and acetylcholinesterase (De la Hoz et al., 1986). The binding capacity of procainamide-Sepharose depends on the purity of the BChE that is applied. An 8 L column of affinity gel is required to bind the BChE in 70 L of citrated human plasma (100,000 units) where the BChE concentration is 0.002 mg/ml and the protein concentration is 30 mg/ml. However, only 150 ml of affinity gel is required to bind the same amount of BChE after it has been purified 400 to 800-fold by ion exchange chromatography at pH 4.0. When the BChE is very pure (50-70% pure) so that only minor increments in purity are being attempted, the affinity gel binds 3 mg BChE per ml gel.
The dependence of binding capacity on BChE purity means that proteins other than BChE bind to the affinity gel. The contaminants bind through ion interactions and are released by low concentrations of sodium chloride (0.2 M). The best binding capacity is obtained with affinity gel containing the maximum number of covalently bound procainamide molecules.
How much endotoxin can a monkey tolerate?
Endotoxin, also called lipopolysaccharide, is the main component of the outer membrane of the cell wall of Gram-negative bacteria such as E. coli. Gram-negative bacteria are ubiquitous in the environment. They are in oral cavities and intestinal tracts of mammals, in ventilation systems, humidifiers, and swimming pools. Endotoxins cause the immune system to release inflammatory cytokines. A high dose of endotoxin can result in multiple organ failure and death.
The endotoxin concentration in our purified BChE preparation is no higher than 1000 EU/ml, which corresponds to 100 ng/ml (Petsch and Anspach, 2000). The BChE protein concentration is 3 mg/ml. A monkey scheduled to receive 60 mg of BChE would be receiving 20 ml of purified BChE and 2 micrograms of endotoxin (equivalent to 20,000 EU). Is this dose of endotoxin likely to be safe when injected iv into monkeys? Rhesus monkeys survived when treated with 150 micrograms endotoxin twice daily for 5 days (Xiao et al., 1998) or with 100 micrograms six times in 4 days (Landman et al., 2003). A lethal dose in rhesus monkeys was 3 × 106 EU/kg (Veloso et al., 1999), which calculates to 1500 micrograms endotoxin for a 5 kg animal. Thus 2 micrograms of endotoxin in our BChE preparation was not expected to cause an adverse reaction in monkeys.
The horse BChE purchased from Sigma had an endotoxin concentration similar to that in our BChE preparation. The Sigma horse BChE has been administered to monkeys without sequelae (Broomfield et al., 1991; Wolfe et al., 1992; Castro et al., 1994; Matzke et al., 1999), supporting our expectation that the level of endotoxin in our BChE preparation can be safely tolerated by monkeys.
The expectation that monkeys would not be harmed by our BChE preparation was confirmed by injecting BChE iv into monkeys. The monkeys had no adverse effects. Since body temperature and endotoxin-induced cytokines were not measured, we cannot rule out the possibility of pyrogenic effects.
Quality of our BChE preparation.
A mean residence time of 93 h for our human BChE preparation compares favorably with the results of Raveh et al who reported a mean residence time of 30-37 hours in the circulation of rhesus monkeys for their purified human BChE (Raveh et al., 1997). No other reports of the residence time of human BChE in monkeys have been published. In mice, the mean residence time for our BChE preparation was 56 h. Human BChE prepared by others was found to have a mean residence time of 33-46 h in mice, 60 h in rats, and 78 h in guinea pigs (Ashani et al., 1991; Kronman et al., 1995; Saxena et al., 1998; Ashani, 2000).
The long residence time of our purified human BChE in monkeys and mice attests to the high quality of our BChE preparation. A long residence time is obtained for BChE that is fully sialylated and that is assembled into tetramers. In our experience, partially desialylated BChE is cleared in less than one hour from the circulation of mice. BChE that is composed of monomers and dimers rather than tetramers is also rapidly cleared (Duysen et al., 2002). The related enzyme, acetylcholinesterase, is rapidly cleared from the circulation when it is desialylated or when it is monomeric, but has a long residence time when it is tetrameric and is maximally sialylated (Saxena et al., 1997; Chitlaru et al., 1998; Saxena et al., 1998; Kronman et al., 2000; Chitlaru et al., 2001).
Another indication of the high quality of our BChE preparation was the finding that monkeys showed no toxic signs when treated with our BChE. This result confirms that our BChE preparation contained only a low level of endotoxin. In conclusion, a method for purifying BChE from up to 100 L of human plasma per batch has been developed. The purified BChE has a low endotoxin content, safe for injections into monkeys.
ACKNOWLEDGEMENTS
Edman degradation of purified BChE was performed by the Protein Structure Core Facility at UNMC, directed by Dr. Laurey Steinke. Supported by NIH grant 1R01DA14023 (to GW and JW) and US Army Medical Research and Materiel Command DAMD17-01-2-0036 (to OL).
REFERENCES
- Allon N, Raveh L, Gilat E, Cohen E, Grunwald J, Ashani Y. Prophylaxis against soman inhalation toxicity in guinea pigs by pretreatment alone with human serum butyrylcholinesterase. Toxicol Sci. 1998;43:121–128. doi: 10.1006/toxs.1998.2463. [DOI] [PubMed] [Google Scholar]
- Altamirano CV, Lockridge O. Association of tetramers of human butyrylcholinesterase is mediated by conserved aromatic residues of the carboxy terminus. Chem Biol Interact. 1999;119-120:53–60. doi: 10.1016/s0009-2797(99)00013-7. [DOI] [PubMed] [Google Scholar]
- Ashani Y. Prospective of human butyrylcholinesterase as a detoxifying antidote and potential regulator of controlled-release drugs. Drug Development Research. 2000;50:298–308. [Google Scholar]
- Ashani Y, Shapira S, Levy D, Wolfe AD, Doctor BP, Raveh L. Butyrylcholinesterase and acetylcholinesterase prophylaxis against soman poisoning in mice. Biochem Pharmacol. 1991;41:37–41. doi: 10.1016/0006-2952(91)90008-s. [DOI] [PubMed] [Google Scholar]
- Axelsen PH, Harel M, Silman I, Sussman JL. Structure and dynamics of the active site gorge of acetylcholinesterase: synergistic use of molecular dynamics simulation and X-ray crystallography. Protein Sci. 1994;3:188–197. doi: 10.1002/pro.5560030204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandeis R, Raveh L, Grunwald J, Cohen E, Ashani Y. Prevention of soman-induced cognitive deficits by pretreatment with human butyrylcholinesterase in rats. Pharmacol Biochem Behav. 1993;46:889–896. doi: 10.1016/0091-3057(93)90218-i. [DOI] [PubMed] [Google Scholar]
- Broomfield CA, Maxwell DM, Solana RP, Castro CA, Finger AV, Lenz DE. Protection by butyrylcholinesterase against organophosphorus poisoning in nonhuman primates. J Pharmacol Exp Ther. 1991;259:633–638. [PubMed] [Google Scholar]
- Carmona GN, Schindler CW, Shoaib M, Jufer R, Cone EJ, Goldberg SR, Greig NH, Yu QS, Gorelick DA. Attenuation of cocaine-induced locomotor activity by butyrylcholinesterase. Exp Clin Psychopharmacol. 1998;6:274–279. doi: 10.1037//1064-1297.6.3.274. [DOI] [PubMed] [Google Scholar]
- Castro CA, Gresham VC, Finger AV, Maxwell DM, Solana RP, Lenz DE, Broomfield CA. Behavioral decrements persist in rhesus monkeys trained on a serial probe recognition task despite protection against soman lethality by butyrylcholinesterase. Neurotoxicol Teratol. 1994;16:145–148. doi: 10.1016/0892-0362(94)90111-2. [DOI] [PubMed] [Google Scholar]
- Chitlaru T, Kronman C, Velan B, Shafferman A. Effect of human acetylcholinesterase subunit assembly on its circulatory residence. Biochem J. 2001;354:613–625. doi: 10.1042/0264-6021:3540613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitlaru T, Kronman C, Zeevi M, Kam M, Harel A, Ordentlich A, Velan B, Shafferman A. Modulation of circulatory residence of recombinant acetylcholinesterase through biochemical or genetic manipulation of sialylation levels. Biochem J. 1998;336(Pt 3):647–658. doi: 10.1042/bj3360647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das PK, Liddell J. Purification and properties of human serum cholinesterase. Biochem J. 1970;116:875–881. doi: 10.1042/bj1160875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De la Hoz D, Doctor BP, Ralston JS, Rush RS, Wolfe AD. A simplified procedure for the purification of large quantities of fetal bovine serum acetylcholinesterase. Life Sci. 1986;39:195–199. doi: 10.1016/0024-3205(86)90530-8. [DOI] [PubMed] [Google Scholar]
- Duysen EG, Bartels CF, Lockridge O. Wild-type and A328W mutant human butyrylcholinesterase tetramers expressed in Chinese hamster ovary cells have a 16-hour half-life in the circulation and protect mice from cocaine toxicity. J Pharmacol Exp Ther. 2002;302:751–758. doi: 10.1124/jpet.102.033746. [DOI] [PubMed] [Google Scholar]
- Ellman GL, Courtney KD, Andres V, Jr., Feather-Stone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- Gabrielson J, Weiner D. Pharmacokinetic and Pharmacodynamic Data Analysis. Concepts and Applications. Swedish Pharmaceutical Press; Stockholm: 1994. [Google Scholar]
- Gaffney PJ., Jr. Human serum cholinesterase. I. Partial purification and nature of the heterogeneity of this system. Biochim Biophys Acta. 1970;207:465–476. [PubMed] [Google Scholar]
- Grunwald J, Marcus D, Papier Y, Raveh L, Pittel Z, Ashani Y. Large-scale purification and long-term stability of human butyrylcholinesterase: a potential bioscavenger drug. J Biochem Biophys Methods. 1997;34:123–135. doi: 10.1016/s0165-022x(97)01208-6. [DOI] [PubMed] [Google Scholar]
- Hoffman RS, Morasco R, Goldfrank LR. Administration of purified human plasma cholinesterase protects against cocaine toxicity in mice. J Toxicol Clin Toxicol. 1996;34:259–266. doi: 10.3109/15563659609013786. [DOI] [PubMed] [Google Scholar]
- Karnovsky MJ, Roots L. A “direct-coloring” thiocholine method for cholinesterases. J Histochem Cytochem. 1964;12:219–221. doi: 10.1177/12.3.219. [DOI] [PubMed] [Google Scholar]
- Kronman C, Chitlaru T, Elhanany E, Velan B, Shafferman A. Hierarchy of post-translational modifications involved in the circulatory longevity of glycoproteins. Demonstration of concerted contributions of glycan sialylation and subunit assembly to the pharmacokinetic behavior of bovine acetylcholinesterase. J Biol Chem. 2000;275:29488–29502. doi: 10.1074/jbc.M004298200. [DOI] [PubMed] [Google Scholar]
- Kronman C, Velan B, Marcus D, Ordentlich A, Reuveny S, Shafferman A. Involvement of oligomerization, N-glycosylation and sialylation in the clearance of cholinesterases from the circulation. Biochem J. 1995;311(Pt 3):959–967. doi: 10.1042/bj3110959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landman RE, Puder JJ, Xiao E, Freda PU, Ferin M, Wardlaw SL. Endotoxin stimulates leptin in the human and nonhuman primate. J Clin Endocrinol Metab. 2003;88:1285–1291. doi: 10.1210/jc.2002-021393. [DOI] [PubMed] [Google Scholar]
- Lockridge O. Genetic variants of human serum cholinesterase influence metabolism of the muscle relaxant succinylcholine. Pharmacol Ther. 1990;47:35–60. doi: 10.1016/0163-7258(90)90044-3. [DOI] [PubMed] [Google Scholar]
- Lockridge O, Adkins S, La Du BN. Location of disulfide bonds within the sequence of human serum cholinesterase. J Biol Chem. 1987a;262:12945–12952. [PubMed] [Google Scholar]
- Lockridge O, Bartels CF, Vaughan TA, Wong CK, Norton SE, Johnson LL. Complete amino acid sequence of human serum cholinesterase. J Biol Chem. 1987b;262:549–557. [PubMed] [Google Scholar]
- Lockridge O, La Du BN. Comparison of atypical and usual human serum cholinesterase. Purification, number of active sites, substrate affinity, and turnover number. J Biol Chem. 1978;253:361–366. [PubMed] [Google Scholar]
- Lockridge O, La Du BN. Loss of the interchain disulfide peptide and dissociation of the tetramer following limited proteolysis of native human serum cholinesterase. J Biol Chem. 1982;257:12012–12018. [PubMed] [Google Scholar]
- Lynch TJ. Production of butyrylcholinesterase. Pharmavene, Inc.; Gaithersburg, MD: Patent number US5272080 1993
- Lynch TJ, Mattes CE, Singh A, Bradley RM, Brady RO, Dretchen KL. Cocaine detoxification by human plasma butyrylcholinesterase. Toxicol Appl Pharmacol. 1997;145:363–371. doi: 10.1006/taap.1997.8187. [DOI] [PubMed] [Google Scholar]
- Malmstrom BG, Levin O, Boman HG. Chromatography of human serum cholinesterase. Acta Chem Scand. 1956;10:1077–1082. [Google Scholar]
- Matzke SM, Oubre JL, Caranto GR, Gentry MK, Galbicka G. Behavioral and immunological effects of exogenous butyrylcholinesterase in rhesus monkeys. Pharmacol Biochem Behav. 1999;62:523–530. doi: 10.1016/s0091-3057(98)00183-x. [DOI] [PubMed] [Google Scholar]
- Nicolet Y, Lockridge O, Masson P, Fontecilla-Camps JC, Nachon F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J Biol Chem. 2003;278:41141–41147. doi: 10.1074/jbc.M210241200. [DOI] [PubMed] [Google Scholar]
- Petsch D, Anspach FB. Endotoxin removal from protein solutions. J Biotechnol. 2000;76:97–119. doi: 10.1016/s0168-1656(99)00185-6. [DOI] [PubMed] [Google Scholar]
- Ralston JS, Main AR, Kilpatrick BF, Chasson AL. Use of procainamide gels in the purification of human and horse serum cholinesterases. Biochem J. 1983;211:243–250. doi: 10.1042/bj2110243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveh L, Grauer E, Grunwald J, Cohen E, Ashani Y. The stoichiometry of protection against soman and VX toxicity in monkeys pretreated with human butyrylcholinesterase. Toxicol Appl Pharmacol. 1997;145:43–53. doi: 10.1006/taap.1997.8160. [DOI] [PubMed] [Google Scholar]
- Raveh L, Grunwald J, Marcus D, Papier Y, Cohen E, Ashani Y. Human butyrylcholinesterase as a general prophylactic antidote for nerve agent toxicity. In vitro and in vivo quantitative characterization. Biochem Pharmacol. 1993;45:2465–2474. doi: 10.1016/0006-2952(93)90228-o. [DOI] [PubMed] [Google Scholar]
- Rochu D, Renault F, Masson P. Detection of unwanted protein-bound ligands by capillary zone electrophoresis: the case of hidden ligands that stabilize cholinesterase conformation. Electrophoresis. 2002;23:930–937. doi: 10.1002/1522-2683(200203)23:6<930::AID-ELPS930>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Saxena A, Ashani Y, Raveh L, Stevenson D, Patel T, Doctor BP. Role of oligosaccharides in the pharmacokinetics of tissue-derived and genetically engineered cholinesterases. Mol Pharmacol. 1998;53:112–122. doi: 10.1124/mol.53.1.112. [DOI] [PubMed] [Google Scholar]
- Saxena A, Hur RS, Luo C, Doctor BP. Natural monomeric form of fetal bovine serum acetylcholinesterase lacks the C-terminal tetramerization domain. Biochemistry. 2003;42:15292–15299. doi: 10.1021/bi030150x. [DOI] [PubMed] [Google Scholar]
- Saxena A, Raveh L, Ashani Y, Doctor BP. Structure of glycan moieties responsible for the extended circulatory life time of fetal bovine serum acetylcholinesterase and equine serum butyrylcholinesterase. Biochemistry. 1997;36:7481–7489. doi: 10.1021/bi963156d. [DOI] [PubMed] [Google Scholar]
- Sharma SK. Endotoxin detection and elimination in biotechnology. Biotechnol Appl Biochem. 1986;8:5–22. [PubMed] [Google Scholar]
- Simon S, Krejci E, Massoulie J. A four-to-one association between peptide motifs: four C-terminal domains from cholinesterase assemble with one proline-rich attachment domain (PRAD) in the secretory pathway. Embo J. 1998;17:6178–6187. doi: 10.1093/emboj/17.21.6178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Shen ML, Pang YP, Lockridge O, Brimijoin S. Cocaine metabolism accelerated by a re-engineered human butyrylcholinesterase. J Pharmacol Exp Ther. 2002;302:710–716. doi: 10.1124/jpet.302.2.710. [DOI] [PubMed] [Google Scholar]
- Surgenor DM, Ellis D. Preparation and properties of serum and plasma proteins. Plasma cholinesterase. J Am Chem Soc. 1954;76:6049–6051. [Google Scholar]
- Veloso D, Smith JI, Cosgriff TM. Rhesus differential susceptibility to endotoxin is not associated with activation of plasma prekallikrein. Immunopharmacology. 1999;43:265–271. doi: 10.1016/s0162-3109(99)00098-3. [DOI] [PubMed] [Google Scholar]
- Wolfe AD, Blick DW, Murphy MR, Miller SA, Gentry MK, Hartgraves SL, Doctor BP. Use of cholinesterases as pretreatment drugs for the protection of rhesus monkeys against soman toxicity. Toxicol Appl Pharmacol. 1992;117:189–193. doi: 10.1016/0041-008x(92)90236-l. [DOI] [PubMed] [Google Scholar]
- Xiao E, Xia-Zhang L, Barth A, Zhu J, Ferin M. Stress and the menstrual cycle: relevance of cycle quality in the short- and long-term response to a 5-day endotoxin challenge during the follicular phase in the rhesus monkey. J Clin Endocrinol Metab. 1998;83:2454–2460. doi: 10.1210/jcem.83.7.4926. [DOI] [PubMed] [Google Scholar]