

Abstract
Two proteins, gp15 and gp3 (gp for gene product), are required to complete the assembly of the T4 tail. gp15 forms the connector which enables the tail to bind to the head, whereas gp3 is involved in terminating the elongation of the tail tube. In this work, genes 15 and 3 were cloned and overexpressed, and the purified gene products were studied by analytical ultracentrifugation, electron microscopy, and circular dichroism. Determination of oligomerization state by sedimentation equilibrium revealed that both gp15 and gp3 are hexamers of the respective polypeptide chains. Electron microscopy of the negatively stained P15 and P3 (P denotes the oligomeric state of the gene product) revealed that both proteins form hexameric rings, the diameter of which is close to that of the tail tube. The differential roles between gp15 and gp3 upon completion of the tail are discussed.
Completion of the assembly of the tail of bacteriophage T4 requires two gene products, gp15 and gp3 (7, 10). Both 15-lacking and 3-lacking tails have an unstable tail sheath, lack the connector, and are consequently incompetent to join the head (10, 11). Tails lacking 3 thus lack 15 as well (10). The tail sheath subunit, gp18, of the 15- and/or 3-lacking tail is more easily dissociated into monomeric gp18 than that of the 15+and/or 3+ tail. Burst-size measurements for restrictive host cells infected with a mix of wild-type T4 and a given amber mutant indicated that gp15 is a structural component of the T4 tail (22). It is a protein with a molecular weight of 31,499 and is reported to form the connector, which constitutes the extension of the tail tube, with a length of around 7 nm at the head-proximal end of the tail (10, 12, 13, 20, 25). Snustad (22) did not describe the type of function of gp3, whether it is catalytic or stoichiometric, in his paper, presumably due to the leakage of 3am mutants, although in vitro complementation assays for both gp3 and gp15 which used sucrose density gradient fractions at a number of concentrations indicated that gene 3 and gene 15 products function noncatalytically (27). Nevertheless, gp3 was, for some time, thought to function catalytically due to the failure of identifying gene 3 products either in the T4 tail (12) or in the 3+ tail (23 deficient, 15 deficient, or 18 deficient) (13). However, recent results clearly demonstrated that it is a component of the tail (26). The role of gp3 is presumably to terminate the elongation of the tube by binding to the tube protein gp19 and to gp29, the ruler molecule (1, 11, 26). The study of in vivo assembly of the tail by pulse-chase analysis by Ferguson and Coombs indicated that gp15 is incorporated into the tail approximately at the same time as gp19 and gp18 (8).
The intergenic cis-trans test for positional effects by Stahl and coworkers indicated that genes 13, 14, and 15 are cotranscribed (23). Clustering of the genes often reflects strong interaction and cooperative functions of the gene products they encode (12). While gp15 is a tail component, gp13 and gp14 are reported to be components of the neck of the head and are required for the tail to bind to the head (6, 7). Osmotic shock by rapid dilution of T4 virion from 1.58 M CsCl to buffer without CsCl disrupts the head and yields a tail with a neck (6). The neck is a knob-like structure on top of the tail with a length of 15 nm and a width of 16 nm. They observed a similar knob-like structure at the top of the tail after prolonged cultivation of lysis-inhibited bacteria infected with head-deficient mutants. It thus predicts that gp13 and/or gp14 may interact with gp15 in vitro.
Two contradictory results on the nucleotide sequence of gene 3 at the 3′ end have been reported. The nucleotide sequence reported by Koch et al. predicts that the total number of amino acids in gp3 is 176, whereas one insertion of a nucleotide in Lipinska et al.'s nucleotide sequence gives 8 additional amino acid residues due to the frameshift (14, 17). A recent paper on gp3 by Vianelli et al. favored the longer sequence based on the measurement of the number of cysteines (26). We therefore reexamined the nucleotide sequence and analyzed overexpressed gp3 by mass spectrometry.
The detailed characterization of gp15 and gp3 is crucial in elucidating the roles of these two proteins in the completion and stabilization of the tail and in head-tail junction. In the present study, gene 15 and gene 3 were cloned in expression vectors and overexpressed separately and together. Both of the gene products, gp15 and gp3, were purified to homogeneity and characterized by analytical ultracentrifugation, electron microscopy (EM), and circular dichroism (CD).
MATERIALS AND METHODS
Bacterial strains.
Escherichia coli XL1-Blue was used for the construction and the isogenic control of plasmids pCC801 [pET29a(+) with cloning of gene 15] and pLZ-T4g3 [pET29a(+) with cloning of gene 3]. E. coli BL21(DE3), carrying the gene for T7 RNA polymerase in λDE3 integrated into the chromosome of BL21, was transformed with pCC801 and pLZ-T4g3 for the overexpression of gp15 and/or gp3.
Plasmids.
pBluescript II SK(−) was obtained from Stratagene. pET29a(+) was obtained from Novagen, Inc.
Media, chemicals, and buffers.
Luria-Bertani (LB) broth contains 1% (wt/vol) tryptone peptone, 0.5% (wt/vol) Bacto yeast extract, and 1% (wt/vol) NaCl. LB plates contain 1% (wt/vol) tryptone peptone, 0.5% (wt/vol) Bacto yeast extract, 1% (wt/vol) Bacto agar, and 1% (wt/vol) NaCl. The final concentrations of antibiotics were 100 μg/ml for ampicillin and 50 μg/ml for kanamycin. IPTG (isopropyl-1-thio-β-d-galactosidase) was used at a concentration of 1 mM for induction. Phenylmethylsulfonyl fluoride (PMSF) was dissolved in isopropanol at a concentration of 100 mM. The working concentration of PMSF was 0.5 mM. 4-(2-Aminoethyl)-benzenesulfonyl fluoride hydrochloride (AEBSF; Boehringer Mannheim) was used at a 0.5 mM concentration. Sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) staining solution contains 0.2% (wt/vol) Coomassie brilliant blue G-250 (Nacalai Tesque) and 10% (vol/vol) acetic acid. Buffer A is 50 mM sodium phosphate (pH 7.0). Buffer B contains 50 mM Tris · HCl (pH 8.0) and 0.2 M NaCl. Buffer C contains 50 mM Tris · HCl and 50 mM N,N-methylenebisacrylamide · Tris propane (pH 7.5).
Plasmid construction for expression of gp15 and gp3 separately or together.
Gene 15 of bacteriophage T4 was amplified by PCR from T4D DNA with 2 primers, 5′-GGATTCGATTAGGAGAAGTGATGT-3′ (BamHI site in bold) and 5′-CGTCGACCTATGTTCTTGGGCCAG-3′ (SalI site in bold). The 846-bp PCR product was cloned into the SmaI site in pBluescript II SK(−). The resulting plasmid was digested with BamHI and SalI and then inserted into the expression vector pET29a(+). The resulting plasmid, pCC801, was transformed into E. coli BL21(DE3) for expression of gp15. A 0.6-kb DNA fragment encoding gp3 was amplified by PCR from the chromosomal DNA of T4D with 2 primers, 5′-TTGGTACCTAAAGCATTATGAG-GAATA-3′ (KpnI site in bold) and 5′-TTGGATCCTTATAACACACAAAACTGA-3′ (BamHI site in bold). The PCR product was cloned into the SmaI site in the pBluescript II SK(−). The resulting plasmid was digested with KpnI and BamHI and then inserted into the expression vector pET29a(+). The plasmid pLZ-T4g3, thus constructed, was transformed into E. coli BL21(DE3) for expression of gp3. For coexpression of gp15 and gp3, pLZ-T4g3 was digested with KpnI and BamHI and the gene 3 fragment was cloned into pCC801. The resulting plasmid pLZ-T4g3g15 was transformed into E. coli BL21(DE3).
DNA sequencing.
The CEQ2000 DNA analysis system (Beckman Coulter) was used for DNA sequencing in combination with a dye terminator cycle sequencing kit. Primers 5′-CTTAAGTCGTGGATAGGAATG-3′ and 5′-GTCTCACGATCATAGTCAATTCC-3′ were used to amplify gene 3 from the T4D genome for DNA sequence determination.
SDS-PAGE.
SDS-PAGE was carried out according to the method described by Laemmli (15) in a vertical minislab gel (9 by 7.5 cm).
Overexpression and isolation of proteins.
E. coli BL21(DE3) cells containing pCC801, pLZ-T4g3, and pLZ-T4g3g15 were grown at 37°C in LB with 50 μg of kanamycin/ml. IPTG was induced when the optical density at 660 nm reached 0.3 for gp15, 0.5 for gp3, and 0.5 for the coexpression system. The cells were continuously cultivated for 4 h afterwards. The cells were then harvested by centrifugation, and the resulting cell pellet was frozen and stored at −80°C. Those cells were resuspended in buffer A and sonicated on ice for 5 min twice (output power, 5; duty cycle, 50%; Branson Sonifier 250). Right after the sonication, PMSF was added to the crude extract at a final concentration of 0.5 mM. The extract was centrifuged at 24,000 × g for 20 min to remove cell debris.
(i) Purification of gp15.
All purification steps were carried out at 4°C except for gel filtration. The supernatant of the lysed cells was fractionated by (NH4)2SO4 at a concentration between 20 and 30% saturation. After centrifugation at 24,000 × g for 20 min, the precipitate was resuspended in buffer A and dialyzed twice against 100 volumes of the same buffer. The dialyzed sample was loaded onto a DEAE-toyopearl (Tosoh) anion-exchange column (2.5 by 18 cm) equilibrated with the same buffer. gp15 was eluted with buffer A containing NaCl from 0 to 0.5 M, followed by washing with buffer A. Fractions were monitored by absorption at 280 nm, collected, and examined on an SDS-12.5% polyacrylamide gel. Fractions with gp15 were combined and concentrated with Amicon ultrafiltration cells (model 8200, 62-mm diameter; model 8010, 25-mm diameter) with YM-10 ultrafilters (Millipore). The concentrated solution was applied to a BioGel A-5m (Bio-Rad) gel filtration column (2.5 cm by 1 m) equilibrated with buffer B. The protein was eluted out by the same buffer at a flow rate of 1.0 ml/min. Each fraction from the gel filtration chromatography was examined by SDS-12.5% PAGE. To inhibit the activity of serine proteases, 0.5 mM AEBSF was added to those fractions containing gp15. The purified gp15 was stored at −80°C.
(ii) Purification of gp3.
All purification steps were carried out at 4°C except for anion-exchange chromatography and for gel filtration chromatography. Ammonium sulfate was added to the supernatant of the lysed cells at a final concentration between 25 and 35% saturation. After centrifugation at 24,000 × g for 20 min, the precipitate was resuspended in buffer C and dialyzed twice against 100 volumes of the same buffer. The dialyzed sample was applied onto a Poros HQ/L column (7.5 by 100 mm; Perseptive Biosystems). Proteins were eluted with buffer C with an NaCl gradient from 0 to 1.0 M. Fractions containing gp3 were combined and concentrated (the protocols are the same as those for gp15). The concentrated sample was applied to a Superdex-200 gel filtration column (HR 10/30; Amersham Pharmacia Biotech) equilibrated with buffer B. The protein was eluted by the same buffer at a flow rate of 0.3 ml/min. Each fraction from the gel filtration chromatography was examined by SDS-12.5% PAGE. To inhibit the activity of serine proteases, 0.5 mM AEBSF was added to those fractions containing P3. The purified P3 was stored at −80°C.
(iii) Isolation of coexpressed P15 and P3.
The purification protocols were the same as those for P3, except that ammonium sulfate was added to the supernatant of the lysed cells at a final concentration between 15 and 35% saturation.
Amino-terminal sequence determination.
Samples were separated from the cell extract by SDS-10% PAGE. Right after the electrophoresis, the protein bands were transferred to a polyvinylidene difluoride membrane (Millipore). Protein bands were visualized by Coomassie brilliant blue R (Sigma). The band of gp15 or gp3 was cut out and applied on a protein sequencer (Shimadzu PPSQ-21). The first 7 amino-terminal residues were determined.
Mass spectrometry.
The molecular mass of gp3 was measured with a Shimadzu Axima-CFR matrix-assisted laser desorption ionization-time of flight mass spectrometer (Genomics Research Center for Shimadzu Biotech).
Analytical ultracentrifugation.
Prior to analysis by analytical ultracentrifugation, P15 or P3 samples were dialyzed against buffer B. The dialysate was used as a reference solution. Both sedimentation velocity and sedimentation equilibrium experiments were performed on an Optima XL-I analytical ultracentrifuge (Beckman) in a 4-hole An60Ti rotor at 20°C with standard double-sector centerpieces and quartz windows. Concentration profiles of samples were monitored by absorbance at 280 nm. The sedimentation velocity experiments were carried out at A280s of 0.2, 0.4, and 0.6 for P15 and of 0.5, 0.8, and 1.0 for P3 at a rotor speed of 40,000 rpm. Scans were recorded rapidly without intervals between successive scans. The sedimentation coefficient was calculated by the second-moment method and was then converted to s20,w (sedimentation coefficient under standard conditions, 20°C in water) with a solvent density of 1.008 g/ml and a solvent viscosity of 1.036 cP. The sedimentation equilibrium experiments were carried out at A280s of 0.1, 0.3, and 0.5 for P15 and 0.2, 0.4, and 0.6 for P3 at rotor speeds of 5,000, 7,000, and 9,000 rpm. Scans were recorded every 2 h, and the equilibrium of the system was judged by the superimposition of the last three scans. The total nine data sets for P15 or P3 were globally fitted to a single-species model to determine the molecular weight. The partial specific volumes, 0.73 cm3/g for gp15 and 0.74 cm3/g for gp3, were based on the amino acid composition of the proteins. The partial specific volume, solvent viscosity, and solvent density were calculated by the Sednterp program (16) (J. Philo et al., unpublished data).
Estimation of the secondary structure by CD.
A solution of P15 or P3 was dialyzed against buffer B prior to application on the CD spectropolarimeter. The dialysate was used as a reference solution. The concentrations of P15 and P3 were 0.20 and 0.17 mg/ml, respectively. The far-UV CD spectra were measured at room temperature by a J-720 spectropolarimeter (Jasco) in a 1-mm-path quartz cell. The CD spectra were analyzed by CONTIN (19) in order to estimate the secondary structures.
EM.
P15 (0.30 mg/ml) or P3 (0.34 mg/ml) in buffer B was applied on carbon-coated copper grids and negatively stained with 2% uranyl acetate. Tube-baseplate (1/50 molar ratio) was added to the samples as a standard. The samples were observed on an H-7500 EM (Hitachi) with a magnification of 40 × 103. Pictures were taken with an Amount camera (C4742-95; Hamamatsu). Twenty samples of each protein were taken from the EM graphs to measure the inner and outer diameter.
EM image analysis.
Suitable micrographs of both P3 and P15 rings were selected and digitalized at 14-μm intervals on a Zeiss PHODIS scanner. Individual ring images were boxed, and 10 images of each protein were averaged based on the reference-free method (18) with the SPIDER software package (9). The ROBEM software (R. Ashmore and T. S. Baker, unpublished data) was used for the calculation of the power spectrum of averaged images and for generating sixfold-symmetry-filtered images.
RESULTS
Genes 3 and 15 were cloned and overexpressed. The expressed gene products were purified and characterized in order to elucidate the roles of these two gene products during the assembly of the tail.
Molecular mass of gp3.
Prior to the present study, some previous contradictory information concerning the nucleotide sequence of gene 3 at the 3′ end region had to be clarified. The nucleotide sequence of gene 3 was independently reported by Koch et al. (14) and Lipinska et al. (17), with a difference at the 3′ end. Namely, the sequence of the latter had one extra nucleotide in the stop codon which caused a frameshift and consequently gave an 8-residue-longer amino acid sequence. Later, Vianelli et al. reported results favoring the longer sequence by Lipinska et al. (17) based on their determination of the cysteine content of gp3 (26). However, as our nucleotide sequence of cloned gene 3 coincided with that of Koch et al., we went on to directly sequence gene 3 in the genome of phage T4D, which also coincided with the shorter sequence of Koch et al. Furthermore, in order to exclude any possibility of translational editing or modification, we have carried out the molecular weight measurement of the overexpressed gp3 by mass spectrometry as shown in Fig. 1. The total mass of 19,585 Da for gp3 definitely supported the shorter sequence by Koch et al. The longer sequence would give the molecular weight of 20,529. We therefore concluded that gp3 consists of 175 amino acid residues as reported by Koch et al. (the initial Met of gp3 was cleaved as described below).
FIG. 1.

Molecular weight analysis of gp3 by matrix-assisted laser desorption ionization-time of flight (mass spectrometry). The spectrum is dominated by a mass peak at an m/z of 19,585 corresponding to the putative shorter gp3 amino acid sequence (calculated molecular weight = 19,581).
Amino-terminal sequence determination of overexpressed gp15 and gp3.
Both gp15 and gp3 were overexpressed in soluble fractions (Fig. 2, lane 3), although the expression of the latter was somewhat low. Expressed proteins were purified by ammonium sulfate fractionation, anion-exchange chromatography, and gel filtration (see Materials and Methods). Both gp15 and gp3 were thus purified to homogeneity as shown in Fig. 2, lanes 4 and 5. To confirm that the purified proteins were our targets, the two proteins were identified by amino-terminal sequence analysis with Edman degradation. The determined amino-terminal sequences of the two proteins were MFGYFYN and SQALQQ, which are identical to those of gp15 and gp3, respectively, as deduced from the nucleotide sequences (14, 17, 20). The N-terminal Met of gp3 had been cleaved off by processing in E. coli, which is in accord with the empirical rule that the N-terminal Met is removed if the side chain of the second residue is small and has no charge (3, 21). Purified proteins thus obtained and identified were used for characterization.
FIG. 2.

Expression and purification of gp15 and gp3. After SDS-PAGE, the gel was stained with Coomassie brilliant blue G. Lane 1, T4 tail used as a molecular mass marker; lanes 2 and 3, expression of gp15 and gp3 from a coexpression vector before and after IPTG induction, respectively (both gp15 and gp3 were expressed in soluble fractions); lanes 4 and 5, purified gp15 and gp3 after gel filtration, respectively (the two proteins were expressed from the respective expression vectors [see Materials and Methods for details]); lanes 6 and 7, peak fractions of gp15 (fraction no. 39) and gp3 (fraction no. 46) from gel filtration (Superdex 200 column) chromatography, respectively (gp15 and gp3 were eluted in different fractions, indicating that they were not interacting or only very weakly interacting); lane 8, molecular mass standards. The bands of gp15 and gp3 are indicated by bold arrows.
Oligomerization states of P15 and P3.
To explore the state of association of the isolated gp15 and gp3 in solution, the molecular weights of the two proteins were determined by analytical ultracentrifugation (see Materials and Methods). Prior to the molecular weight measurement by sedimentation equilibrium, a sedimentation velocity assay was carried out to examine the homogeneity of the purified proteins. A single symmetric boundary was observed for both gp15 and gp3 (Fig. 3), which indicated that they are single-oligomer species. The associated states of the gene products are hereafter designated P15 and P3, respectively. The sedimentation coefficients, s20,w, of P15 and of P3 were calculated to be 7.76 and 6.20 S, respectively, by using the second-moment method. Sedimentation equilibrium revealed that the molecular weights were 196,200 ± 4,300 for P15 (Fig. 4A) and 119,100 ± 3,300 for P3 (Fig. 4B). Based on the calculated molecular weight of 31,499 for gp15 and 19,581 for gp3 (initial Met cleaved) expected from the nucleotide sequences (14, 20), the number of subunits in the complexes were estimated to be 6.23 ± 0.14 and 6.08 ± 0.17, respectively. It was thus concluded that both gp15 and gp3 are homohexamers of each constituent subunit.
FIG. 3.

Sedimentation velocity of P15 (A) and P3 (B). The moving boundaries of the data with the absorbance at 280 nm of 0.4 (A) and of 0.8 (B) at the rotor speed of 40,000 rpm are shown. The s20,w was determined to be 7.76 S for P15 and 6.20 S for P3 by using the second-moment method. Both specimens revealed only single species.
FIG. 4.

Sedimentation equilibrium of P15 (A) and P3 (B). For each oligomeric protein, nine data sets, with three initial concentrations and three different speeds, were obtained and globally fitted to a single-species model. The rotor speeds were 5,000, 7,000, and 9,000 rpm, and the initial loading absorbance values at 280 nm were 0.1, 0.3, and 0.5 for P15 and 0.2, 0.4, and 0.6 for P3, respectively. A concentration profile for P15 (initial absorbance of 0.3 at 280 nm, measured at 9,000 rpm) (A) and that for P3 (initial absorbance of 0.6 at 280 nm, measured at 5,000 rpm) (B) are shown. The obtained molecular weights were 196,200 ± 4,300 for P15 and 119,100 ± 3,300 for P3, respectively, indicating that both complexes are homohexamers.
Observation of P15 and P3 under EM.
The hexameric subunit structure of P15 and P3 prompted us to examine the shape of the oligomers by EM. Both P15 and P3 were negatively stained and examined by EM. The obtained images of the oligomers are shown in Fig. 5, where some tube-baseplates are included to indicate the dimension. Both P15 and P3 appear as rings. We measured the inner and outer diameters for both protein complexes in the electron micrographs (Table 1). The dimensions of P15 and P3 are similar to those of the tail tube (5). Some tube-baseplates occasionally have a ring at the tip of the tube (Fig. 5A, inset), which is presumably a detached P15 from the tube.
FIG. 5.

EM images of P15 (A) and P3 (B). Samples were negatively stained by 2% uranyl acetate on a carbon-coated collodion membrane on copper grids and were observed in an H-7500 EM (Hitachi) with a magnification of 40 × 103 (see text for details). A small amount of tube-baseplates was added as a control for comparison. Ring structures are occasionally observed on the tip of some tube-baseplates (inset of panel A; the ring is indicated with a black arrowhead). The black arrows point to P15 rings (A) and similar P3 structures (B). Bars, 50 nm.
TABLE 1.
Dimensions of P15 and P3 ringsb
| Ring | Inner diam (nm) | Outer diam (nm) |
|---|---|---|
| P15 | 4.0 ± 0.5 | 13.0 ± 0.9 |
| P3 | 3.0 ± 0.4 | 10.0 ± 0.6 |
| Tail tubea | 3.0 | 8.0 |
| Tail sheatha | 8.0 | 21.0 |
Taken from Coombs and Arisaka (5).
Values for P15 and P3 are means ± standard deviations.
To determine the rotational symmetry of both P3 and P15 rings, individual ring images were boxed, 10 images of each ring were averaged, and a power spectrum along the ring axis was calculated. The power spectra clearly showed the sixfold symmetry (Fig. 6). This rotational symmetry agrees with the symmetry of both the tail tube and sheath.
FIG. 6.

Rotationally averaged P3 (A) and P15 (D) rings and sixfold-filtered P3 (B) and P15 (E) rings. The rotational power spectra of P3 (C) and P15 (F) both clearly showed sixfold symmetry.
Secondary structure of P15 and P3.
Far-UV CD spectra of P15 and P3 were measured, and the content of the secondary structure was estimated by a program, CONTIN (19). The P15 complex contains 14% α-helix, 47% β-sheet, and 13% β-turn (Fig. 7). The secondary structure of P3 was calculated to have 6% α-helix, 39% β-sheet, and 27% β-turn (Fig. 7). The results indicate that both proteins are rich in β-structure.
FIG. 7.

Far-UV CD spectra of P15 and P3. The secondary structure of the P15 complex was estimated by CONTIN to contain 14% α-helix, 47% β-sheet, and 13% β-turn (dotted line). P3 was calculated to contain 6% α-helix, 39% β-sheet, and 27% β-turn (solid line).
DISCUSSION
Elongation of the tail of bacteriophage T4 is terminated by gp3 followed by the formation of a connector made of gp15. Both gp3 and gp15 stabilize the tail sheath and make the tail ready for joining the head.
In the present study, we purified both gp15 and gp3 from cloned and overexpressed genes. Using analytical ultracentrifugation, we measured the sedimentation coefficients of both P15 and P3, which were 7.8 and 6.2 S, respectively. As we found that both P15 and P3 form hexameric ring structures from EM, Kirkwood's theory as adapted for sedimentation coefficient by Bloomfield et al. (4) was applied to calculate the expected sedimentation coefficients for P15 and P3 in order to confirm that these complexes also form ring structures in solution. Assuming that a monomeric protein is a sphere and the hexameric ring is constituted by six beads in a plane, we have
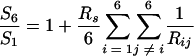 |
where S1 and S6 are sedimentation coefficients of the monomer and hexamer, respectively, Rij represents the center-to-center distance of the spheres, and Rs is related to f1 by the equation
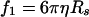 |
In this case, as S1 is not available, Rs was estimated by assuming that the monomer is a sphere:
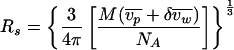 |
where M is the molecular weight of the monomer, 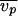 is the partial specific volume of the protein,
is the partial specific volume of the protein, 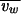 is the specific volume of water, NA is Avogadro's number, and δ is the hydration. For P15 and P3, the hydration was calculated to be 0.39 and 0.38 g/g of protein, respectively. The hydration and the partial specific volume of each protein were calculated by using Sednterp 1.0 (16) (Philo et al., unpublished). The calculated S6 for P15 and P3 were 8.8 and 6.1 S, respectively, which is in good agreement with the measured values and in contrast to the values for spheres, namely 10.2 and 7.2 S, respectively.
is the specific volume of water, NA is Avogadro's number, and δ is the hydration. For P15 and P3, the hydration was calculated to be 0.39 and 0.38 g/g of protein, respectively. The hydration and the partial specific volume of each protein were calculated by using Sednterp 1.0 (16) (Philo et al., unpublished). The calculated S6 for P15 and P3 were 8.8 and 6.1 S, respectively, which is in good agreement with the measured values and in contrast to the values for spheres, namely 10.2 and 7.2 S, respectively.
It is not obvious at this stage whether the hexameric gp15 (P15) binds to the tip of the tube or whether the monomeric gp15 binds onto the tip to form the hexamer in infected cells. However, it is likely that the hexamer is an active intermediate, as Yoshida (27) has shown that the complementation activity of gp15 was detected in the sucrose density gradient of a 10-deficient lysate as an 8-S component. This value of sedimentation coefficient is in agreement with our presently obtained value of 7.8 S for purified P15. Likewise, it is not certain whether the hexameric gp3 (P3) binds to the tube in vivo, but Yoshida's observation that gp3 sedimented as two species with sedimentation coefficients of 7 S (unstable) and 6 S (stable) (27) also suggests that P3 is an active precursor, considering that purified P3 has a sedimentation coefficient of 6.2 S. In comparison to P15, P3 is less stable upon storage and readily aggregates, which might be related with the unstable species of 7 S in the sucrose density gradient which was observed by Yoshida (see above). It is noted that there was some discrepancy between the reported length of the connector, which was thought to be composed solely of gp15 (13), i.e., 7 nm, and the expected thickness of P15, which is around 4.2 nm. However, if we take the thickness of P3, which is around 3.6 nm, as part of the connector, the total thickness would be 7.8 nm, which is close to 7 nm.
Negatively stained P15 and P3 were examined by EM, and the size and shape were compared with those of the tail tube and sheath (Table 1). The P3 hexameric ring has inner and outer diameters of 3.0 and 10.0 nm, respectively, which are close to those of the tail tube (Table 1). This is in agreement with the idea that gp3 recognizes a newly created binding site on top of the tail tube consisting of gp19 and the C terminus of gp29, the ruler molecule. Polymerization of gp19 is assumed to stop when gp29 has fully extended (1, 11). Vianelli et al. have further indicated that the absence of gp3 tends to cause further polymerization of gp19, the tail tube protein, suggesting the capping function of gp3 to prevent further elongation of the tail tube (26).
P15 also forms a hexameric ring, of which the inner and outer diameters are slightly larger than those of P3 but smaller than those of the tail sheath (Table 1). It is known that, even though the tail sheath is stabilized by gp3 and gp15, the sheath subunit gp18 can be dissociated from the tube-baseplate by prolonged dialysis against low-ionic-strength buffer (2, 24). After dissociation of gp18 from the tail this way, the tube-baseplate and dissociated gp18 can be separated by differential centrifugation. When the supernatant and the precipitate were separately subjected to SDS-gel electrophoresis, gp15 was found in the supernatant with monomeric gp18. On the other hand, gp3 was found in the precipitate with tube-baseplate (G. Ozawa and F. Arisaka, unpublished data). This observation indicates that gp3 binds to the tube-baseplate more strongly than does gp15. During tail contraction, the bonds between the gp18 and gp19 subunits are disrupted, but the sheath and the tube must be tightly connected at the proximal end to the head (2). Whether the head-joining of the tail induces some conformational change to gp3 and/or gp15 to make the tail sheath highly stable against dissociation or some other proteins are involved in the stabilization of gp18 and gp19 at the head-proximal remains to be elucidated.
As gp3 and gp15 form similar hexameric ring structures, it was likely that they interact with each other in vitro. Therefore, we mixed P15 and P3 to see if they form a complex by analytical ultracentrifugation. However, no association of P15 and P3 was detected (data not shown). There are a number of possibilities. One of the possibilities is that gp15 may associate as a monomer with a preformed hexamer of gp3. It is also possible that the hexamer formation of gp15 and binding to P3 may be kinetically competing and it is only in the absence of gp3 that gp15 forms a complex. In order to test these hypotheses, coexpression of genes 3 and 15 was carried out with the expectation that coexpression instead of mixing in vitro might facilitate the association of gp15 and gp3. However, coexpression did not seem to facilitate association. During the course of purification, the peak fractions of P15 and P3 did not coincide either in anion exchange or in gel filtration chromatography, indicating that they do not form a complex (Fig. 2, lanes 6 and 7). Immunoblotting of 15am T4 tails with anti-gp3 antiserum revealed that gp3 is assembled into the T4 tail prior to the addition of gp15 (26). Therefore, it is possible that gp3 may have to bind to gp19 before gp15 binds to gp3 in vivo. The association between gp3 and gp19 may lead to a conformational change of gp3, which is required for gp15 or P15 to bind to gp3 or to the tube through gp3.
The tail fails to join the head in the absence of gp13 and gp14 as well as gp3 and gp15 (7, 10). Coombs and Eiserling have observed that some knob-like structure, which contains gp13 and gp14, is formed on top of the tail during the prolonged cultivation of the cells infected by head-deficient mutants (6). The knob-like structure was similar to the knob which was observed in the tails of phage particles that had been treated by osmotic shock (6). Stahl et al. reported that gene 15 is cotranscribed with gene 13 and gene 14 as indicated by intergenic polarity effects test (23). These results indicate that gp15 may interact with the neck proteins gp13 and/or gp14 upon joining the head. That interaction may change the conformation of P15 to the tight binding form, which stabilizes the tail sheath from dissociation.
Acknowledgments
This work was supported in part by a grant to F.A. from the Ministry of Education, Science, Sports, and Culture of Japan.
We thank P. R. Chipman for taking some of the electron micrographs and M. C. Morais for advice and help on analysis of EM images. We thank Y. Takeda for technical assistance.
REFERENCES
- 1.Abuladze, N. K., M. Gingery, J. Tsai, and F. A. Eiserling. 1994. Tail length determination in bacteriophage T4. Virology 199:301-310. [DOI] [PubMed] [Google Scholar]
- 2.Arisaka, F., J. Tschopp, R. Van Driel, and J. Engel. 1979. Reassembly of the bacteriophage T4 tail from the core-baseplate and the monomeric sheath protein P18: a co-operative association process. J. Mol. Biol. 132:369-386. [DOI] [PubMed] [Google Scholar]
- 3.Ben-Bassat, A., K. Bauer, S. Y. Chang, K. Myambo, A. Boosman, and S. Chang. 1987. Processing of the initiation methionine from proteins: properties of the Escherichia coli methionine aminopeptidase and its gene structure. J. Bacteriol. 169:751-757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bloomfield, V., W. O. Dalton, and K. E. Van Holde. 1967. Frictional coefficients of multisubunit structures. I. Theory. Biopolymers 5:135-148. [DOI] [PubMed] [Google Scholar]
- 5.Coombs, D. H., and F. Arisaka. 1994. T4 tail structure and function, p. 259-281. In J. D. Karam et al. (ed.), Molecular biology of bacteriophage T4. American Society for Microbiology, Washington, D.C.
- 6.Coombs, D. H., and F. A. Eiserling. 1977. Studies on the structure, protein composition and assembly of the neck of bacteriophage T4. J. Mol. Biol. 116:375-405. [DOI] [PubMed] [Google Scholar]
- 7.Edgar, R. S., and I. Lielausis. 1968. Some steps in the assembly of bacteriophage T4. J. Mol. Biol. 32:263-276. [DOI] [PubMed] [Google Scholar]
- 8.Ferguson, P. L., and D. H. Coombs. 2000. Pulse-chase analysis of the in vivo assembly of the bacteriophage T4 tail. J. Mol. Biol. 297:99-117. [DOI] [PubMed] [Google Scholar]
- 9.Frank, J., M. Radermacher, P. Penczek, J. Zhu, Y. Li, M. Ladjadj, and A. Leith. 1996. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol. 116:190-199. [DOI] [PubMed] [Google Scholar]
- 10.King, J. 1968. Assembly of the tail of bacteriophage T4. J. Mol. Biol. 32:231-262. [DOI] [PubMed] [Google Scholar]
- 11.King, J. 1971. Bacteriophage T4 tail assembly: four steps in core formation. J. Mol. Biol. 58:693-709. [DOI] [PubMed] [Google Scholar]
- 12.King, J., and U. K. Laemmli. 1973. Bacteriophage T4 tail assembly: structural proteins and their genetic identification. J. Mol. Biol. 75:315-337. [DOI] [PubMed] [Google Scholar]
- 13.King, J., and N. Mykolajewycz. 1973. Bacteriophage T4 tail assembly: proteins of the sheath, core and baseplate. J. Mol. Biol. 75:339-358. [DOI] [PubMed] [Google Scholar]
- 14.Koch, T., N. Lamm, and W. Ruger. 1989. Sequencing, cloning and overexpression of genes of bacteriophage T4 between map positions 74.325 and 77.184. Nucleic Acids Res. 17:4392.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed]
- 16.Laue, T. M., B. D. Shah, T. M. Ridgeway, and S. L. Pelletier. 1992. Computer-aided interpretation of analytical sedimentation data for proteins, p. 90-125. In S. E. Harding, A. J. Rowe, and J. C. Horton (ed.), Analytical ultracentrifugation in biochemistry and polymer science. Royal Society of Chemistry, Cambridge, United Kingdom.
- 17.Lipinska, B., A. S. Rao, B. M. Bolten, R. Balakrishnan, and E. B. Goldberg. 1989. Cloning and identification of bacteriophage T4 gene 2 product gp2 and action of gp2 on infecting DNA in vivo. J. Bacteriol. 171:488-497. [DOI] [PMC free article] [PubMed]
- 18.Penczek, P., M. Radermacher, and J. Frank. 1992. Three-dimensional reconstruction of single particles embedded in ice. Ultramicroscopy 40:33-53. [PubMed] [Google Scholar]
- 19.Provencher, S. W., and J. Glockner. 1981. Estimation of globular protein secondary structure from circular dichroism. Biochemistry 20:33-37. [DOI] [PubMed] [Google Scholar]
- 20.Selivanov, N. A., A. G. Prilipov, and V. V. Mesyanzhinov. 1989. Nucleotide sequences of bacteriophage T4 genes 13, 14 and 15. Nucleic Acids Res. 17:3583.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sherman, F., J. W. Stewart, and S. Tsunasawa. 1985. Methionine or not methionine at the beginning of a protein. Bioessays 3:27-31. [DOI] [PubMed] [Google Scholar]
- 22.Snustad, D. P. 1968. Dominance interactions in Escherichia coli cells mixedly infected with bacteriophage T4D wild-type and amber mutants and their possible implications as to type of gene-product function: catalytic vs. stoichiometric. Virology 35:550-563. [DOI] [PubMed] [Google Scholar]
- 23.Stahl, F. W., J. M. Crasemann, C. Yegian, M. M. Stahl, and A. Nakata. 1970. Co-transcribed cistrons in bacteriophage T4. Genetics 64:157-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tschopp, J., F. Arisaka, R. van Driel, and J. Engel. 1979. Purification, characterization and reassembly of the bacteriophage T4D tail sheath protein P18. J. Mol. Biol. 128:247-258. [DOI] [PubMed] [Google Scholar]
- 25.Vanderslice, R. W., and C. D. Yegian. 1974. The identification of late bacteriophage T4 proteins on sodium dodecyl sulfate polyacrylamide gels. Virology 60:265-275. [DOI] [PubMed] [Google Scholar]
- 26.Vianelli, A., G. R. Wang, M. Gingery, R. L. Duda, F. A. Eiserling, and E. B. Goldberg. 2000. Bacteriophage T4 self-assembly: localization of gp3 and its role in determining tail length. J. Bacteriol. 182:680-688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoshida, F. 1973. Studies on two proteins which terminate the bacteriophage T4 tail. M.S. thesis. Massachusetts Institute of Technology, Cambridge.