

Abstract
The widespread use of antibiotics has encouraged the development of drug resistance in pathogenic bacteria. In order to overcome this problem, the modification of existing antibiotics and/or the identification of targets for the design of new antibiotics is currently being undertaken. Bifunctional penicillin-binding proteins (PBPs) are membrane-associated molecules whose transpeptidase (TP) activity is irreversibly inhibited by β-lactam antibiotics and whose glycosyltransferase (GT) activity represents a potential target in the antibacterial fight. In this work, we describe the expression and the biochemical characterization of the soluble extracellular region of Streptococcus pneumoniae PBP1b (PBP1b*). The acylation efficiency for benzylpenicillin and cefotaxime was characterized by stopped-flow fluorometry and a 40-kDa stable TP domain was generated after limited proteolysis. In order to analyze the GT activity of PBP1b*, we developed an electrophoretic assay which monitors the fluorescence signal from PBP1b*-bound dansylated lipid II. This binding was inhibited by the antibiotic moenomycin and was specific for the GT domain, since no signal was observed in the presence of the purified functional TP domain. Binding studies performed with truncated forms of PBP1b* demonstrated that the first conserved motif of the GT domain is not required for the recognition of lipid II, whereas the second motif is necessary for such interaction.
Both gram-positive and gram-negative bacteria are surrounded by a cell wall, composed mainly of peptidoglycan, a strong net-like polymer responsible for maintaining the shape and size of the bacterial cell and for resisting the high intracellular osmotic pressure. Peptidoglycan consists of repeating β-1,4-linked N-acetylglucosaminyl-N-acetylmuramyl units cross-linked through short peptide chains.
The final steps of peptidoglycan biosynthesis occur extracellularly, are associated with the plasmic membrane, and are catalyzed by penicillin-binding proteins (PBPs) (10). Class A PBPs bear two enzymatic functions, namely glycosyltransfer (GT) and transpeptidation (TP) (Fig. 1). The former is responsible for the elongation of the glycan strands using the disaccharide N-acetylglucosaminyl-N-acetylmuramyl pentapeptide as a substrate anchored to the membrane by a 55-carbon undecaprenyl chain (lipid II) while the latter cross-links the peptide chains attached to the glycan strands. Such bifunctional PBPs have been identified in both gram-positive and gram-negative bacteria. The N-terminal domains of these enzymes contain the GT activity while the C-terminal domains possess the TP activity. In addition to bifunctional PBPs, all bacterial species contain class B PBPs which harbor only the TP activity, together with N- and C-terminal domains to which no function has yet been associated. Another enzymatic activity peculiar to some PBPs is d,d-carboxypeptidation (9). All class A and B PBPs are inserted into the plasma membrane through a single helix, which is preceded by a short N-terminal region located in the cytoplasm; both GT and TP activities are localized in the extracellular space (4).
FIG. 1.

Schematic diagrams of S. pneumoniae PBP1b. (A) Topology of the native PBP1b protein. The solid and hatched boxes indicate the N-terminal cytoplasmic region and the membrane anchor, respectively. The GT and TP domains are represented together with their conserved motifs; active serine 460 is indicated by an asterisk. x represents any amino acid. The arrows delineate the proteolytic PBP1b TP domain: the N- and C-terminal residues experimentally identified are indicated in italics. The grey box represents the C-terminal domain, whose predicted N- and C-terminal residues are Ile 711 and Pro 791, respectively. The dotted box indicates the polyserine tail. (B) Schematic diagrams of the PBP1b-derived constructs. A stop codon had been inserted after the P791 in order to delete the Ser-rich extension. The peptide at the GST-GT junction of the PBP1b* construct includes sequences specific for the thrombin and Tev proteases (italic type) and the N-terminal amino acids of GT (boldface type). The His6 tag is placed on the N terminus of the shorter forms, PBP1b*ATL and PBP1b*LIK.
The biosynthesis of peptidoglycan is essential for bacterial survival; disruption of peptidoglycan may lead to cell lysis, suggesting why the most-effective antibiotics currently in use are inhibitors of peptidoglycan biosynthesis (2). Indeed, penicillin and other β-lactam antibiotics are specific inhibitors of TP, d,d-carboxypeptidases, and d,d-endopeptidases due to their structural similarity to the natural substrates, the stem peptides (33). β-Lactams form a covalent complex with the active serine of the TP domain, preventing the cross-linking of stem peptides and weakening the peptidoglycan structure. Furthermore, the glycopeptide antibiotic vancomycin binds to the d-alanyl-d-alanine moiety of stem peptides, impeding their subsequent recognition by TPs and carboxypetidases; the GT reaction may also be inhibited by glycopeptides through steric hindrance (2, 8). Extensive use of these cell wall biosynthesis inhibitors has promoted the development of bacterial strains highly resistant to such drugs, hindering antibiotic-based treatments.
Streptococcus pneumoniae is responsible for a high proportion of cases of pneumonia, acute otitis media, acute sinusitis, bacteremia, and meningitis which lead to more than 1 million deaths per year, mostly of young children in developing countries. This gram-positive pathogen possesses six PBPs, including three class A (PBP1a, 1b, and 2a) and two class B (PBP2x and 2b) molecules as well as a d,d-carboxypeptidase, PBP 3 (14). Individual deletion of the pbp2x or pbp2b genes is lethal for S. pneumoniae (18). However, neither the pbp1a, pbp1b, nor pbp2a gene is required for growth when deleted individually, but the presence of at least pbp1a or pbp2a is essential for cell viability (16, 27).
β-Lactam-resistant S. pneumoniae strains contain the same PBP pattern as the R6 strain (the penicillin-sensitive reference strain), albeit with decreased antibiotic affinity. Low-affinity PBP variant genes conferring resistance are mostly acquired by homologous recombination and have a mosaic structure, in which parts of genes are replaced by equivalent parts from drug-resistant bacteria (7, 19, 22). PBP2b and PBP2x constitute the most-common resistance determinants, and they confer low-level resistance to piperacillin and cefotaxime, respectively (13). Low-affinity variants of PBP2a confer a higher level of β-lactam resistance (15), whereas modification of PBP1a is required for high-level β-lactam resistance (1, 15, 25, 29).
The efficiency of β-lactam antibiotics in combating bacterial infections stresses the value of PBPs as targets in antibiotherapy. Targeting the GT activity is an attractive alternative to TP classical inhibition. However, the only GT activity inhibitor known, moenomycin (flavomycin), cannot be used in human therapy due to its poor absorption and long half-life (11). The major difficulty in studying class A PBPs has been the limited availability of lipid II, the GT reaction substrate. Terrak et al. and van Heijenoort succeeded in purifying radiolabeled lipid II, allowing significant progress in the functional characterization of the GT activity of Escherichia coli PBPs (32, 34). Nevertheless, the purification of significant amounts of lipid II still remains the bottleneck for class A PBP studies. Very recently, two groups have succeeded in the total synthesis of lipid II (31, 35), which provides a priceless biochemical tool which should allow rapid progress in the enzymatic characterization of PBPs (30).
In this work, we describe the biochemical characterization of PBP1b*, one of three pneumococcal PBPs with bifunctional activity. While the TP domain was characterized by stopped-flow fluorometry, limited proteolysis, and [3H]benzylpenicillin binding, we developed a novel fluorescence-based assay for the GT activity which exploits the recognition of dansylated lipid II by PBP1b* in native gel conditions. N-terminally truncated forms of PBP1b* were also investigated with this assay, and the binding site for lipid II was then mapped. Finally, moenomycin binding to the GT domain of PBP1b* was also demonstrated.
MATERIALS AND METHODS
Materials.
Trypsin, thrombin, glutathione, penicillin G, cefotaxime, and phenylmethylsulfonyl fluoride were purchased from Sigma (Saint Quentin Fallavier, France). IPTG (isopropyl-β-d-thiogalactopyranoside), ampicillin, and 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) were purchased from Euromedex (Mundolsheim, France). Complete protease inhibitor cocktail was obtained from Roche (Meylan, France). Dansylated lipid II was generously provided by Eli Lilly (Indianapolis, Ind.). Moenomycin was a gift from Aventis (Frankfurt/Main, Germany).
Construction of vectors encoding the different PBP1b* forms.
The uncapsulated R6 strain of S. pneumoniae was anaerobically propagated in glucose-buffered broth (Diagnostics Pasteur) for 16 h at 30°C. Genomic DNA was extracted from 5 ml of culture with the High Pure PCR template preparation kit (Roche) by following the manufacturer's instructions; 100 ng of genomic DNA was recovered in 200 μl of water. Restriction and modification DNA enzymes were from Promega (Charbonnières, France). DNA sequencing was performed by Genome Express (Grenoble, France).
The same protocol was used for all amplification and cloning procedures. The primers used are listed in Table 1. Genomic DNA from the S. pneumoniae R6 strain was used as a template (2 ng) to amplify the extracellular region of the pbp1b gene. The PCR product was cloned into the pCR-Script vector (Stratagene) following the manufacturer's instructions. After transformation into E. coli XL1-Blue supercompetent cells, clones containing the pbp1b gene were selected as white colonies on IPTG-X-Gal-ampicillin-containing agar plates. Gene pbp1b was subcloned into a pGEX-4T1 vector (Amersham Biosciences); this construct, pDG1b0, encodes the complete periplasmic form of PBP1b, amino acids G74 to R821, fused to the glutathione S-transferase (GST) protein.
TABLE 1.
Primers used for S. pneumoniae PBP1b* constructs
| Plasmid | Function or description | Primer and sequence (5′-3′) |
|---|---|---|
| pDG1b0 | PBP1b* total periplasmic; (G74-R821), GST fusion | Upstream, CGGGATCCGGGATTGCTTTGGGATACGGAGTGGa Downstream, GGCTCGAGTTATCGTCTCGCCCTTGAAGAAGAAb,c |
| pDG1b1 | Insertion of stop codon after P791; PBP1b* ΔC-ter (G74-P791), GST fusion | 1bstop A, GTATTGTGGGGAGTCTACCATAACCATGGAGCTCCAGCTCCAGCc,d1bstop B, GCTGGAGCTGGAGCTCCATGGTTATGGTAGACTCCCCACAATACc,d |
| pDG1b2 | Mutagenesis of K158N and R162P; PBP1b*NPΔC-ter (G74-P791), GST fusion | 1bNP5, GGTGTAGTACCCAACGCGGTGATCCCGGCGACCTTGGGGAAATTTGe,f1bNP3, CAAATTTCCCCAAGGTCGCCGGGATCACCGCGTTGGGTACTACACCe,f |
| pDG1b3 | Insertion of Tev protease site; PBP1b*NPΔC-ter (G74-P791), GST fusion, Tev protease site | Upstream, CCGCGTGGATCCGAGAATCTTTATTTTCAGGGCGGGTCAGGGATTGCTTTGGGATACGa,g Downstream, CCGCTCGAGTTATGGTAGACTCCCCACAATACTb |
| pDG1b4 | PBP1b*ATL (A163-P791), N-terminal His6 tag | Upstream, GGCATATGGACCATCATCATCATCATGCGACCTTGGGGAAATTTGTAGGTh,i Downstream, GGCTCGAGTTATGGTAGACTCCCCACAATACTb,c |
| pDG1b5 | PBP1b*LIK (L184-P791), N-terminal His6 tag | Upstream, GGCATATGCACCATCATCATCATCATCTAATTAAACAGCAGGTGGTTGGGh,i Downstream, GCCTCGAGTTATGGTAGACTCCCCACAATACTb,c |
Boldface type indicates the BamHI site.
Boldface type indicates the XhoI site.
The stop codon is underlined.
Boldface type indicates the NcoI site.
Boldface type indicates the NciI site.
The Asn and Pro codons are underlined.
The Tev protease site is underlined.
Boldface type indicates the NdeI site.
The His6 tag is underlined.
Site-directed mutagenesis was performed with the QuikChange site-directed mutagenesis kit (Stratagene) by following the manufacturer's instructions in order to insert a stop codon after P791 and delete the serine-rich C-terminal tail (Table 1 lists the sequences of the complementary mutagenic primers). After transformation into E. coli XL1-Blue supercompetent cells, mutant clones were verified by diagnostic restriction digestion and gave rise to the pDG1b1 construct.
To eliminate two secondary thrombin cleavage sites, two point mutations were introduced in the GT domain. Using the pDG1b1 construct as a template, K158 and R162 were mutated into Asp and Pro, respectively, by the same method as described above (Table 1). The resulting construct, pDG1b2, encodes the fusion protein GST-PBP1b*NPΔCter. Lastly, to avoid further problems with thrombin cleavages, a fragment coding for a Tev protease site was introduced between the GST and PBP1b* coding regions (pDG1b3, which was derived from pDG1b2). The protein encoded by this construct was used in all experiments and, without further specifications, will be referred to as PBP1b*.
N-terminally truncated forms of PBP1b* (PBP1b*ATL, residues A163 to P791, and PBP1b*LIK, residues L184 to P791) were expressed as N-terminal His6-tagged fusions (pDG1b4 and pDG1b5 constructs). The primers listed in Table 1 were used to amplify the genes, which were further cloned in the pET30b expression vector.
Protein purification procedures. (i) GST-fused PBP1b*.
Overnight cultures of E. coli MC1061 transformed with the pDG1b3 plasmid were used to inoculate (1:50) 1 liter of Luria-Bertani medium supplemented with 100 μg of ampicillin/ml. Upon achieving an optical density at 600 nm of 1 at 37°C, protein expression was induced with 1 mM IPTG while incubating for 16 h at 15°C. Purification steps were all performed either at 4°C or on ice. After cells were centrifuged at 6,000 × g for 15 min, the pellet was resuspended in 80 ml of a solution of 50 mM Tris-HCl (pH 8.0), 200 mM NaCl, 1 mM EDTA (buffer A), containing 1 tablet of Complete protease inhibitor cocktail (Roche). The cells were then disrupted by sonication, the cell lysate was centrifuged at 31,000 × g for 20 min, and the resulting supernatant was loaded onto a 5-ml glutathione-Sepharose column (Amersham Biosciences) previously equilibrated in buffer A. GST-PBP1b* was eluted with 10 mM reduced glutathione in buffer A. Extensive dialysis against buffer A was performed to eliminate reduced glutathione from the protein solution. GST-PBP1b* was incubated with 15 U of Tev protease per mg of fusion protein in an overnight incubation at room temperature, and the cleaved product was reloaded onto the glutathione-Sepharose column. The resulting flowthrough, containing the isolated PBP1b*, was dialyzed against 25 mM Tris-HCl (pH 8.8) and 50 mM NaCl. Anion-exchange chromatography (Resource Q; Amersham Biosciences) was performed at a flow rate of 2 ml/min with an NaCl gradient (0 to 250 mM) to elute PBP1b*.
(ii) His-tagged PBP1b*ATL and PBP1b*LIK.
Overnight cultures of E. coli BL21(DE3) transformed with pDG1b4 and pDG1b5 plasmids were used to inoculate (1:50) 1 liter of Luria-Bertani medium supplemented with 30 μg of kanamycin/ml. Cell lysis and sonication steps were performed as previously described but in the absence of 1 mM EDTA. The supernatant was loaded at a flow rate of 1 ml/min onto a 7-ml chelating-Sepharose column (Amersham Biosciences) previously loaded with 100 mM NiSO4 and equilibrated in 50 mM Tris-HCl (pH 8.0) and 200 mM NaCl (buffer B). Two extensive wash steps (10 column volumes) with buffer B containing 20 and 50 mM imidazole, respectively, preceded the elution of the fusion protein with 100 mM imidazole in buffer B.
Generation of the TP domain of PBP1b*.
Purified PBP1b* in 50 mM Tris-HCl (pH 8.0), 200 mM NaCl, and 1 mM EDTA was incubated with trypsin at a protease/PBP1b* ratio of 1:100 (wt/wt) for different periods of time at room temperature. Following incubation, the proteolyzed samples were subjected to sodium dodecyl sulfate (SDS)-12.5% polyacrylamide gel electrophoresis (PAGE). On a preparative scale, the TP domain was produced by using a trypsin/PBP1b* ratio of 1:60 (wt/wt) for 1 h at room temperature and then the protease activity was inhibited by 1 mM phenylmethylsulfonyl fluoride. The cleaved sample was dialyzed against 25 mM Tris-HCl (pH 8.8) and 1 mM EDTA and loaded onto a MonoQ column previously equilibrated with the same buffer. The purified TP domain was eluted with a 0 to 500 mM NaCl gradient.
Determination of kinetic parameters.
PBPs interact with β-lactam antibiotics according to the three-step scheme:
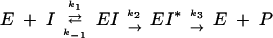 |
where E is the PBP enzyme, I is the β-lactam antibiotic, EI is the Michaelis-Menten complex, EI* is the acyl-enzyme covalent complex, and P is the product of the reaction (degraded β-lactam antibiotic). K, the dissociation constant of the enzyme-substrate complex, is equal to k−1/k1.
The k2/K parameter, accounting for the acylation efficiency, was determined by following the decrease of the intrinsic fluorescence of the protein in the presence of antibiotic at 37°C using a Biologic SFM3 stopped-flow apparatus. The excitation wavelength was 280 nm, and the emission was measured in the 305 to 360 nm range. PBP1b* at a concentration of 0.6 μM in 50 mM Tris-HCl (pH 8.0) and 200 mM NaCl was incubated with β-lactam antibiotics at concentrations ranging from 20 to 100 μM.
The covalent binding of β-lactam to the PBP1b* TP domain was assayed by using [3H]benzylpenicillin (20 Ci/mmol, 1 mCi/ml; Amersham Biosciences). Purified TP (123 μM) in 25 mM Tris-HCl (pH 8.8), 1 mM EDTA, 200 mM NaCl was incubated at 37°C with [3H]benzylpenicillin (5 μM) for 15 min, at which time the reaction was completed. The samples were then subjected to SDS-PAGE. [3H]benzylpenicillin bound to PBP1b* was monitored by using two different procedures. The gel was stained with Coomassie blue, destained, incubated with Amplify (Amersham), dried, and either exposed to a film for 16 h or cut around the protein bands. In the latter case, the radioactivity of the gel slice was measured by using a liquid scintillation analyzer (Packard model 2100TR) after mixing with 10 ml of liquid scintillation counting cocktail (Picofluor 15; Packard).
The deacylation reaction obeys the following equation: −k3t = {[EI*]t/[EI*]0}, where [EI*]0 is the initial concentration of acyl-enzyme and [EI*]t is its concentration time at t. Determination of k3 was performed as follows: 5 μM purified PBP1b* was labeled with 5 μM [3H]benzylpenicillin for 15 min at 37°C. Cold benzylpenicillin (15 mM) was added, and the incubation was continued at 37°C. Samples were removed at various times, loaded onto an SDS-containing gel, and after denaturing electrophoresis, the amount of radioactivity was measured in the protein bands as described above.
Lipid II binding assay.
Dansylated lipid II was incubated with the different forms of PBP1b* for 15 min at room temperature (see Results for the quantities of respective components) before analysis by native PAGE. After migration, the fluorescence emitted by the dansyl group of lipid II was detected by illuminating the gel with UV (Bio-Rad UV transilluminator): both free and bound lipid II forms could be detected. The gel was then stained with Coomassie blue to visualize the protein pattern.
Moenomycin protection assay.
PBP1b* (18 μM) was incubated with 0.5 mM moenomycin for 1 h at room temperature. Chymotrypsin was added at protease/PBP1b* ratios of 1:12,000 and 1:6,000 (wt/wt) for 1 h at room temperature. Following incubation, the proteolyzed samples were subjected to SDS-12.5% PAGE. A 16-kDa protein band specifically obtained in the presence of moenomycin was submitted to N-terminal sequencing.
RESULTS
PBP1b sequence and topology.
PBP1b from S. pneumoniae contains a cluster of 28 hydrophobic residues (I46 to A73), which probably functions as a membrane anchor (Fig. 1A). This sequence immediately follows a short region (M1 to V45) likely localized in the cytoplasm, with no sequence identity within other class A PBPs. The extracellular region (G74 to R821), most of which is expressed by the pDG1b3 vector described in this work (G74 to P791), harbors 5 conserved GT motifs (E145DxxFxxHxG, G176xSTxTQQ, R198KxxE, K215xxILxxYxN, and R294RxxVL) as well as the three classical TP motifs (S460xxK, S516xN, and K651TG) (15). The GT domain displays no similarity to other known GTs; indeed, bifunctional PBPs have been classified as GT family 51 (http://afmb.cnrs-mrs.fr/CAZY). The TP domain displays some similarity, albeit low (approximately 15%), to class A β-lactamases (17). Interestingly, PBP1b contains an 11-residue polyserine tail which may play a role either in contacting other proteins involved in peptidoglycan metabolism or in protein turnover (Fig. 1A and 2) (10, 15).
FIG. 2.

Alignment sequence of class A high-Mr PBPs S. pneumoniae (Spn) PBP1b, PBP2a, and PBP1a and E. coli (Eco) PBP1b (beginning at residue 125). The conserved motifs (numbered 1 to 5) in the GT domain are underlined and in boldface. The active-site conserved motifs in the TP domain are underlined. The 4 N-terminal sequences of the tryptic products of S. pneumoniae PBPs generating TP domains are underlined and in italics: PBP1a (S264), PBP2a (D264 and I301), and PBP1b (Q323 and D337). The C-terminal residues of these products are in boldface (5, 6). The diamond indicates the putative C terminus of TP domains based on S. pneumoniae PBP2x structure (28). For E. coli PBP1b, the italic residues correspond to the permissive site delineating the GT and TP domains (20).
Expression and purification of PBP1b*.
The extracellular form of PBP1b purified in this work harbors several changes compared to the wild-type R6 sequence (Fig. 1B). With the goal of generating a protein which is compact and suitable for crystallographic studies, a stop codon was inserted after P791 in order to delete the serine-rich C-terminal tail. In addition, early attempts to cleave the GST tag from GST-PBP1b* with thrombin led to secondary cleavage sites within the GT domain (K158 and R162). To suppress these internal digestion sites, a first approach included the mutation of these residues into asparagine and proline, respectively. Furthermore, a second construct was made in which the thrombin cleavage site was followed by a Tev site. Cleavage with Tev protease released the full-size PBP1b* protein which was used in this study.
GST-PBP1b* is expressed in E. coli in high amounts as a highly soluble cytoplasmic protein. PBP1b*, which is recovered after the second glutathione-Sepharose step, migrates in SDS-PAGE with an apparent molecular mass higher than 67 kDa, in agreement with the calculated mass for PBP1b* (78.5 kDa) (Fig. 3, lane 1). After affinity, ion-exchange, and gel filtration chromatographies, the yield was approximately 10 to 15 mg of PBP1b* per liter of E. coli culture. This behavior contrasts with those of other class A PBP extracellular regions, which were insoluble in the absence of detergents (4, 6, 21, 36).
FIG. 3.

Trypsin cleavage of PBP1b* performed at room temperature with a trypsin/PBP1b* ratio of 1:100 (wt/wt). Proteins were separated by SDS-12.5% PAGE and stained with Coomassie blue. Numbers at the left indicate sizes of standard molecular mass markers. Lanes: 1, undigested PBP1b*; 2, molecular mass markers; 3, 20-min digestion; 4, 45-min digestion; 5, 90-min digestion; 6, fluorogram of a dried gel from SDS-12.5% PAGE of TP protein (5 μg, 123 μM) labeled with [3H]benzylpenicillin (5 μM) prior to the migration. QDFL/DYLY and R/AGY are the N-terminal sequences of the 40- and 13-kDa species, respectively.
Determination of TP domain kinetic parameters.
The kinetic properties of the TP domain of PBP1b* were determined for a cephalosporin and a penicillin. Second-order rate constants of acylation were measured by stopped-flow fluorescence, and k2/K values for cefotaxime and benzylpenicillin are reported in Table 2. The k3 deacylation rate of PBP1b* for benzylpenicillin given in Table 2 is in the same range as the values reported for other pneumococcal PBPs (5, 6, 24).
TABLE 2.
Kinetic parameters of S. pneumoniae PBPs
| Protein |
k2/K (M−1 s−1) for:
|
Benzylpenicillin
|
Reference | ||
|---|---|---|---|---|---|
| Cefotaxime | Benzylpenicillin | k3 (s−1) | Half-life (h) | ||
| PBP1b* | 22,181a | 19,970b | 5.62 × 10−5c | 3.44d | This work |
| PBP2a* | 7,700 | <3,800 | 3.2 × 10−5 | 6 | 6 |
| PBP1a* | 11,933 | 34,200 | 1.0 × 10−5 | 14 | 5 |
| S-PBP2x* | 209,000 | 99,000 | 3.5 × 10−5 | 5.8 | 24 |
| R-PBP2x* | 3,300 | <3,800 | 4.2 × 10−5 | 4.6 | 24 |
Standard deviation, 1,850 M−1s−1.
Standard deviation, 461 M−1s−1.
Standard deviation, 0.85 × 10−5s−1.
Standard deviation, 0.5 h.
Limited trypsin proteolysis of PBP1b* releases the TP domain.
Proteolysis of purified PBP1b* with a trypsin/protein ratio of 1:100 (wt/wt) results in the production of two major protein fragments of approximately 40 and 13 kDa as well as a minor product of around 32 to 35 kDa (Fig. 3, lanes 3 to 5). Only the two higher-molecular-mass fragments are competent for [3H]benzypenicillin binding (Fig. 3, lane 6) showing that they encompass the TP domain. N-terminal sequencing revealed that the 40-kDa band was composed of two species, one starting at position QDFLP and the other one at DYLYF, corresponding to trypsin digestion after K322 and R340, respectively (Fig. 2 and 3). The size of these protein fragments implies that trypsin cleavage also occured in the C-terminal region of PBP1b*. Moreover, the 13-kDa fragment has the N-terminal sequence AGYSN, corresponding to digestion after R686. This digestion product encompasses the C terminus of PBP1b*, leading to a fragment unable to bind [3H]benzypenicillin, as demonstrated in Fig. 3, lane 6. The N- and C-terminal extremities of the 40-kDa product have been confirmed by mass spectrometry, taking into account the site specificity of trypsin. The measured mass of 40,391 Da corresponds to fragment Q323 to R686 (theoretical mass of 40,386 Da). In conclusion, the 40-kDa tryptic product corresponds to the TP domain of PBP1b*, although it is 24 residues shorter than the predicted TP domain from the PBP2x structure (Fig. 2) (28).
Lipid II binding on the GT domain of PBP1b*.
Lipid II is the natural substrate for the GT activity of class A PBPs (30, 31, 35). The lipid II molecule used in this work has a dansyl group linked to the amine moiety of the lysine side chain. In order to assay the recognition of lipid II by the GT domain of PBP1b, protein and substrate were allowed to interact for 15 min at room temperature before analysis by native PAGE (Fig. 4). The dansylated lipid II was then revealed by illuminating gels with UV soon after the completion of the electrophoresis (Fig. 4A, lanes 1 to 3, and B). Subsequently, gels were stained with Coomassie blue in order to visualize the protein (lipid II is not stained by Coomassie blue) (Fig. 4A, lanes 4 and 6, and C). Lipid II migrates with the dye front as indicated by the position of the fluorescent band in lane 2 of Fig. 4A. PBP1b* alone doesn't emit fluorescence (Fig. 4A, lane 1), but when mixed with a sevenfold molar excess of lipid II, it causes the shift of the lipid II fluorescence band to the top of the gel, suggesting that lipid II may be bound to PBP1b* (Fig. 4A, lane 3). This is confirmed by the observation of the same gel stained with Coomassie blue. In the presence of lipid II, a proportion of PBP1b* molecules is shifted towards the position on the gel which corresponds to that of the fluorescent shifted lipid II (Fig. 4A, lanes 3 and 6). The shift of lipid II in the presence of PBP1b* is consequently the result of an interaction between these two components. It is important to note that all of lipid II was bound to PBP1b* (as seen by a total shift of the fluorescence) but that not all of the PBP1b* molecules bound lipid II, despite a sevenfold molar excess of the latter. This observation suggests that molecules of lipid II may form micelles or vesicles and that PBP1b* binds to its substrate, but not in an equimolar way, thus impairing further calculations of affinity constant values.
FIG. 4.

Lipid II binding to PBP1b* and TP1b. The binding was performed at room temperature for 15 min before loading onto native PAGE. (A) Lanes: 1 and 4, PBP1b* (38 μM); 2 and 5, lipid II (250 μM); 3 and 6, PBP1b* (38 μM) and lipid II (250 μM). The same gel was observed under UV (lanes 1 to 3) before staining with Coomassie blue (lanes 4 to 6). The arrow indicates the position of the complex PBP1b*-lipid II. (B) Lanes: 1, PBP1b* (38 μM); 2, PBP1b* (38 μM) and lipid II (250 μM); 3, lipid II (250 μM); 4, PBP1b* (38 μM), lipid II (250 μM), and moenomycin (4 mM); 5, PBP1b* (38 μM), lipid II (250 μM), and moenomycin (7.3 mM). The gel was observed under UV. (C) Lanes: 1, TP1b (0.19 mM); 2, TP1b (0.19 mM) and lipid II (0.52 mM). The gel was stained with Coomassie blue.
Moenomycin is an inhibitor of GT activity due to its structural similarity to the substrate lipid II. In order to map the relative binding site of moenomycin and lipid II within the GT domain of PBP1b*, a competition between the two molecules was set up by using the assay described above (Fig. 4B). As previously observed, in the absence of moenomycin, all of the lipid II binds to PBP1b* and comigrates with the protein on top of the gel (Fig. 4B, lane 2); in the presence of an excess of moenomycin, all of the fluorescent signal migrates with the dye front (Fig. 4B, lanes 4 and 5). This result suggests that moenomycin and lipid II compete for the same site on the GT domain of PBP1b*.
In order to verify the specificity of lipid II binding to the GT domain, the ligand binding test was performed on the TP domain isolated from PBP1b*. On a Coomassie blue-stained native gel, no TP protein shift is observed in the presence of lipid II (Fig. 4C), and no modification was observed in the migration of fluorescent lipid II (data not shown), indicating that no binding occured between the lipid II molecule and the TP domain.
We were further interested in the more-precise characterization of the lipid II binding site within the GT domain, in particular the identification of the conserved motifs involved in binding of the substrate. Two shorter forms of PBP1b were constructed: PBP1b*ATL and PBP1b*LIK, which are deleted from the first GT motif (E145DxxFxxHxG) and from the first and second motifs (G176xSTxTQQ), respectively (Fig. 1B). The lipid II binding test for these two variants was performed as described above (Fig. 5). A complete migration shift of the dansylated lipid II was observed for PBP1b* and PBP1b*ATL, while only a small proportion of lipid II was associated with PBP1b*LIK. In conclusion, lipid II binds to PBP1b*ATL despite the deletion of the first GT motif (E145DxxFxxHxG), but deletion of the first two GT motifs greatly interferes with binding. This observation indicates that the second motif (G176xSTxTQQ) is required for lipid II recognition, whereas the first motif (E145DxxFxxHxG) may play a secondary role in substrate binding or protein stabilization.
FIG. 5.

Lipid II binding to PBP1b variants. The binding was performed at room temperature for 15 min before loading onto native PAGE and observation under UV. Lanes: 1, PBP1b* (18 μM); 2, PBP1b* (18 μM) and lipid II (3.7 μM); 3, lipid II (37 μM); 4, PBP1b*ATL (18 μM); 5, PBP1b*ATL (18 μM) and lipid II (3.7 μM); 6, PBP1b*LIK (18 μM); 7, PBP1b*LIK (18 μM) and lipid II (3.7 μM).
Moenomycin binding to the GT domain of PBP1b*.
In an attempt to further map the moenomycin binding site within the GT domain, a limited proteolysis study of PBP1b* was performed in the absence or presence of moenomycin. Chymotrypsin was added after a 15-min preincubation of PBP1b* with 0.5 mM moenomycin at various protease/PBP1b* ratios for 1 h at room temperature (Fig. 6). This study revealed that in the presence of the antibiotic, PBP1b* is more susceptible to proteolysis, suggesting that the binding of moenomycin may induce conformational changes within the GT domain which exposes previously buried regions. Notably, a 16-kDa band which is not present in the experiment performed in the absence of moenomycin displays the N-terminal sequence D85KVRV, thus corresponding to a region within the GT domain which becomes exposed (Fig. 6, lanes 6 and 7).
FIG. 6.

Effect of moenomycin binding to PBP1b*. Chymotrypsin digestion of PBP1b* (18 μM) for 1 h at room temperature in the presence (+) or absence (−) of 0.5 mM (final concentration) moenomycin. Proteins were separated by SDS-15% PAGE and stained with Coomassie blue. Numbers at the left indicate sizes of standard molecular mass markers. Lanes: 1, molecular mass markers; 2 and 5, no chymotrypsin; 3 and 6, chymotrypsin/PBP1b* ratio of 1:12,000; 4 and 7, chymotrypsin/PBP1b* ratio of 1:6,000. The arrow indicates the 16-kDa sequence within the GT domain referred to in the text.
DISCUSSION
In this work, we have expressed and characterized the bifunctional periplasmic region of S. pneumoniae PBP1b (PBP1b*) deprived of its cytoplasmic domain and transmembrane anchor. PBP1b* is overexpressed in the cytosol and highly soluble, unlike other class A PBPs from a variety of organisms, including other pneumococcal PBPs. Indeed, the GT domains of PBP1a* and PBP2a* from S. pneumoniae were shown to be aggregated and membrane bound, respectively (5, 6). Among the small number of bifunctional PBPs that have been biochemically studied, a common feature is observed: the presence of a membrane interaction site in the GT domain, distinct from the N-terminal transmembrane anchor helix. The membrane association sites in E. coli PBP1b and in S. pneumoniae PBP2a* encompass only the first GT motif, whereas in Mycobacterium leprae PBP1, the region comprising the third, fourth, and fifth GT motifs contains the secondary membrane binding site (4, 6, 21, 36). Monofunctional GT proteins from Staphylococcus aureus and Ralstonia eutropha deprived from their transmembrane anchors were also shown to aggregate (26, 37). This putative membrane association of the GT domain from class A PBPs is consistent with the membrane localization of the substrate lipid II. The high solubility of purified S. pneumoniae PBP1b* appears to be an exception. PBP1b* is the first bifunctional PBP, containing the complete extracellular region, to be overexpressed and purified in a soluble form. Recently, PBP1* from Mycobacterium tuberculosis has been obtained in soluble form but with the second, third, and part of the fourth GT motif deleted (30).
Benzylpenicillin and cefotaxime acylation efficiencies of PBP1b* are both in the same range, whereas other S. pneumoniae PBPs behave differently in respect to these β-lactams. PBP2x* presents the highest acylation efficiency value for cefotaxime, 9-, 17-, and 27-fold the values measured for PBP1b*, PBP1a*, and PBP2a*, respectively (Table 2) (5, 6, 24). This pattern correlates well with the observation that PBP2x is the most important cefotaxime resistance determinant which confers, when altered, low-level resistance to this antibiotic (15). An in-depth investigation of the cephalosporin resistance conferred by PBP2x has been performed at the molecular level through the three-dimensional structure resolution of S-PBP2x* from the β-lactam-sensitive strain R6 and from the resistant clinical isolate strain Sp328 (3, 12, 28) as well as through the analysis of amino acid mutations in the PBP2x variant sequence from different drug-resistant clinical isolates (23, 24). Resistance to β-lactams in streptococcal species is acquired by homologous recombination of pbp genes between resistant and sensitive strains; the high-level cefotaxime resistance transferred from Streptococcus mitis to S. pneumoniae necessitates the successive alterations of PBP2x, PBP2a, PBP1a, and PBP1b (15). One would expect that the in vitro acylation efficiency values for cefotaxime of R6 S. pneumoniae PBPs would follow this hierarchy. This is not the case, since PBP1b* presents an acylation efficiency for a cefotaxime value superior to the PBP1a* and PBP2a* values but close to 10-times lower than the value for S-PBP2x* (Table 2). The different roles of the various PBPs in cell growth and some functional redundancy may explain why the sensitivity scale is not reflected in the hierarchy of mutation development in PBPs and on selective pressure. Finally, the PBP1b* deacylation rate for benzylpenicillin is in the same range as for the other S. pneumoniae PBPs. Measurement of the kinetic parameters of PBP1b* from the R6 strain is a first step for in-depth understanding regarding the role of PBP1b in β-lactam resistance acquisition.
The functional TP domain of PBP1b*, which is the target for β-lactams, was delineated by limited proteolysis with trypsin. This led to the identification of the PBP1b* interdomain hinge region which links the GT and TP domains. Such a boundary region has been located in S. pneumoniae PBP1a* and PBP2a* and in E. coli PBP1b (5, 6, 20). Protein sequence alignment of these bifunctional class A PBPs indicate that these enzymes share a conserved domain organization (Fig. 2). The TP domain thus produced is soluble, highly stable, and functional, since it is able to bind [3H]benzylpenicillin. Notably, a variety of attempts to express the TP domain with the boundary regions determined by proteolysis but in the absence of the GT domain led to high overexpression of insoluble proteins. This confirms previous observations by our group and others that correct folding of the TP domain requires that it be expressed downstream of the GT domain (A. M. Di Guilmi and A. Dessen, unpublished data) (32).
Two distinct groups have performed the total synthesis of lipid II (31, 35). The availability of large quantities of the natural substrate will allow advances in GT activity characterization. In order to test the specific binding of lipid II onto PBP1b*, an electrophoretic assay which takes advantage of the fluorescence of dansylated lipid II was developed. A shift of the fluorescent band was observed in the presence of complete PBP1b* (GT and TP) but not in the presence of the isolated TP domain, leading to the conclusion that lipid II binds specifically to the GT domain of PBP1b*. This shift does not appear to be the result of the integration of PBP1b* into lipidic vesicles, as no shift was observed with Coomassie blue staining when similar assays were performed in the presence of detergent {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate [CHAPS]} and of an unrelated lipid (dioleoyl phosphatidylcholine) in place of lipid II (data not shown). The results suggest that lipid II interacts in a specific manner with the GT domain.
Moenomycin is an antibiotic whose structure is reminiscent of that of lipid II, suggesting that moenomycin competes with lipid II for the binding to PBP. The electrophoretic assay developed in this work shows, indeed, the absence of lipid II binding to PBP1b* in the presence of moenomycin. Notably, this inhibition is competitive, since the same assay was used to verify that lipid II is not inserted into moenomycin micelles and is thus fully available for protein binding (data not shown).
Two N-terminal PBP1b* truncations lacking the first (E145DxxFxxHxG) and second (G176xSTxTQQ) GT motifs were tested for lipid II binding with the electrophoretic assay. The similarity of behavior of full-length PBP1b* and PBP1b*ATL (lacking the first motif) suggests that this motif plays a minor or no role in substrate recognition. However, the only partial shift displayed by PBP1b*LIK (lacking the first and second motifs) suggests that the second motif (G176xSTxTQQ) is essential for substrate binding. Notably, the GT activity of E. coli PBP1b has been extensively analyzed, revealing that glutamate 233 (EDxxFxxHxG), which is located in the first motif, is central to the catalyzed GT reaction (32). It is conceivable that in PBP1b* from S. pneumoniae, this motif is involved in catalysis but not in substrate binding. Further analysis, including the testing of the ability of PBP1b* to polymerize glycan chains, will shed light on this question.
Lastly, the binding effect of moenomycin on the GT domain of PBP1b* was tested by limited chymotrypsin treatment of the full-length molecule. In the presence of 500 μM moenomycin, PBP1b* becomes more sensitive to the protease, generating a variety of minor bands (corresponding to the full-length molecule cleaved at different sites), a major band which corresponds to the TP domain, and a minor band of approximately 16 kDa whose N-terminal sequence corresponds to an internal site in the GT domain. It seems probable that the binding of moenomycin generates major conformational changes within the GT domain, exposing a variety of regions which make stabilizing contacts in the apo structure.
In conclusion, we have expressed and purified the first soluble class A PBP and have developed a fluorescence-based assay to characterize it biochemically. The TP domain of this bifunctional protein can only be produced by limited proteolysis of the full-length molecule and is then generated in soluble, active form. The GT domain is able to bind lipid II and moenomycin in a competitive manner, and recognition of lipid II is efficient despite the deletion of the first GT motif. The assay presented in this work will be useful in further GT characterization of the PBPs.
Acknowledgments
We are very grateful to Larry C. Blaszczak (Infectious Diseases Division, Lilly Research Laboratories) for the generous gift of dansylated lipid II. We thank Jean-Pierre Andrieu (IBS, Grenoble, France) for N-terminal sequencing, Laurent Chesnel (IBS) for determination of PBP1b* k2/K values, and André Zapun (IBS) and Nicolas Mouz (Protein'eXpert, Grenoble, France) for encouragement and helpful discussions.
REFERENCES
- 1.Coffey, T. J., M. Daniels, L. K. McDougal, C. G. Dowson, F. C. Tenover, and B. G. Spratt. 1995. Genetic analysis of clinical isolates of Streptococcus pneumoniae with high-level resistance to expanded-spectrum cephalosporins. Antimicrob. Agents Chemother. 39:1306-1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dessen, A., A. M. Di Guilmi, T. Vernet, and O. Dideberg. 2001. Molecular mechanisms of antibiotic resistance in Gram-positive athogens. Curr. Drug Targets Infect. Disord. 1:63-77. [DOI] [PubMed] [Google Scholar]
- 3.Dessen, A., N. Mouz, E. Gordon, J. Hopkins, and O. Dideberg. 2001. Crystal structure of PBP2x from a highly penicillin-resistant Streptococcus pneumoniae clinical isolate: a mosaic framework containing 83 mutations. J. Biol. Chem. 276:45106-45112. [DOI] [PubMed] [Google Scholar]
- 4.Di Guilmi, A. M., A. Dessen, O. Dideberg, and T. Vernet. 2002. Bifunctional penicillin-binding proteins: focus on the glycosyltransferase domain and its specific inhibitor moenomycin. Curr. Pharm. Biotechnol. 3:63-75. [DOI] [PubMed] [Google Scholar]
- 5.Di Guilmi, A. M., N. Mouz, J. P. Andrieu, J. Hoskins, S. R. Jaskunas, J. Gagnon, O. Dideberg, and T. Vernet. 1998. Identification, purification, and characterization of transpeptidase and glycosyltransferase domains of Streptococcus pneumoniae penicillin-binding protein 1a. J. Bacteriol. 180:5652-5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Di Guilmi, A. M., N. Mouz, L. Martin, J. Hoskins, S. R. Jaskunas, O. Dideberg, and T. Vernet. 1999. Glycosyltransferase domain of penicillin-binding protein 2a from Streptococcus pneumoniae is membrane associated. J. Bacteriol. 181:2773-2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dowson, C. G., A. Hutchison, and B. G. Spratt. 1989. Extensive re-modelling of the transpeptidase domain of penicillin-binding protein 2B of a penicillin-resistant South African isolate of Streptococcus pneumoniae. Mol. Microbiol. 3:95-102. [DOI] [PubMed] [Google Scholar]
- 8.Fraimow, H. S., and P. Courvalin. 2000. Resistance to glycopeptides in gram-positive pathogens, p. 621-634. In V. A. Fischetti et al. (ed.), Gram-positive pathogens. American Society for Microbiology, Washington, D.C.
- 9.Ghuysen, J. M. 1994. Molecular structures of penicillin-binding proteins and beta-lactamases. Trends Microbiol. 2:372-380. [DOI] [PubMed] [Google Scholar]
- 10.Goffin, C., and J.-M. Ghuysen. 1998. Multimodular penicillin-binding proteins: an enigmatic family of orthologs and paralogs. Microbiol. Mol. Biol. Rev. 62:1079-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldman, R. C., and D. Gange. 2000. Inhibition of transglycosylation involved in bacterial peptidoglycan synthesis. Curr. Med. Chem. 7:801-820. [DOI] [PubMed] [Google Scholar]
- 12.Gordon, E., N. Mouz, E. Duee, and O. Dideberg. 2000. The crystal structure of the penicillin-binding protein 2x from Streptococcus pneumoniae and its acyl-enzyme form: implication in drug resistance. J. Mol. Biol. 299:477-485. [DOI] [PubMed] [Google Scholar]
- 13.Grebe, T., and R. Hakenbeck. 1996. Penicillin-binding proteins 2b and 2x of Streptococcus pneumoniae are primary resistance determinants for different classes of beta-lactam antibiotics. Antimicrob. Agents Chemother. 40:829-834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hakenbeck, R., T. Briese, H. Ellerbrok, G. Laible, C. Martin, C. Metelmann, H. M. Schier, and S. Tornette. 1988. Antibiotic inhibition of bacterial cell surface assembly and function, p. 390-399. In P. Actor, L. Daneo-Moore, M. L. Higgins, M. R. J. Salton, and G. D. Shockman (ed.), Targets of β-lactams in Streptococcus pneumoniae. American Society for Microbiology, Washington, D.C.
- 15.Hakenbeck, R., A. Konig, I. Kern, M. van der Linden, W. Keck, D. Billot-Klein, R. Legrand, B. Schoot, and L. Gutmann. 1998. Acquisition of five high-Mr penicillin-binding protein variants during transfer of high-level β-lactam resistance from Streptococcus mitis to Streptococcus pneumoniae. J. Bacteriol. 180:1831-1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoskins, J., P. Matsushima, D. L. Mullen, J. Tang, G. Zhao, T. I. Meier, T. I. Nicas, and S. R. Jaskunas. 1999. Gene disruption studies of penicillin-binding proteins 1a, 1b, and 2a in Streptococcus pneumoniae. J. Bacteriol. 181:6552-6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joris, B., J. M. Ghuysen, G. Dive, A. Renard, O. Dideberg, P. Charlier, J. M. Frere, J. A. Kelly, J. C. Boyington, P. C. Moews, et al. 1988. The active-site-serine penicillin-recognizing enzymes as members of the Streptomyces R61 DD-peptidase family. Biochem. J. 250:313-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kell, C. M., U. K. Sharma, C. G. Dowson, C. Town, T. S. Balganesh, and B. G. Spratt. 1993. Deletion analysis of the essentiality of penicillin-binding proteins 1A, 2B and 2X of Streptococcus pneumoniae. FEMS Microbiol. Lett. 106:171-175. [DOI] [PubMed] [Google Scholar]
- 19.Laible, G., B. G. Spratt, and R. Hakenbeck. 1991. Interspecies recombinational events during the evolution of altered PBP 2x genes in penicillin-resistant clinical isolates of Streptococcus pneumoniae. Mol. Microbiol. 5:1993-2002. [DOI] [PubMed] [Google Scholar]
- 20.Lefevre, F., M. H. Remy, and J. M. Masson. 1997. Topographical and functional investigation of Escherichia coli penicillin-binding protein 1b by alanine stretch scanning mutagenesis. J. Bacteriol. 179:4761-4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mahapatra, S., S. Bhakta, J. Ahamed, and J. Basu. 2000. Characterization of derivatives of the high-molecular-mass penicillin-binding protein (PBP) 1 of Mycobacterium leprae. Biochem. J. 350:75-80. [PMC free article] [PubMed] [Google Scholar]
- 22.Martin, C., C. Sibold, and R. Hakenbeck. 1992. Relatedness of penicillin-binding protein 1a genes from different clones of penicillin-resistant Streptococcus pneumoniae isolated in South Africa and Spain. EMBO J. 11:3831-3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mouz, N., A. M. Di Guilmi, E. Gordon, R. Hakenbeck, O. Dideberg, and T. Vernet. 1999. Mutations in the active site of penicillin-binding protein PBP2x from Streptococcus pneumoniae. Role in the specificity for beta-lactam antibiotics. J. Biol. Chem. 274:19175-19180. [DOI] [PubMed] [Google Scholar]
- 24.Mouz, N., E. Gordon, A. M. Di Guilmi, I. Petit, Y. Pétillot, Y. Dupont, R. Hakenbeck, T. Vernet, and O. Dideberg. 1998. Identification of a structural determinant for resistance to beta-lactam antibiotics in Gram-positive bacteria. Proc. Natl. Acad. Sci. USA 95:13403-13406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Munoz, R., C. G. Dowson, M. Daniels, T. J. Coffey, C. Martin, R. Hakenbeck, and B. G. Spratt. 1992. Genetics of resistance to third-generation cephalosporins in clinical isolates of Streptococcus pneumoniae. Mol. Microbiol. 6:2461-2465. [DOI] [PubMed] [Google Scholar]
- 26.Paik, J., D. Jendrossek, and R. Hakenbeck. 1997. A putative monofunctional glycosyltransferase is expressed in Ralstonia eutropha. J. Bacteriol. 179:4061-4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paik, J., I. Kern, R. Lurz, and R. Hakenbeck. 1999. Mutational analysis of the Streptococcus pneumoniae bimodular class A penicillin-binding proteins. J. Bacteriol. 181:3852-3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pares, S., N. Mouz, Y. Petillot, R. Hakenbeck, and O. Dideberg. 1996. X-ray structure of Streptococcus pneumoniae PBP2x, a primary penicillin target enzyme. Nat. Struct. Biol. 3:284-289. [DOI] [PubMed] [Google Scholar]
- 29.Reichmann, P., A. Konig, A. Marton, and R. Hakenbeck. 1996. Penicillin-binding proteins as resistance determinants in clinical isolates of Streptococcus pneumoniae. Microb. Drug Resist. 2:177-181. [DOI] [PubMed] [Google Scholar]
- 30.Schwartz, B., J. A. Markwalder, S. P. Seitz, Y. Wang, and R. L. Stein. 2002. A kinetic characterization of the glycosyltransferase activity of Escherichia coli PBP1b and development of a continuous fluorescence assay. Biochemistry 41:12552-12561. [DOI] [PubMed] [Google Scholar]
- 31.Schwartz, B., J. A. Markwalder, and Y. Wang. 2001. Lipid II: total synthesis of the bacterial cell wall precursor and utilization as a substrate for glycosyltransfer and transpeptidation by penicillin binding protein (PBP) 1b of Escherichia coli. J. Am. Chem. Soc. 123:11638-11643. [DOI] [PubMed] [Google Scholar]
- 32.Terrak, M., T. K. Ghosh, J. van Heijenoort, J. Van Beeumen, M. Lampilas, J. Aszodi, J. A. Ayala, J. M. Ghuysen, and M. Nguyen-Disteche. 1999. The catalytic, glycosyl transferase and acyl transferase modules of the cell wall peptidoglycan-polymerizing penicillin-binding protein 1b of Escherichia coli. Mol. Microbiol. 34:350-364. [DOI] [PubMed] [Google Scholar]
- 33.Tipper, D. J., and J. L. Strominger. 1965. Mechanism of action of penicillins: a proposal based on their structural similarity to acyl-d-alanyl-d-alanine. Proc. Natl. Acad. Sci. USA 54:1133-1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Heijenoort, J. 2001. Formation of the glycan chains in the synthesis of bacterial peptidoglycan. Glycobiology 11:25R-36R. [DOI] [PubMed] [Google Scholar]
- 35.van Nieuwenhze, M. S., S. C. Mauldin, M. Zia-Ebrahimi, B. E. Winger, W. J. Hornback, S. L. Saha, J. A. Aikins, and L. C. Blaszczak. 2002. The first total synthesis of lipid II: the final monomeric intermediate in bacterial cell wall biosynthesis. J. Am. Chem. Soc. 124:3656-3660. [DOI] [PubMed] [Google Scholar]
- 36.Wang, C.-C., D. E. Schultz, and R. A. Nicholas. 1996. Localization of a putative second membrane association site in penicillin-binding protein 1b of Escherichia coli. Biochem. J. 316:149-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang, Q. M., R. B. Peery, R. B. Johnson, W. E. Alborn, W.-K. Yeh, and P. L. Skatrud. 2001. Identification and characterization of a monofunctional glycosyltransferase from Staphylococcus aureus. J. Bacteriol. 183:4779-4785. [DOI] [PMC free article] [PubMed] [Google Scholar]