

Abstract

The reduction of β-amino ketones with samarium(II) iodide has been investigated. Either the syn or anti 1,3-amino alcohols can be obtained as the major product due to a divergence in selectivity with different N-protecting groups.
The development of methodology for the efficient generation of relative stereochemistry in acyclic systems continues to be a subject of major importance in synthetic organic chemistry. Recently, a protocol for the reduction of β-hydroxy and certain β-alkoxy ketones with SmI2 was developed in our laboratories. Anti 1,3-diols and certain anti 1,3-alkoxy alcohols can be produced with high selectivity in this electron-transfer reduction.1 The product-determining event was suggested to be protonation of an intermediate samarium carbanion in a favored conformation of a chelate formed by complexation of a second samarium between the β-oxygen and the carbonyl oxygen. The β-oxygen was also shown to be responsible for a significant rate enhancement in these reductions. We record herein the results of an investigation of β-nitrogen substituents in such reductions. In this context, it shou1d be noted that 1,3-amino alcohol fragments are of interest due to their presence in several antibiotics and other biologically active natural products.2 Thus, several reduction methods for accessing such structures have previously been developed.3 For example, Narasaka achieved good syn diasteroselectivity in the LiAlH4–NaOMe reduction of O-benzyl oximes derived from β-hydroxy ketones.4 Pilli and co-workers have also developed a stereoselective route to anti and syn N-aryl-β-amino alcohols via reduction with Et3BHLi and Zn(BH4)2, respectively.5
The first set of β-amino ketones used in this study was prepared by the sequence shown in Scheme 1. Allylation of cyclohexylcarboxaldehyde with methallyl-tri-n-butylstannane gave the corresponding homoallylic alcohol.6 Conversion to the mesylate and displacement with sodium azide gave the desired homoallylic azide 1 in 57% overall yield. Reduction of azide 1 was accomplished with LiAlH4 in THF at 0 °C. The resulting amine was then protected as either the trifluoroacetamide, acetamide, tert-butyl carbamate, or benzyl carbamate using standard protocols. Subsequent ozonolysis provided the corresponding N-protected amino ketones in good yields (Scheme 1).
Scheme 1.

Preparation of N-Acyl β-Amino Ketones
Each of these N-acyl-β-amino ketones (2a–d) was first reduced with NaBH4 to afford a mixture containing roughly equal amounts of the 1,3-syn and 1,3-anti diastereomers. These mixtures were then used to develop an analytical HPLC separation of the reduction products expected from each substrate. Reductions with SmI2 were then investigated. In a typical reaction, the ketone and 20 equiv of MeOH were dissolved in THF and added to a freshly prepared solution (0.1–0.5 M) of SmI2 in THF at 0 °C.
The results of these experiments (Table 1) showed that high yields and reasonable levels of stereoselectivity are obtained for the reduction of these protected β-amino ketones. However, at this point the stereochemistry of the products was not known.
Table 1.
Reduction of N-Acyl β-Amino Ketones Using SmI2-MeOH
 |
| entry | P | time (h) | yield of 3 (%) | ratio (syn:anti) |
|---|---|---|---|---|
| 1 | TFA | 4.75 | 86 | 90:10 |
| 2 | Ac | 6 | 96 | 89:11 |
| 3 | Boc | 4 | quant | 81:19 |
| 4 | Cbz | 7 | 84 | 84:16 |
The structure of the major diastereomer formed in these reactions was unambiguously established by an X-ray crystal structure determination of the major product obtained from 2a. This was followed by chemical correlation of that material with the major products obtained from 2b–d. Thus, removal of the trifluoroacetate7 (K2CO3/MeOH/H2O) from both the major (syn) and minor (anti) diastereomers of 3a provided samples of the free amino alcohols.
These were then compared with the amino alcohols obtained by removal of the protecting groups present in the major diastereomers of 3b–d. In all cases, the major diastereomer was 1,3-syn. The observed stereochemical outcome is initially quite surprising given the anti selectivity previously observed in reductions of both β-hydroxy and β-alkoxy ketones under these conditions. A different class of protected β-amino ketones was also synthesized using the methodology of Kobayashi.8 This reaction was applied successfully to the synthesis of a number of N-aryl-β-amino ketones (Scheme 2). Again, each of these substrates (4a–f) was reduced with NaBH4 to secure mixtures containing comparable amounts of the expected 1,3-syn and 1,3-anti reduction products; these were then used to develop analytical HPLC separations for each substrate as before.
Scheme 2.

Preparation of N-Aryl β-Amino Ketones
Under our standard conditions, these N-aryl-β-amino ketones were also reduced to the corresponding 1,3-amino alcohols in excellent yield (90–99%). Stereochemical assignments for the products of these reactions were again made unambiguously through use of the X-ray crystal structure determination of 3a. This involved the removal of the o-MeOPh (catalytic amount of silver nitrate and ammonium persulfate in aqueous CH3CN)9 or p-MeOPh (CAN in aqueous CH3CN)10 groups from each of the reduction products 5a–e.11 The resulting free β-amino alcohols were then directly compared (13C NMR) to the free β-amino alcohols that were obtained from the major (syn) and minor (anti) diastereomers of 3a. It was found that a complete reversal of stereoselectivity for the reduction occurs between the two classes of substrates: whereas the N-acyl derivatives all reduced to afford the 1,3-syn diastereomer as the major product, the N-aryl derivatives all afforded the 1,3-anti diastereomer as the major product.
Thus, these N-aryl β-amino ketones are reduced in the same sense (1,3-anti diastereoselectivity) as the previously studied β-hydroxy and β-alkoxy ketones. We suggest that the overall mechanism is as previously proposed for those cases. However, the present situation seems more complex than with the oxygen series. Reductions of two N-alkyl-β-amino ketones were examined in order to determine the extent to which these reductions paralleled those of the β-alkoxy ketones.
In contrast to the corresponding O-benzyl derivative, the N-benzyl derivative was reduced but with a low level of stereoselectivity (3.5:1). The N-methyl derivative also reduced, but, in contrast to previous observations in the oxygen series, afforded a 1:1 mixture of diastereomers. Reductions of such β-methoxy ketones in our previous study showed very high levels of stereoselectivity; note eq 2. Although the substituents differ in the cases shown (cyclohexyl vs phenyl) in eqs 1 and 2, inspection of Table 2 shows that the level of asymmetric induction realized is relatively independent of this substituent. Cases in which this substituent was a phenyl group were not examined in the present study due to the low stability of these materials toward β-elimination.
Table 2.
Reduction of N-Aryl β-Amino Ketones
 |
| entry | R | P | yield of 5 (%) | ratio (anti:syn) |
|---|---|---|---|---|
| 1 | n-C3H7 | o-MeOPh | 95 | 97:3 |
| 2 | C6H11 | o-MeOPh | 100 | 97:3 |
| 3 | PhCH[unk]CH | o-MeOPh | 90 | 97:3 |
| 4 | n-C3H7 | p-MeOPh | 95 | 94:6 |
| 5 | C6H11 | p-MeOPh | 98 | >99:1 |
| 7 | n-C3H7 | Ph | 96 | 85:15 |
Thus N-substitution by an electron-rich aryl ring seems to be required to realize high levels of stereoselectivity in  these reductions. In this context, reduction of substrate 4f, which lacks any additional electron-releasing substituents on the aryl ring other than nitrogen, leads to a somewhat lower level of stereoselectivity than is observed with the methoxy-substituted compounds. Moreover, since both the ortho and para methoxy compounds are reduced with similarly high levels of diastereoselectivity, a chelation motif involving the nitrogen and the methoxy group itself cannot be involved as a control element in determining the stereochemical outcome.8,12 However, it does seem plausible based upon the available data that complexation of the electron-rich aryl ring by samarium may influence the stereochemical outcome of the reduction in these cases.
these reductions. In this context, reduction of substrate 4f, which lacks any additional electron-releasing substituents on the aryl ring other than nitrogen, leads to a somewhat lower level of stereoselectivity than is observed with the methoxy-substituted compounds. Moreover, since both the ortho and para methoxy compounds are reduced with similarly high levels of diastereoselectivity, a chelation motif involving the nitrogen and the methoxy group itself cannot be involved as a control element in determining the stereochemical outcome.8,12 However, it does seem plausible based upon the available data that complexation of the electron-rich aryl ring by samarium may influence the stereochemical outcome of the reduction in these cases.
At first glance, the syn stereochemistry observed in reduction of the N-acyl derivatives would seem to preclude a mechanism involving chelation as a control element; however, these results are easily accommodated by such a mechanism. Since samarium(II) is believed to be a hard oxophilic Lewis acid,13 it is reasonable to expect that the oxygen of the amidate, rather than the nitrogen, is involved in initial coordination to samarium. The reduction could then occur via the formation of an intermediate eight-membered ring chelated structure as shown in Scheme 3. Initial coordination of samarium to the oxygen of the amidate is followed by a one-electron transfer to the carbonyl to give III. A second equivalent of Sm(II) donates another electron to the resulting radical leading to the samarium carbanion IV. Finally, protonation of this anion then leads to the syn amino alcohol V in the stereochemistry-determining step.
Scheme 3.

The syn stereochemical outcome predicted by this scheme relies upon extrapolation of the known conformational preferences in eight-membered carbocyclic rings14 to this case involving samarium. The A value associated with the substituent “R” in structure IV is known to be very large in eight-membered ring carbocycles.
In summary, the reduction of β-amino ketones using samarium(II) iodide provides a versatile synthetic method for the synthesis of 1,3-amino alcohols. High levels of stereoselectivity and excellent yields are possible in the reduction of N-aryl β-amino ketones, while a variety of N-acyl derivatives are reduced in good yield and with moderate levels of 1,3-syn stereoselectivity. One especially attractive feature of the process is that by employing different protecting groups, one can easily access both syn and anti diastereomers of these β-amino alcohols.
Supplementary Material
Figure 1.
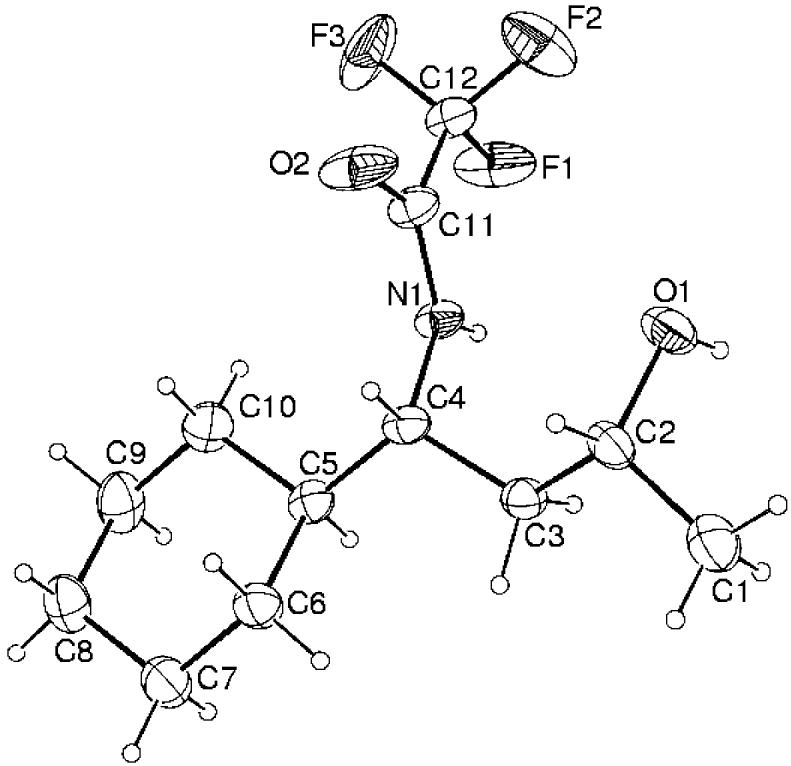
Structure of the major diastereomer of N-trifluoroacetamide 3a by X-ray analysis.
Acknowledgment
Financial assistance provided by the National Institutes of Health (Grant GM-28961) is gratefully acknowledged.
Footnotes
Supporting Information Available: Full experimental details and analytical data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Keck GE, Wager CA, Sell T, Wager TT. J Org. Chem. 1999;64:2172. [Google Scholar]; (b) Keck GE, Wager CA. Org. Lett. 2000;2:2307. doi: 10.1021/ol006072c. [DOI] [PubMed] [Google Scholar]
- 2.Nogradi M. VCH: Wienheim; Germany: 1987. Stereoselective Synthesis. Chapters 3, 5, 6. [Google Scholar]
- 3.Barluenga J, Aguilar E, Fustero S, Olano B, Viado A. J. Org. Chem. 1992;57:1219. and references therein. [Google Scholar]
- 4.Narasaka K, Ukaji Y, Yamazaki S. Bull. Chem. Soc. Jpn. 1986;59:525. [Google Scholar]
- 5.(a) Pilli RA, Russowsky D, Dias LC. Chem. Commun. 1987:1053. [Google Scholar]; (b) Pilli RA, Russowsky D, Dias LC. J. Chem. Soc., Perkin Trans. 1. 1990:1213. [Google Scholar]
- 6.Keck GE, Krishnamurthy D, Grier MC. J. Org. Chem. 1993;58:6543. For an asymmetric version of this reaction, see: [Google Scholar]
- 7.Boger DL, Yohannes D. J. Org. Chem. 1989;54:2498. [Google Scholar]
- 8.Kobayashi S, Ueno M, Suzuki R, Ishitani H, Kim H-S, Wataya Y. J. Org. Chem. 1999;64:6833. doi: 10.1021/jo990877k. [DOI] [PubMed] [Google Scholar]
- 9.Saito SS, Hatanaka K, Yamamoto H. Org. Lett. 2000;2:1891. doi: 10.1021/ol000099e. [DOI] [PubMed] [Google Scholar]
- 10.Kronenthal DR, Han CY, Taylor MK. J. Org. Chem. 1982;47:2765. [Google Scholar]
- 11. The stereochemistry of product 5f has not been unambiguously established but is assumed on the basis of the results with the other N-aryl compounds and the similarity of the 1H and 13C NMR data.
- 12.Ishitani H, Ueno M, Kobayashi S. J. Am. Chem. Soc. 2000;122:8180. For previous proposals of such a complexation mode in reactions involving o-hydroxy anilines and derivatives, see: and references therein. [Google Scholar]
- 13.Molander GA. Org. React. 1994;46:211. [Google Scholar]
- 14.Still WC, Galynker I. Tetrahedron. 1981;37:3981. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.