

Abstract

Reactions of the bifunctional allylstannane 2-(chloromethyl)-3-(tributylstannyl)propene with aldehydes have been examined. These generally occur in high yields using Lewis acid promoters and the products can be isolated and purified without incident. Good yields and high enantioselectivities are also realized in catalytic asymmetric allylations (CAA reactions) using the previously described BITIP catalyst system. Protection of the free hydroxyl can be accomplished without cyclization to the derived tetrahydrofuran, although this transformation is also facile. The utility of the incorporated allyl chloride functionality allows for the obvious use of such products in reactions with nucleophiles. Use of these products in a less obvious connective strategy is demonstrated in the synthesis of the C12–C27 segment of bryostatin 1 where a connective, or “lynchpin”, double-allylation process was employed. The β-hydroxy allyl chloride obtained from an initial chelation-controlled allylation of aldehyde 16 was converted to allylstannane 19 and applied in a second allylation reaction, thus allowing for a highly convergent synthesis of the bryostatin C ring backbone in a stereoselective fashion.
Introduction
The Lewis acid promoted additions of storable nucleophiles to aldehydes has proven to be an extremely powerful bond construction. Allylstannanes have proven particularly useful in this regard. The stereochemistry of these reactions is of both interest and practical importance, and examples in which stereochemistry is controlled by stereoelectronic effects, by substrate preorganization via chelation, by the use of stoichiometric chiral reagents, and by Lewis acid chirality are all known.1 Our own studies initially utilized the parent allyltributylstannane,2 but substituted allylstannanes3 can also be employed to good advantage. Synthetic studies currently underway in our laboratories have prompted us to examine variations on the allylation methodology further by using 2-substituted allylstannanes possessing reactive functionality, which can serve as a handle for subsequent chemistry.4 Herein, we report the results of allylation studies using 2-(chloromethyl)-3-(tributylstannyl)propene 1 in both BINOL titanium 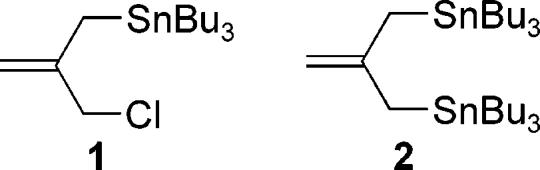 tetraisopropoxide (BITIP) catalyzed asymmetric additions to aldehydes1b and diastereoselective reactions leading to the synthesis of the C17–C27 segment of the bryostatins, a class of potent anticancer agents.5,6
tetraisopropoxide (BITIP) catalyzed asymmetric additions to aldehydes1b and diastereoselective reactions leading to the synthesis of the C17–C27 segment of the bryostatins, a class of potent anticancer agents.5,6
Results and Discussion
An analysis of substructures found in the bryostatins and other natural products being studied in our laboratories prompted research into the development of an allylstannane-based isobutene synthon that could serve as a coupling agent between two different aldehydes in a stereoselective double allylation procedure as outlined in eq 1. Our past experience in the synthesis of functionalized piperidines7 led us to initially consider the known reagent 2 as a suitable equivalent to such a synthon.

It is well-known that the additional allylic stannyl group creates a dramatic difference in reactivity between 2 and the subsequent mono-allylstannane intermediate obtained after the initial addition reaction. This difference in reactivity has allowed for iterative additions to electrophiles without isolation of intermediates.8 Our own research interests, however, required that the sensitive mono-stannane intermediate be isolated and the hydroxyl group protected prior to the second addition reaction. We were also concerned that the increased reactivity of the bis-stannane reagent might adversely affect the degree of stereoselectivity obtained under both chiral Lewis acid controlled and substrate controlled additions. We began to address these requirements and concerns in a study where the bis-stannane reagent was employed in the catalytic asymmetric addition reaction using the BITIP catalyst.
Reaction of allylstannane 2 with benzaldehyde gave the desired product 3 and the methallylation product 4 in low and variable yields (Scheme 1). Product 4 could arise by one of two ways. Protiodestannylation of 3 could occur to give the corresponding methallyl adduct, or alternatively protiodestannylation of the tin reagent 2 could take place giving methallyl tributyltin followed by addition of that reagent to benzaldehyde to give product 4. In the former scenario, both 3 and 4 would be produced with the same enantiomeric excess, which is not necessarily true for the second pathway. The observed ee values suggest that both pathways contribute, but that the major pathway to 4 is via protiodestannylation of 2. In fact, NMR studies found that this highly reactive stannane reagent undergoes rapid hydrolysis in the presence of (1,1′-binaphthalene)-2,2′-diol, and to some extent in 2-propanol. This unfortunate competing side reaction would also explain the low levels of product formation as such a process would rob the system of protons which are thought to be important for catalyst turnover.
Scheme 1.

Asymmetric Allylation Using Allylstannane 2
Isolation and purification of the sensitive allylstannane product was also quite problematic. The allylstannane functionality was prone to acid catalyzed hydrolysis making it necessary to carefully treat all glassware with triethylamine to eliminate all residual acid sources. Likewise, purification by silica gel chromatography resulted in complete decomposition to give the methallylation product unless the silica gel was pretreated with triethylamine. Given the sensitive nature of this reagent and of the product from the initial allylation reaction, we elected to pursue a different approach in which the second tributylstannyl substitutent would be introduced after the first allylation reaction. Thus allylstannane 1 was synthesized and investigated as a more robust surrogate to the bis-stannane reagent 2 through eventual conversion of the allyl chloride to the allylstannane. This approach had the added benefit of providing for additional useful synthetic applications, in that the allylic chloride product could clearly be used in reactions with a wide variety of nucleophiles (e.g. enolates, dithianes, amines, etc.) which would lead to other convergent approaches to more complex materials.
Recently reported syntheses of allylstannane 1 have accessed the reagent through a two-step process beginning with 2-methyl-1-propen-3-ol.9 The reported processes require silica gel chromatography at each step and the overall yield averaged approximately 45%. A more simple and direct synthesis was sought that avoided chromatography as a means of purification. Our approach to allylstannane 1 was to selectively displace one of the chloride groups of 3-chloro-2-chloromethyl-1-propene 5 with 1 equiv of lithium tributyltin (Scheme 2). This strategy originated from the assumption that the steric effect from the bulky stannane group would disfavor a second chloride substitution. However, when the reaction was carried out in THF, it was found by NMR analysis of the crude reaction mixture that a statistical mixture of starting material, product 1, and bis-stannane 2 formed, suggesting the displacement of the second chloride is a competitive process. After consideration of possible transition states for these two processes, we considered that the presence of an electron donating stannane group in the transition state (7) may facilitate the second chloride substitution by enhancing the SN1 nature of the process relative to the transition state (6) for the first displacement. In an attempt to overcome this problem, experiments were performed in which the polarity of the solvent was lowered in order to suppress the impact such an electronic effect would have on the system. In our study, we employed hexanes as a cosolvent to reduce the overall polarity of the reaction medium, and found the best result could be obtained when a 1:1 mixture of THF/hexanes was used. Under these conditions, the desired mono-displaced product 1 could be isolated in up to 60% yield. Only a trace amount of bisstannane was formed. The reaction has been performed on multigram scale and the product can be easily isolated through simple distillation. It should be noted that nonpolar solvents also reduce the reactivity of bis-chloride 5. Therefore, a higher ratio of hexanes is not suggested for the reaction, as a significant amount of starting material will remain unreacted under such conditions.
Scheme 2.

Synthesis of Allylstannane 1
With the reagent in hand, experiments were done to ascertain its utility in BITIP catalyzed asymmetric additions to aldehydes. Although it can reasonably be assumed that the incorporation of an electron withdrawing group on the allylstannane nucleophile would lower its reactivity, the degree of impact was unknown. In one case, however, we had shown that a mildly electron withdrawing group at the 2-position was well tolerated by the BITIP catalyst system.4a
Reactions of this allylstannane with benzaldehyde were carried out to determine the optimal experimental conditions. The outcome of the experiments showed that reactions carried out in CH2Cl2 using BITIP catalyst prepared by “method A”2b gave superior results; however, catalyst prepared by “method B” gave results that were not significantly different. A catalyst loading of 20 mol % was initially used while determining optimal reaction conditions, but then lowered to 10 mol % once reaction parameters were established with no noticeable negative effect on reaction outcome. The results of reactions using a variety of aldehydes under “method A” condition are summarized in Table 1. In most cases, both aromatic and aliphatic aldehydes gave the corresponding β-hydroxy allyl chlorides in good yields and with high levels of enantioselectivity.
Table 1.

Isolated Yields and Enantiomeric Excess for the Reaction of 1 with Aldehydes
A number of synthetic transformations can be explored utilizing the allyl chloride functionality found in the resulting product. For example, employing the allyl chloride in an intramolecular cyclization event gave a chiral methylenetetrahydrofuran ring having a substitution pattern that can be found in a number of compounds of biological interest (eq 2).10

The concept of exploiting the bifunctional nature of allylstannane 1 as a linchpin for two different aldehydes was realized while engaged in the synthesis of the C17–C27 (C-ring) segment of bryostatin 1.11 The structural features found in this segment also served as a vehicle for evaluating substrate controlled diastereoselective addition reactions using the stannane reagent.
The key features of our approach are shown retrosynthetically in Scheme 3. Our approach to this segment was aimed at providing an efficient synthesis of intermediate 11 (Scheme 3). We envisioned from the outset that the hemiketal moiety present at C19 would be established from the corresponding hydroxy ketone, most probably as its mixed methoxy ketal. We also planned to introduce the prenyl moiety present at C16–C18 via reaction of an aldehyde with a prenyl organometallic reagent. Finally, we planned to introduce the unsaturated ester present at C21 via a Horner–Emmons reaction, most probably after the ring closure had occurred. A (protected) hydroxyl substituent was thus needed at C21. The stereochemistry of the C21 center was inconsequential since it was destined for oxidation, thus we could choose its configuration in a way that best accommodated our planned synthetic route. By placing an (R)-configuration at the C21 position, as in 13, a quasi C2 symmetry around the C23 center with regard to the arrangement of the flanking hydroxyl stereocenters is produced. If we consider that the C23 hydroxyl could be obtained by reduction of the corresponding ketone, as in 14, then it can be seen that this material might be considered to arise from two consecutive aldol reactions, or alternatively, through the use of allylstannane additions as a surrogate for the aldol process. With the C21 position configured as (R), it can be seen that the vicinal pairs (C20–C21 and C25–C26) in the allyl addition product 15 are both syn. This is the stereochemical result that would be established by a chelation controlled addition to an α-alkoxy aldehyde.2a Based on this retrosynthetic strategy, our key intermediate 12 may be disassembled into the lactate derived aldehyde 16, allylstannane 1, and aldehyde 17. In this way, through a linchpin provided by the allylstannane 1, an advanced skeleton of our target can be constructed in a highly convergent fashion.
Scheme 3.

Proposed Approach to the Bryostatin C-Ring
The reaction between stannane 1 and the lactate derived aldehyde 16 under MgBr2·OEt2 mediated conditions afforded the desired alcohol 18 in high yield and with a 30:1 diastereomeric ratio (Scheme 4).2a Protection of the resulting alcohol is required prior to further elaboration. Although we had found that such alcohols cyclized readily, it was also found that 18 could be either silyated or acylated without cyclization. After protection of the resulting β-hydroxyl group, in this case as a pivalate, the allyl chloride was converted to the allylstannane by reaction with Bu3SnLi to give compound 19.
Scheme 4.

Double-Allylation Approach
The second allylation now involved a nucleophile with a preexisting stereocenter. Although this center is quite remote, we had no previous examples of how that might influence the stereochemical outcome of the upcoming addition reaction. As the C21 hydroxyl group would be used to establish the chirality of the crucial C23 stereocenter, the chelation controlled addition of 19 to aldehyde 17 had to proceed with a high degree of stereoselectivity.
To our gratification, the coupling reaction between 17 and 19 under chelation controlled conditions did indeed establish the C21 center with a high (20:1) level of stereoselectivity, thereby affording the C19–C27 carbon skeleton. To help confirm the stereochemistry of the newly generated C21 center, the reaction was also performed using BF3·OEt2 to promote coupling under Felkin-Anh control. The major product 21 isolated from this reaction had the opposite stereochemistry from the product obtained under chelation controlled conditions, which indirectly provides evidence to support our assignment of the C21 stereochemistry of 20. The stereochemistry of this center was ultimately established through further chemistry and spectroscopy; these experiments are detailed in the Supporting Information.12
Completion of the C17–C27 segment of the bryostatins is shown in Scheme 5. Oxidative cleavage of the double bond and subsequent syn-reduction of the resulting hydroxy ketone under the conditions developed by Prasad13 afforded the diol in high yields as a single isomer, thus providing key intermediate 23 with all stereocenters in place. The diol unit in 23 was then protected as its acetonide, from which the syn-diol relationship was confirmed by 13C NMR.14
Scheme 5.

Completion of the Bryostatin C-Ring
Deprotection and oxidation of the primary hydroxyl group furnished the corresponding aldehyde, which was treated with an in situ generated prenyl indium reagent15 to complete the carbon backbone of our target molecule. Oxidation of the resulting alcohol was found to be problematic. With most commonly used oxidants, such as TPAP, (COCl)2/DMSO/NEt3, Dess–Martin, and SO3·Py/DMSO, the reaction did not proceed to any extent resulting in the recovery of starting material. Only moderate success was achieved using PCC oxidation conditions where a 40% yield of the ketone was isolated. Fortunately, a solution was found in which oxidation could be attained in greater than 85% yields using a 3:2:2 mixture of DMSO/Ac2O/NEt3, conditions considered most suitable for oxidizing hindered alcohols.16 Operationally, the steps of TPAP oxidation of compound 25, prenyl indium addition, and DMSO/Ac2O/NEt3 oxidation were routinely performed without purification of the intermediates giving compound 27 in an overall yield of 60%.
Thus far, as demonstrated by the successful synthesis of ketone 27, we had developed a highly efficient approach to the complete carbon backbone of the C17–C27 segment of the bryostatins, with all the requisite stereocenters set and functionality in place. The completion of our synthesis only required cyclization to form the C ring and the construction of the C21 exocyclic olefin.
In theory, the C ring can be generated by acetonide deprotection, which should also trigger concomitant internal ketal formation. In practice, however, cyclization after removal of the acetonide proved quite problematic. Treatment of compound 27 with acidic methanol under a variety of conditions did not provide any cyclized product. Although the acetonide was efficiently removed under acidic conditions, the resulting diol failed to undergo cyclization. More forcing conditions resulted in elimination of the C21 alcohol to give the corresponding PMB enol ether. Additional studies in which the C21 hydroxyl group was inverted in an effort to minimize unfavorable steric interactions with other ring substituents gave only marginal yields of cyclized product.
A major breakthrough came when attempts were made to accelerate the cyclization process by preforming the dimethoxy ketal from ketone 27. For this transformation, forcing conditions utilizing TMSOMe and a catalytic amount of TMSOTf in CH2Cl2 were adopted.17 Surprisingly, ketone 28 was isolated in good yields from the reaction. In addition to its potential utility in our synthesis, this transformation is remarkable in that it is the net result of five different reactions, which include deprotection of the acetonide, cyclization to form the internal hemiketal, formation of the methoxy mixed ketal, elimination of the C21 alcohol, and deprotection of the PMB ether. An unfortunate and surprising aspect of this transformation, however, is that the C21 oxygen substituent (bryostatin numbering), which had been intended to allow for installation of the unsaturated ester via an Emmons reaction, was lost in this process. This forced us to revise our strategy for completion of the C ring segment by intercepting the chemistry previously developed by Evans and by Wender for installation of the unsaturated ester.
Thus, with ketone 28 in hand, our synthesis of the C ring segment of the bryostatins was completed following a known procedure.11g An aldol condensation of the enolate derived from 28 with methyl glyoxylate, followed by treatment of the resulting alcohol with MsCl and DBU to effect elimination gave enone 30 in excellent yield and as a single isomer with regard to the olefin geometry. The 1H NMR spectrum of 30 revealed a significant difference in the chemical shift between the two allylic protons at the C22 position. The equatorial proton has a chemical shift at δ 3.31, which is indicative of it residing in the deshielding cone of the C21 unsaturated ester. As expected, this proton also shows a weak coupling (J = 2.1 Hz) with the C23 axial proton. On the other hand, the C22 axial proton has a chemical shift at δ 2.87, and exhibits a strong coupling (J = 12.5 Hz) with the C23 proton. All the spectral data strongly support the assigned structure. It is believed that the A-strain from the C20 ketone controls the geometry of the newly created olefin. Finally, a Luche reduction18 from the less hindered face of the C20 ketone completed the synthesis of our target compound 11 in high yield and as a single stereoisomer.
Conclusions
The application of 2-substituted allylstannane 1 in the stereoselective allylation of aldehydes and in a synthesis of the C17–C27 segment of the bryostatins has been presented. The reagent was found to be compatible with the BITIP catalyst and was successfully used in the enantioselective synthesis of attractively functionalized building blocks, namely certain β-hydroxy allyl chlorides. These reactions were found to occur with very high enantiomeric excess and in excellent chemical yields.
The inherent utility of the incorporated allyl chloride substituent was demonstrated in a convergent and flexible construction of the C ring subunit of the bryostatins via a linchpin approach. Here successive chelation controlled reactions were utilized effectively. The research presented in this regard represents a preliminary effort in the bryostatin area.19 There is still plenty of room for improvement, which is evident by the fact that two of the preestablished stereocenters (C20 and C21) were destroyed during the cyclization step of this approach due to an unexpected elimination process that we were unable to circumvent. Consequently, based on the knowledge we gained from this study, new strategies for the synthesis of this segment of the bryostatins, with considerably better efficiency, have been developed and will be reported separately.
Experimental Section
Preparation of 4,4-Dibutyl-2-(chloromethyl)-4-stan-naoct-1-ene (1)
To a stirring solution of diisopropylamine (1.70 mL, 13.0 mmol) in 30 mL of THF at 0 °C was added n-BuLi (5.0 mL, 12 mmol, 2.5 M in hexanes). After 5 min, tributyltinhydride (3.14 g, 10.8 mmol) was added. The resulting solution was stirred at 0 °C for 15 min and then added via syringe pump over 1 h to a stirring solution of 3-chloro-2-(chloromethyl)prop-1-ene (1.50 g, 12.0 mmol) in 50 mL of hexanes at −78 °C. The mixture was stirred at −78 °C for 1 h then quenched by the addition of 5 mL of water. The mixture was diluted with 120 mL of 10% EtOAc/hexanes and washed with 100 mL of water. The organic phase was dried over anhydrous Na2SO4 and then concentrated by rotary evaporation. The residue was distilled under reduced pressure. The fraction at 100–120 °C/0.4 mmHg was collected to give 2.60 g (63%) of stannane 1 as a light yellow oil, which is sufficiently pure for most uses. A portion of the distilled material (260 mg) was further purified by flash chromatography on a silica gel column (1.5 × 28 cm), eluting with hexanes to provide 230 mg of analytically pure 1 as a colorless oil. Analytical data: Rf 0.63 (hexanes); 1H NMR (300 MHz, CDCl3)δ 4.84 (dd with Sn satellites, J = 2.4, 1.0 Hz, JH–Sn = 17.1 Hz, 1H), 4.71 (dd with Sn satellites, J = 2.4, 1.0 Hz, JH–Sn = 17.9 Hz, 1H), 3.95 (d with Sn satellites, J = 1.0 Hz, JH–Sn = 6.7 Hz, 2H), 1.99 (d with Sn satellites, J = 1.0 Hz, JH–Sn = 57.8 Hz, 2H), 1.54−1.43 (m, 6H), 1.36−1.24 (m, 6H), 1.00−0.77 (m, 9H); 13C NMR (75 MHz, CDCl3) δ 145.9, 110.1, 50.4, 29.3, 27.6, 16.1, 13.9, 9.9; IR (film) 2957, 2925, 1625, 1461 cm−1. Anal. Calcd for C16H33ClSn: C, 50.63; H, 8.76. Found: C, 50.74; H, 8.92.
Representative Example for BITIP Catalyzed Allylation Reactions. Preparation of (4S)-2-(Chloromethyl)-9,9-dimethyl-9-siladec-1-en-7-yn-4-ol (9e)
BITIP catalyst was prepared according to method A. To a stirring solution of (R)-(+)-1,1′-bi-2-naphthol (122 mg, 0.426 mmol) in methylene chloride (25 mL) was added 4 Å molecular sieves (1.3 g) and a 1.5 M solution of titanium isopropoxide in methylene chloride (284 μL, 0.426 mmol). The resulting mixture was heated at reflux for 1 h and then cooled to room temperature before aldehyde 8e (658 mg, 4.26 mmol) was added. The mixture was stirred for 5 min and then cooled to −78 °C and allylstannane 1 (2.87 mL, 3.24 g, 8.53 mmol) was added dropwise. The reaction was stirred an additional 15 min at −78 °C before transferring to a freezer at −20 °C where it was allowed to react for 5 days. The mixture was then filtered through a pad of Celite (to remove the molecular sieves) with the aid of ethyl acetate (50 mL), and the filtrate was washed with saturated aqueous sodium bicarbonate solution (2 × 50 mL). The organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. Purification of this material was accomplished by flash chromatography eluting with a gradient of 100 mL each of 5%, 15%, and 30% EtOAc/hexanes, collecting 8 mL fractions. The product-containing fractions were combined and concentrated under reduced pressure to give 9e (991 mg, 95% yield) as a clear colorless oil. Analytical data: Rf 0.71 (30% EtOAc/hexanes); [α]20D –7.6 (c 1.04, CHCl3); 500 MHz 1H NMR (CDCl3)δ 5.28 (s, 1H), 5.09 (app. d, J = 1.0 Hz, 1H), 4.10 (ABq, JAB = 11.7 Hz, Δv = 13.1 Hz, 2H), 4.01−3.96 (m, 1H), 2.45−2.35 (m, 3H), 2.29 (dd, J = 15.1, 8.8 Hz, 1H), 1.97 (d, J = 3.9 Hz, 1H), 1.76−1.63 (m, 2H), 0.15 (s, 9H); 75 MHz 13C NMR (CDCl3)δ 142.1, 117.5, 106.6, 85.6, 68.8, 48.2, 41.3, 35.5, 16.6, 0.06; IR (neat) 3394 (broad), 2957, 2174, 1433, 1250, 1069, 910, 843, 760 cm−1. The ee was determined to be >99% ee by HPLC analysis using a Chiralcel OD-H column, using a mobile phase of 1% i-PrOH/hexanes and a flow rate of 0.5 mL/min, which gave retention times for the major and minor enantiomers of 21.9 and 24.0 min, respectively. HRMS (CI at 120 eV) calcd for C12H22ClOSi (M + 1) 245.11284, found 245.11393.
Preparation of (4R)-2-Methylene-4-(2-phenylethyl)-3,4,5-trihydrofuran (10)
To a 10 mL round-bottom flask was added KH as a 30% w/w suspension in mineral oil (23.2 mg, 0.17 mmol). A nitrogen line was inserted and the flask subjected to continuous nitrogen purge. Freshly distilled hexanes was added with stirring. The heterogeneous solution was allowed to settle and the hexanes was removed by pipet, taking care not to remove any KH. This procedure was repeated two more times. Then the nitrogen line was removed and KI (20 mg, 0.12 mmol) was added and the flask sealed with a septum. The flask was charged with dry THF (0.5 mL). Compound 9d (26 mg, 0.12 mmol) was added as a solution in THF (250 μL) via cannula washing with THF (0.5 mL). The mixture was stirred at room temperature for 18 h before quenching by the addition of saturated aqueous sodium bicarbonate solution. The mixture was diluted with diethyl ether (20 mL) and washed with saturated aqueous sodium bicarbonate solution (10 mL) and water (10 mL). The combined aqueous wash was extracted with diethyl ether (10 mL), and the combined organic layers were dried over anhydrous magnesium sulfate, filtered, and concentrated under reduced pressure. Purification of the crude product was accomplished by flash chromatography eluting with 50 mL each of a gradient of 5% and 10% EtOAc/hexanes. The product-containing fractions were combined and concentrated to give 10 (17.6 mg, 81% yield) as a clear, colorless oil. Analytical data: Rf 0.53 (20% EtOAc/hexanes); [α]20D – 18.3 (c 0.63, CHCl3); 300 MHz 1H NMR (CDCl3)δ 7.12−7.16 (m, 5H), 4.97 (m, 1H), 4.91 (m, 1H), 4.32 (ABq, JAB = 13.3 Hz, Δv = 3.8 Hz, 2H), 3.94 (dddd, J = 8.1, 8.1, 5.8, 5.8 Hz, 1H), 2.83−2.60 (m, 3H), 2.27−2.17 (m, 1H), 2.02−1.67 (m, 2H); 75 MHz 13C NMR (CDCl3)δ 148.3, 141.9, 128.4, 128.3, 125.8, 104.1, 79.1, 70.7, 38.6, 36.7, 32.3; IR (neat) 2927, 2496, 1056, 883, 747, 700 cm−1. HRMS (CI at 120 eV) calcd for C13H17O (M + 1) 189.12794, found 189.12657.
Preparation of (2R,3R)-5-(Chloromethyl)-2-[(phenylmethoxy)methoxy]hex-5-en-3-ol (18)
To a solution of aldehyde 16 (1.37 g, 7.06 mmol) in 60 mL of CH2Cl2 was added MgBr2·OEt2 (2.55 g, 9.88 mmol). The resulting mixture was stirred at room temperature for 10 min before it was cooled to −15 °C. The stannane 1 (3.22 g, 8.47 mmol) was then added. The mixture was gradually warmed to room temperature over a period of 1 h and stirred at room temperature for an additional 20 min, then quenched by the addition of 20 mL of saturated aqueous NaHCO3 solution. The mixture was diluted with 60 mL of CH2Cl2 and then washed with water (100 mL) and with brine (100 mL). The organic phase was dried over anhydrous Na2SO4 and concentrated by rotary evaporation. The residue was purified by flash chromatography on a silica gel column (2.8 × 22 cm), eluting with 15% EtOAc/hexanes to give 1.80 g (90%) of 18 as a colorless oil. Analytical data: Rf 0.20 (20% EtOAc/hexanes); [α]20D –18.4 (c 2.60, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.39−7.26 (m, 5H), 5.30 (br s, 1H), 5.10 (dd, J = 2.2, 1.3 Hz, 1H), 4.88 (d, J = 7.0 Hz, 1H), 4.83 (d, J = 7.0 Hz, 1H), 4.68 (d, J = 11.8 Hz, 1H), 4.62 (d, J = 11.8 Hz, 1H), 4.15 (dd, J = 11.7, 1.0 Hz, 1H), 4.10 (dd, J = 11.7,1.0 Hz, 1H), 3.76−3.71 (m, 2H), 2.63 (d, J = 3.2 Hz, 1H), 2.53 (appt. d, J = 15.3 Hz, 1H), 2.30 (ddd, J = 14.6, 9.3, 0.8 Hz, 1H), 1.25 (d, J = 6.2 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 142.5, 137.6, 128.7, 128.1 (two carbons), 117.3, 94.0, 77.7, 73.2, 70.1, 48.6, 37.0, 16.9; IR (film) 3458, 3031, 2935, 1651, 1494 cm−1; HRMS calcd for C15H22ClO3 (M+) 285.1258, found 285.1260. Anal. Calcd for C15H21ClO3: C, 63.26; H, 7.43. Found: C, 62.73; H, 7.30.
Preparation of (1R)-1-{(1R)-1-[(Phenylmethoxy)methoxy]ethyl}-5,5-dibutyl-3-methylene-5-stannanonyl 2,2-Dimethylpropanoate (19)
To a solution of alcohol 18 (550 mg, 1.93 mmol) in 3 mL of CH2Cl2 was added PivCl (4.76 mL, 38.7 mmol) followed by pyridine (1.68 mL, 19.3 mmol) and DMAP (24.5 mg, 0.2 mmol). The resulting solution was stirred at room temperature for 24 h. It was then diluted with 100 mL of 10% EtOAc/hexanes and washed with 1 N aqueous HCl solution (50 mL), saturated aqueous NaHCO3 solution (50 mL), and brine (50 mL), then dried over anhydrous Na2SO4 and concentrated by rotary evaporation. The residue was purified by flash chromatography on a silica gel column (2.8 × 22 cm), eluting with 8% EtOAc/hexanes to give 575 mg (81%) of the product as a colorless oil. Analytical data: Rf 0.50 (20% EtOAc/hexanes); [α]20D –5.47 (c 1.96, CHCl3); 1H NMR (300 MHz, CDCl3) δ7.37−7.26 (m, 5H), 5.16 (br s, 1H), 5.12 (ddd, J = 10.4, 4.5, 3.0 Hz, 1H), 5.00 (m, 1H), 4.84 (d, J = 7.0 Hz, 1H), 4.81 (d, J = 7.1 Hz, 1H), 4.67 (d, J = 11.7 Hz, 1H), 4.62 (d, J = 11.8 Hz, 1H), 4.16 (dd, J = 11.8, 0.9 Hz, 1H), 4.02 (d, J = 11.8 Hz, 1H), 3.88 (qd, J = 6.5, 4.6 Hz, 1H), 2.65 (dd, J = 14.7, 2.0 Hz, 1H), 2.45 (dd, J = 14.9, 10.4 Hz, 1H), 1.22 (d, J = 6.5 Hz, 3H), 1.18 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 178.1, 141.5, 137.9, 128.7, 128.0, 127.9, 117.7, 94.0, 74.0, 72.4, 69.8, 48.0, 39.1, 33.7, 27.4, 16.2; IR (film) 2975, 1726, 1480 cm−1; Anal. Calcd for C20H29ClO4: C, 65.12; H, 7.92. Found: C, 65.30; H, 8.00.
To a stirring solution of diisopropylamine (273 mg, 2.70 mmol) in 12 mL of THF was added n-BuLi (0.99 mL, 2.48 mmol, 2.5 M in hexanes) at 0 °C. It was stirred at 0 °C for 5 min, then Bu3SnH (721 mg, 2.48 mmol) was added. The resulting solution was stirred at 0 °C for 15 min, then added via syringe pump over 30 min into a solution of the above product (830 mg, 2.25 mmol) in 12 mL of THF stirred at −78 °C. The mixture was stirred at −78 °C for an additional 30 min then quenched by adding 4 mL of water. The resulting mixture was diluted with 100 mL of hexanes and washed with water (50 mL). The organic phase was dried over anhydrous Na2SO4 and concentrated by rotary evaporation. The residue was purified by flash chromatography on a silica gel column (2.8 × 20 cm), eluting with 4% EtOAc/hexanes to give 1.10 g (78%) of 19 as a colorless oil. Analytical data: Rf 0.37 (5% EtOAc/hexanes); [α]20D –8.7 (c 2.00, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.37−7.26 (m, 5H), 5.14 (ddd, J = 7.6, 5.5, 4.5 Hz, 1H), 4.83 (d, J = 7.0 Hz, 1H), 4.80 (d, J = 7.0 Hz, 1H), 4.67 (d, J = 11.8 Hz, 1H), 4.61 (d, J = 11.7 Hz, 1H), 4.60−4.52 (m, 1H), 4.52−4.43 (m, 1H), 3.87 (qd, J = 6.4, 4.5 Hz, 1H), 2.31−2.19 (m, 2H), 1.85 (d, J = 11.8 Hz, 1H), 1.76 (d, J = 11.7 Hz, 1H), 1.54−1.42 (m, 6H), 1.36−1.22 (m, 6H), 1.20 (s, 9H), 1.19 (d, J = 6.5 Hz, 3H), 0.92−0.80 (m, 15H); 13C NMR (75 MHz, CDCl3) δ 178.0, 145.6, 138.0, 128.6, 128.1, 127.9, 108.0, 94.0, 73.9, 73.1, 69.7, 39.1, 38.4, 29.3, 27.6, 27.5, 18.9, 16.3, 13.9, 9.6; IR (film) 2957, 2927, 1729, 1629, 1459 cm−1. Anal. Calcd for C32H56O4Sn: C, 61.64; H, 9.05. Found: C, 61.61; H, 9.30.
Preparation of 1-{(1R)-1-[(Phenylmethoxy)methoxy]-ethyl}(1R,5R,6R)-5-hydroxy-6-[(4-methoxyphenyl)-methoxy]-3-methylene-7-(1,1,2,2-tetramethyl-1-silapropoxy)heptyl 2,2-Dimethylpropanoate (20)
To a stirring solution of aldehyde 17 (496 mg, 1.54 mmol) in 20 mL of CH2Cl2 at −15 °C was added MgBr2·OEt2 (795 mg, 3.08 mmol). The resulting mixture was stirred at −15 °C for 15 min. A solution of the stannane reagent 19 (960 mg, 1.54 mmol) in 5.0 mL of CH2Cl2 was then added. The mixture was gradually warmed to room temperature over a period of 1 h and then stirred at room temperature for 20 min. It was quenched by the addition of saturated aqueous sodium bicarbonate solution (10 mL). The resulting mixture was stirred at room temperature for 10 min, then diluted with 100 mL of CH2Cl2 and washed with water (60 mL), then with brine (60 mL). The organic phase was dried over anhydrous Na2SO4 and concentrated by rotary evaporation. The residue was purified by flash chromatography on a silica gel column (2.8 × 20 cm), eluting with 15% EtOAc/hexanes to give 948 mg (93%) of 20 as a colorless oil. Analytical data: Rf 0.28 (20% EtOAc/hexanes); [α]20D +0.76 (c 2.88, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.36−7.24 (m, 7H), 6.90−6.84 (m, 2H), 5.09 (ddd, J = 8.0, 5.3, 4.5 Hz, 1H), 4.89 (br s, 1H), 4.86 (br s, 1H), 4.81 (d, J = 7.0 Hz, 1H), 4.78 (d, J = 7.0 Hz, 1H), 4.68 (d, J = 11.1 Hz, 1H), 4.65 (d, J = 11.6 Hz, 1H), 4.59 (d, J = 11.6 Hz, 1H), 4.50 (d, J = 11.2 Hz, 1H), 3.90−3.70 (m, 3H), 3.79 (s, 3H), 3.75 (dd, J = 10.5, 6.5 Hz, 1H), 3.38 (ddd, J = 5.5, 5.5, 3.5 Hz, 1H), 2.43 (d, J = 6.0 Hz, 1H), 2.42−2.21 (m, 4H), 1.19 (s, 9H), 1.19 (d, J = 6.3 Hz, 3H), 0.90 (s, 9H), 0.07 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 178.1, 159.5, 142.2, 138.0, 130.7, 129.7, 128.6, 128.0, 127.9, 115.3, 114.0, 94.0, 80.9, 74.1, 72.9, 72.7, 69.7, 69.1, 63.0, 55.4, 39.9, 39.0, 36.4, 27.5, 26.1, 18.4, 16.3, −5.25, −5.27; IR (film) 3528, 2954, 2931, 1727, 1613, 1514, 1460 cm−. Anal. Calcd for C37H58O8Si: C, 67.44; H, 8.87. Found: C, 67.32; H, 8.94.
Preparation of 1-{(1R)-1-[(Phenylmethoxy)methoxy]-ethyl}(3S,1R,5R,6R)-3,5-dihydroxy-6-[(4-methoxyphenyl)-methoxy]-7-(1,1,2,2-tetramethyl-1-silapropoxy)heptyl 2,2-Dimethylpropanoate (23)
To a solution of ketone 22 (564 mg, 0.854 mmol) in 12.5 mL of THF and 2.5 mL of water was added diethylmethoxyborane (1.20 mL, 1.20 mmol, 1.0 M in THF). The resulting solution was stirred at room temperature for 30 min, then cooled to −78 °C and NaBH4 (67.8 mg, 1.79 mmol) was added. The reaction was stirred at −78 °C for 9 h, then quenched by the addition of 30% aqueous H2O2 solution (15 mL). The resulting mixture was allowed to stand in a −20 °C refrigerator for 10 h without stirring, then warmed to room temperature and stirred for 10 min. It was diluted with 100 mL of 80% EtOAc/hexanes and then washed with water (50 mL) and with brine (50 mL). The organic phase was dried over anhydrous Na2SO4 and concentrated by rotary evaporation. The residue was purified by flash chromatography on a silica gel column (2.8 × 20 cm), eluting with 35% EtOAc/hexanes to give 520 mg (92%) of diol 23 as a colorless oil. Analytical data: Rf 0.08 (20% EtOAc/hexanes); [α]20D +12.1 (c 2.55, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.37−7.23 (m, 7H), 6.90−6.84 (m, 2H), 5.11 (ddd, J = 8.6, 5.6, 4.4 Hz, 1H), 4.83 (d, J = 7.0 Hz, 1H), 4.79 (d, J = 7.2 Hz, 1H), 4.69 (d, J = 11.2 Hz, 1H), 4.65 (d, J = 11.8 Hz, 1H), 4.60 (d, J = 11.8 Hz, 1H), 4.50 (d, J = 11.4 Hz, 1H), 3.95−3.65 (m, 5H), 3.79 (s, 3H), 3.72 (dd, J = 10.7, 5.5 Hz, 1H), 3.38 (m, 2H), 1.80−1.62 (m, 3H), 1.57 (ddd, J = 14.2, 3.1, 2.9 Hz, 1H), 1.22 (s, 9H), 1.20 (d, J = 6.5 Hz, 3H), 0.90 (s, 9H), 0.07 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 179.4, 159.4, 137.9, 130.7, 129.7, 128.6, 128.0, 127.9, 113.9, 94.0, 81.3, 74.3, 72.8, 72.7, 72.1, 69.8, 68.0, 62.9, 55.4, 39.4, 39.2, 38.3, 27.4, 26.1, 18.4, 16.3, −5.3 (two carbons); IR (film) 3501, 2955, 2932, 1728, 1613, 1514, 1461 cm−1. Anal. Calcd for C36H58O9Si: C, 65.22; H, 8.82. Found: C, 65.30; H, 8.88.
Preparation of (1R,2R)-1-[(6-{(1S)-1-[(4-Methoxyphenyl)methoxy]-3,3-dimethyl-2-oxopent-4-enyl}(4S,6R)-2,2-dimethyl(1,3-dioxan-4-yl))methyl]-2-[(phenylmethoxy)-methoxy]propyl 2,2-Dimethylpropanoate (27)
To a solution of alcohol 25 (316 mg, 0.537 mmol) in 18 mL of CH2Cl2 were added sequentially oven-dried powdered 4 Å molecular sieves (840 mg), NMO (83.0 mg, 0.708 mmol), and TPAP (15.1 mg, 0.0429 mmol). After being stirred at room temperature for 20 min, the resulting dark green mixture was diluted with 30 mL of 30% EtOAc/hexanes and filtered through a pad of Celite (1 cm) and Florisil (4 cm). The pad was washed with 170 mL of 30% EtOAc/hexanes and the filtrate was concentrated by rotary evaporation. The crude aldehyde was used immediately in the subsequent reaction without further purification.
To a mixture of the crude aldehyde (315 mg, 0.537 mmol, theoretical) and prenyl bromide 26 (321 mg, 2.15 mmol) was added 3.5 mL of DMF, followed by indium powder (247 mg, 2.15 mmol). The reaction mixture was stirred at room temperature for 40 min, then diluted with 15 mL of EtOAc and filtered through Celite. The filtrate was further diluted with 50 mL of 30% EtOAc/hexanes, and then washed sequentially with 1 N aqueous HCl solution (50 mL), saturated aqueous NaHCO3 solution (50 mL), and brine (50 mL). The organic phase was dried over anhydrous Na2SO4 and concentrated by rotary evaporation. The resulting crude alcohol was then dissolved in 3 mL of DMSO. To this solution was added 2 mL of Ac2O with stirring. After stirring at room temperature for 10 min Et3N (2 mL) was added, and stirring was continued at room temperature for 17 h. The resulting dark brown solution was diluted with 10 mL of EtOAc and poured into a separatory funnel. It was further diluted with 100 mL of 30% EtOAc/hexanes and washed with water (2 × 60 mL), then with brine (60 mL). The organic phase was dried over anhydrous Na2SO4 and concentrated by rotary evaporation. The residue was purified by flash chromatography on a silica gel column (2.8 × 23 cm), eluting with 15% EtOAc/hexanes to give 225 mg (64% over three steps) of 27 as a colorless oil. Analytical data: Rf 0.56 (30% EtOAc/hexanes); [α]20D +15.4 (c 1.95, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.38−7.19 (m, 7H), 6.88−6.82 (m, 2H), 5.92 (dd, J = 17.3, 10.7 Hz, 1H), 5.24−5.16 (m, 1H), 5.17 (dd, J = 10.8, 0.9 Hz, 1H), 5.15 (dd, J = 17.4, 0.9 Hz, 1H), 4.80 (d, J = 7.0 Hz, 1H), 4.77 (d, J = 7.0 Hz, 1H), 4.62 (s, 2H), 4.52 (d, J = 11.0 Hz, 1H), 4.48 (d, J = 11.0 Hz, 1H), 4.32 (ddd, J = 9.4, 6.5, 4.5 Hz, 1H), 4.23 (d, J = 6.5 Hz, 1H), 3.83 (qd, J = 6.3, 4.1 Hz, 1H), 3.81−3.72 (m, 1H), 3.79 (s, 3H), 1.77−1.64 (m, 2H), 1.35 (s, 6H), 1.33−1.15 (m, 2H), 1.23 (s, 3H), 1.21 (s, 12H), 1.14 (d, J = 6.5 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 209.4, 177.8, 159.5, 141.9, 138.0, 130.3, 129.9, 128.6, 128.1, 127.9, 114.9, 113.9, 99.1, 94.0, 80.1, 74.0, 72.6, 71.7, 70.8, 69.7, 65.1, 55.5, 50.7, 39.1, 36.7, 32.8, 30.2, 27.5, 23.7, 23.4, 19.7, 16.0; IR (film) 2976, 2936, 1729, 1613, 1514, 1459 cm−1. Anal. Calcd for C38H54O9: C, 69.70; H, 8.31. Found: C, 69.48; H, 8.39.
Preparation of (1R,2R)-1-{[(6S)-6-(1,1-Dimethylprop-2-enyl)-6-methoxy-5-oxo(3,4,6-trihydro-2H-pyran-2-yl)]-methyl}-2-[(phenylmethoxy)methoxy]propyl 2,2-Dimethylpropanoate (28)
To a stirring solution of ketone 27 (114 mg, 0.174 mmol) in 1.5 mL of CH2Cl2 at −78 °C was added TMSOMe (0.150 mL, 1.09 mmol) and TMSOTf (6.0 μL, 0.033 mmol). The mixture was stirred at −78 °C for 4 h then warmed to −40 °C and stirred for 8 h. Finally, the solution was further warmed to 0 °C and stirred for an additional 1 h, then 2 mL of saturated aqueous NaHCO3 solution was added, and the resulting mixture was stirred at room temperature for 10 min. The mixture was diluted with 20 mL of CH2Cl2 and washed with saturated aqueous NaHCO3 solution (20 mL), then with brine (10 mL). The organic phase was dried over anhydrous Na2SO4 and concentrated by rotary evaporation. The residue was purified by flash chromatography on a silica gel column (1.3 × 25 cm), eluting with 10% EtOAc/hexanes to give 58 mg (68%) of 28 as a colorless oil. Analytical data: Rf 0.51 (20% EtOAc/hexanes); [α]20D +3.9 (c 2.30, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.38−7.26 (m, 5H), 6.16 (dd, J = 18.0, 10.4 Hz, 1H), 5.25 (ddd, J = 9.8, 4.2, 2.3 Hz, 1H), 5.00 (dd, J = 18.0, 1.4 Hz, 1H), 4.98 (dd, J = 10.6, 1.3 Hz, 1H), 4.82 (s, 2H), 4.65 (s, 2H), 3.95 (qd, J = 6.6, 4.1 Hz, 1H), 3.94−3.86 (m, 1H), 3.22 (s, 3H), 2.58−2.37 (m, 2H), 2.00 (ddd, J = 14.6, 9.4, 2.4 Hz, 1H), 1.96−1.87 (m, 2H), 1.81 (ddd, J = 14.6, 9.8, 3.2 Hz, 1H), 1.19 (s, 9H), 1.18 (d, J = 6.6 Hz, 3H), 1.14 (s, 3H), 1.08 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 206.8, 177.9, 144.3, 137.9, 128.6, 128.1, 127.9, 112.8, 103.8, 93.7, 73.0, 71.6, 69.8, 69.0, 51.9, 44.9, 39.0, 37.2, 35.7, 30.0, 27.4, 22.6, 22.4, 15.4; IR (film) 2975, 2935, 1729, 1477 cm−1; HRMS (CI) calcd for C27H39O6 (MH+ − MeOH) 459.2746, found 459.2731. Anal. Calcd for C28H42O7: C, 68.54; H, 8.63. Found: C, 68.29; H, 8.64.
Supplementary Material
Acknowledgment
Financial support from the National Institutes of Health (through GM-28961) is gratefully acknowledged.
Footnotes
Supporting Information Available: Additional synthetic procedures and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Yamamoto Y, Asao N. Chem. Rev. 1993;93:2207–2293. For reviews, see: [Google Scholar]; (b) Marshall JA. Chem. Rev. 1996;96:31–48. doi: 10.1021/cr950037f. [DOI] [PubMed] [Google Scholar]; (c) Denmark SE, Fu J. Chem. Rev. 2003;103:2763–2794. doi: 10.1021/cr020050h. [DOI] [PubMed] [Google Scholar]
- 2.(a) Keck GE, Boden EP. Tetrahedron Lett. 1984;25:265–268. [Google Scholar]; (b) Keck GE, Tarbet KH, Geraci LS. J. Am. Chem. Soc. 1993;115:8467–8468. [Google Scholar]
- 3.(a) Keck GE, Boden EP. Tetrahedron Lett. 1984;25:1879–1882. [Google Scholar]; (b) Keck GE, Abbott DE. Tetrahedron Lett. 1984;25:1883–1886. [Google Scholar]; (c) Keck GE, Abbott DE, Wiley MR. Tetrahedron Lett. 1987;28:139–142. [Google Scholar]; (d) Keck GE, Krishnamurthy D, Grier MC. J. Org. Chem. 1993;58:6543–6544. [Google Scholar]
- 4.(a) Keck GE, Yu T. Org. Lett. 1999;1:289–291. doi: 10.1021/ol990603j. [DOI] [PubMed] [Google Scholar]; (b) Keck GE, Wager CA, Wager TT, Savin KA, Covel JA, McLaws MD, Krishnamurthy D, Cee VJ. Angew. Chem., Int. Ed. 2001;40:231–234. [PubMed] [Google Scholar]; (c) Keck GE, Covel JA, Schiff T, Yu T. Org. Lett. 2002;4:1189–1192. doi: 10.1021/ol025645d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Pettit GR. J. Nat. Prod. 1996;59:812–821. doi: 10.1021/np9604386. [DOI] [PubMed] [Google Scholar]; (b) Pettit GR. In: Progress in the Chemistry of Organic Natural Products. 57. Herz W, editor. Springer-Verlag; New York: 1991. p. 153. [Google Scholar]; (c) Schuchter LM, Esa AH, May W, Laulis MK, Pettit GR, Hess AD. Cancer Res. 1991;51:682. [PubMed] [Google Scholar]; (d) Kraft AS. J. Natl. Cancer Inst. 1993;85:1790. doi: 10.1093/jnci/85.22.1790. [DOI] [PubMed] [Google Scholar]; (e) Stone RM. Leukemia Res. 1997;21:399. doi: 10.1016/s0145-2126(96)00123-3. [DOI] [PubMed] [Google Scholar]
- 6.Hale KJ, Hummersone MG, Manaviazar S, Frigerio M. Nat. Prod. Rep. 2002;19:413–453. doi: 10.1039/b009211h. For a review, see: [DOI] [PubMed] [Google Scholar]
- 7.Keck GE, Palani A. Tetrahedron Lett. 1993;34:3223–3224. [Google Scholar]
- 8.(a) Sano H, Okara M, Ueno Y. Synthesis. 1984:933–935. [Google Scholar]; (b) Degl'Innocenti A, Dembech P, Mordini A, Ricci A, Seconi G. Synthesis. 1991:267–269. [Google Scholar]
- 9.(a) Kang K-T, Hwang YB, Kim MY, Lee SK, Lee JG. Bull. Korean Chem. Soc. 2002;23:1333–1336. and ref 19 therein. [Google Scholar]; (b) Martín-Matute B, Buñuel E, Méndez M, Nieto-Oberhuber C, Cárdenas DJ, Echavarren AM. J. Organomet. Chem. 2003;687:410–419. [Google Scholar]
- 10.(a) Ochiai M, Fujita E, Arimoto M, Yamaguchi H. Chem. Pham. Bull. 1985;33:989–997. [Google Scholar]; (b) Trost BM, King SA, Schmidt T. J. Am. Chem. Soc. 1989;111:5902–5915. [Google Scholar]; (c) Kakinuma K, Hanson CA, Rinehart KL., Jr. Tetrahedron. 1976;32:217–222. [Google Scholar]; (d) Bartrolí J, Carceller E, Merlos M, García-Rafanell J, Forn J. J. Med. Chem. 1991;34:373–386. doi: 10.1021/jm00105a058. [DOI] [PubMed] [Google Scholar]; (e) Ishibashi Y, Ohba S, Nishiyama S, Yamamura S. Bull. Chem. Soc. Jpn. 1995;68:3643–3649. [Google Scholar]
- 11.(a) Kageyama M, Tamura T, Nantz MH, Roberts JC, Somfai P, Whritenour DC, Masamune S. J. Am. Chem. Soc. 1990;112:7407–7408. For other approaches to the C-ring of the bryostatins, see: [Google Scholar]; (b) DeBrabander J, Vandewalle M. Synthesis. 1994:855–865. [Google Scholar]; (c) Norcross RD, Paterson I. Chem. Rev. 1995;95:2041–2114. [Google Scholar]; (d) Hale KJ, Lennon JA, Manaviazar S, Javaid MH. Tetrahedron Lett. 1995;36:1359–1362. [Google Scholar]; (e) Gracia J, Thomas EJ. J. Chem. Soc., Perkin Trans. 1. 1998;17:2865–2871. [Google Scholar]; (f) Baxter J, Mate EG, Thomas EJ. Tetrahedron. 1998;54:14359. [Google Scholar]; (g) Wender PA, DeBrabander J, Harran PG, Jimenez J-M, Koehler MFT, Lippa B, Park CM, Shiozaki M, Pettit GR. J. Am. Chem. Soc. 1998;120:4534–4535. [Google Scholar]; (h) Obitsu T, Ohmori K, Ogawa Y, Hosomi H, Ohba S, Nishiyama S, Yamamura S. Tetrahedron Lett. 1998;39:7349–7352. [Google Scholar]; (i) Evans DA, Carter PH, Charette AB, Prunet JA, Lautens M. J. Am. Chem. Soc. 1999;121:7540–7552. [Google Scholar]; (j) Hale KJ, Frigerio M, Manaviazar S. Org. Lett. 2003;5:503–505. doi: 10.1021/ol027393m. [DOI] [PubMed] [Google Scholar]; (k) Wender PA, Koehler MFT, Sendzik M. Org. Lett. 2003;5:4549–4552. doi: 10.1021/ol0355332. [DOI] [PubMed] [Google Scholar]; (l) Voight EA, Roethle PA, Burke SD. J. Org. Chem. 2004;69:4534–4537. doi: 10.1021/jo0495081. [DOI] [PubMed] [Google Scholar]
- 12. For additional experimentation confirming the relative stereochemistry of the C21, C23, C25 triad, see the Supporting Information section of this paper.
- 13.Chen K-M, Hardtmann GE, Prasad K, Repic O, Shapiro MJ. Tetrahedron Lett. 1987;28:155–158. [Google Scholar]
- 14.Rychnovsky SD, Yang G, Powers JP. J. Org. Chem. 1993;58:3511–3515. [Google Scholar]
- 15.Araki S, Ito H, Butsugan Y. J. Org. Chem. 1988;53:1831–1833. [Google Scholar]
- 16.Albright JD, Goldman L. J. Am. Chem. Soc. 1967;89:2416–2423. [Google Scholar]
- 17.Tsunoda T, Suzuki M, Noyori R. Tetrahedron Lett. 1980;21:1357–1358. [Google Scholar]
- 18.Luche J-L. J. Am. Chem. Soc. 1978;100:2226–2227. [Google Scholar]
- 19. For the development of chemistry relevant to the asymmetric incorporation of the bryostatin B ring, see ref 3c.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.