

Abstract
Hypoxia is a common neonatal stress that induces insulin resistance and a decrease in body weight gain. Dexamethasone is often used to treat neonatal cardiopulmonary disease, and also leads to insulin resistance and a decrease in body weight gain. The current study addressed the hypothesis that serum concentrations of the adipokines adiponectin and/or resistin are altered during hypoxia and/or dexamethasone therapy in neonatal rats. Rat pups with their lactating dams were exposed to hypoxia (11% O2) from birth and treated with a tapering regimen of dexamethasone from postnatal day (PD) 3–6. Serum adiponectin and resistin were measured on PD7. Hypoxia and dexamethasone independently decreased body weight gain and increased adiponectin levels. The combination of hypoxia and dexamethasone did not further increase adiponectin. Dexamethasone caused a small increase in resistin in normoxic pups, which may facilitate the hyperinsulemic-normoglycemic state we previously described. We also conclude that adiponectin is increased during hypoxia in response to a decrease in the sensitivity to insulin.
Keywords: Adiponectin, resistin, hypoxia, rat, newborn, glucocorticoids
Introduction
Hypoxia, usually caused by cardiopulmonary disease, is a common cause of neonatal distress (1,2). Many studies have focused on the short-term cardiovascular and neurological consequences of neonatal hypoxia (2–6). We have extensively characterized the short-term endocrine and metabolic effects of hypoxia from birth to 7 d of age. Briefly, hypoxia from birth induces a decrease in weight gain without a significant change in body composition (7,8) and a state of relative insulin resistance (7,9,10). Furthermore, we have recently demonstrated that a tapering regimen of the glucocorticoid dexamethasone, administered to mimic clinical practice (11), results in hyperinsulinemia with normoglycemia and decreased weight gain (12). There is also a significant interaction between dexamethasone, which is often used to treat neonatal cardiopulmonary disorders (13), and hypoxia with respect to many parameters of metabolic function (12,14,15).
It is now clear that adipokines—hormones released from adipose tissue—are critically important in endocrine and metabolic regulation (16–20). We have demonstrated that the adipokine leptin is increased by dexamethasone and hypoxia (15). We have also demonstrated that ghrelin, a hormone released by the GI tract that has a wide variety of endocrine and metabolic actions, is increased by dexamethasone therapy (15). The differential actions of leptin and ghrelin on food intake, insulin sensitivity, and metabolic function made it clear that these two factors per se could not account for the changes in growth hormone (7), IGF-I (8), body composition (7,8), weight gain (7,8), lipid profiles (12,14), and other parameters of metabolic function in our previous studies.
The present study measured adiponectin and resistin in response to hypoxia from birth and/or a tapering regimen of dexamethasone therapy in 7-d-old rat pups. Adiponectin, which is elevated in newborns (21,22) and decreases thereafter (23), is an adipokine with anti-inflammatory and anti-atherogenic activity, and also appears to increase insulin sensitivity (16,19). Resistin is increased with diet-induced obesity; it decreases insulin sensitivity and has been associated with glucocorticoid-induced insulin resistance (18, 20). We hypothesized, therefore, that hypoxia and dexamethasone alter adiponectin and resistin levels in neonatal rats.
Results
Figure 1 shows body weights for rats exposed to normoxia or hypoxia from birth to postnatal day (PD) 7 and administered vehicle or dexamethasone in a tapering dose on PD 3–6. Control (normoxia-vehicle) pups gained about 1 g of body weight per day. Normoxic pups given dexamethasone initially lost weight (at PD4) and then gained a little weight back (approx 0.7 g over 2 d). Hypoxia per se resulted in a lower body weight at PD3 compared to normoxia. Interestingly, hypoxic vehicle-treated pups gained weight between PD4 and 6 at a rate not different from normoxic vehicle-treated pups, albeit starting at a lower weight. Hypoxic pups given dexamethasone did not gain weight from PD3 to 6 (similar to normoxic pups given dexamethasone).
Fig. 1.
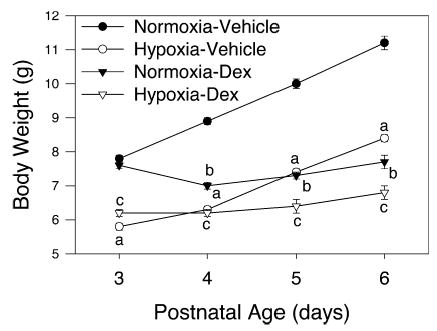
Body weight on PD3–6 (n = 9 per mean/SEM). Hypoxic vehicle-treated pups had significantly decreased body weight on PD3, but gained weight in parallel with normoxic vehicle-treated pups from PD4 to 6 (p < 0.001). Dexamethasone in normoxic and hypoxic pups caused an initial loss in body weight at PD4, but body weight increased thereafter: a, hypoxia-vehicle less than normoxic-vehicle (p < 0.05); b, normoxia-dexamethasone less than normoxia-vehicle (p < 0.05); c, hypoxia-dexamethasone less than normoxia-dexamethasone (p < 0.05).
Figure 2 shows adiponectin levels in 7-d-old rat pups exposed to hypoxia from birth and/or given a tapering regimen of dexamethasone from PD3 to 6. Hypoxia per se and dexamethasone per se each resulted in a significant increase in adiponectin levels. The combination of hypoxia and dexamethasone treatment did not increase adiponectin levels above those observed with dexamethasone alone. Resistin levels (Fig. 3) were not affected by hypoxia, whereas dexamethasone resulted in a small increase in resistin levels in normoxic pups only suggesting a negative interaction.
Fig. 2.
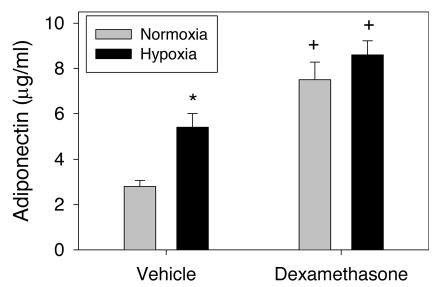
Effects on serum adiponectin of hypoxia from birth without or with dexamethasone treatment (PD3–6) in 7-d-old rat pups (n = 9 per mean/SEM). *Different from normoxia (p < 0.05). +Different from vehicle (p < 0.05).
Fig. 3.
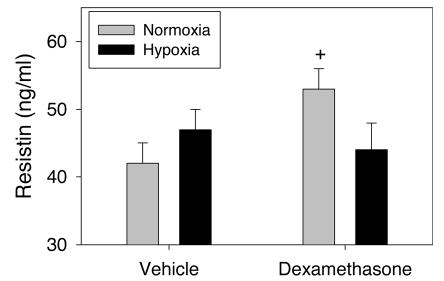
Effects on serum resistin of hypoxia from birth without or with dexamethasone treatment (PD3–6) in 7-d-old rat pups (n = 9 per mean/SEM). +Different from vehicle (p < 0.05).
Discussion
The major finding of this study was that hypoxia and dexamethasone independently increased serum adiponectin levels and decreased body weight gain in neonatal rat pups. The combination of hypoxia and dexamethasone administration did not increase adiponectin above levels observed with dexamethasone alone. Finally, dexamethasone induced a small increase in serum resistin in normoxic, but not hypoxic, rat pups.
We have previously used this model of hypoxia and/or dexamethasone to examine endocrine and metabolic function in neonatal rats (12,14,15). Pertinent to this study was the previous finding that dexamethasone resulted in marked hyperinsulinemia without a change in plasma glucose, indicating a state of relative insulin resistance (12). We also previously demonstrated that both leptin and ghrelin were increased by dexamethasone, but that only leptin was increased by hypoxia (15). Because hypoxia and dexamethasone clearly decrease body weight gain, we thought it was clear that these various humoral factors involved in the control of glucose uptake must have complex interactions, and that other factors must be involved.
Increases in adiponectin are thought to increase insulin sensitivity (16,19). It is possible that the marked increases in adiponectin in the current study were in response to the decrease in insulin sensitivity we have previously demonstrated (7,9,12).
Alternatively, dexamethasone-induced increases in serum adiponectin may be due to enhanced adipocyte differentiation (24). Increased plasma corticosterone during neonatal hypoxia (15) may have had the same effect. It is also important to note that the rat pups had decreased weight gain when treated with dexamethasone and had decreased weight gain when exposed to hypoxia. We have previously demonstrated that body composition in hypoxic rat pups is essentially unchanged—that is, the pups are smaller than their normoxic controls, but not altered with respect to muscle or fat mass (7,8). Therefore, although body mass and gross body composition are not altered, adipocyte differentiation and maturity may have induced changes in adiponectin and/or resistin release.
The findings of growth retardation and increased adiponectin in this study, and insulin resistance in previous experiments using the same treatment paradigm (7,9,12), are similar to a variety of other pathophysiological states including prenatal growth restriction (25) and anorexia or bulimia nervosa (26,27). This “paradoxical” increase in adiponectin may be a response to, rather than a cause of, changes in insulin resistance. The effect of the combination of hypoxia and dexamethasone on adiponectin was not additive suggesting they may work through a similar mechanism. In fact, the small increase in adiponectin during hypoxia may have been mediated by the large increase in corticosterone we have observed previously (15). Because dexamethasone is a very potent glucocorticoid, its effect on adiponectin may be due to a pharmacological, and therefore maximal effect, whereas the smaller effect of hypoxia may have been mediated via the same mechanism by an endogenous increase in corticosterone.
The small increase in resistin induced by dexamethasone was not apparent in hypoxic pups. We have observed a similar attenuation (negative interaction) of the leptin response to dexamethasone in hypoxic rat pups (15). The attenuation of resistin responses to dexamethasone in hypoxic pups may be a mechanism to prevent a magnification of the insulin resistance one would expect with the combination of pharmacological glucocorticoid therapy and hypoxia (12). We are unaware of comprehensive studies analyzing the interaction of hypoxia and glucocorticoid therapy on insulin sensitivity in human newborns. A recent study of fetal insulin secretion (represented by umbilical vein sampling) suggested a similar interaction of glucocorticoid therapy and in utero oxygenation (28).
This initial small study was performed in only eight litters (two per treatment). It is possible that more variability in the data would have been evident if the pups studied were derived from additional litters. Interestingly, the variability in body weight was not much different from previous studies in many more litters (12,15) giving us confidence that pups behaved relatively independently within each litter. Even so, a larger study with more litters and additional hormonal analyses is warranted. Another cave at that is inherent in this experimental model is that dams were exposed to hypoxia and, therefore, that some of the effects observed in pups could be due to maternal factors such as lactation and milk composition. We have previously demonstrated minor changes in milk composition in this model (29). Furthermore, levels of ionized calcium, intact PTH, 25-hydroxy-vitamin D, osteocalcin, and total calcium and phosphorus in hypoxic nursing pups are not altered by hypoxia indicating normal absorption of calcium and vitamin D from milk (30).
This initial study will hopefully lead to a more complete evaluation of the control of adiponectin release, adipocyte differentiation, and the effects of adiponectin in the hypoxic neonatal rat. Because glucocorticoid therapy is used in the treatment of lung disease of the newborn (13), additional studies on the interaction of hypoxia per se and dexamethasone in newborn humans will hopefully lead to new strategies for the treatment of the metabolic and endocrine effects of neonatal cardiopulmonary distress.
Materials and Methods
The experimental protocols were approved by the Institutional Animal Care and Use Committees of Aurora Health Care and the Medical College of Wisconsin. Timed pregnant Sprague–Dawley rats (Harlan, Indianapolis, IN; n = 8) were obtained at 14 d gestation and maintained on a standard sodium diet and water ad libitum in a controlled environment (lights on, 0600–1800 h). Litters were born on the afternoon of gestational d 21, after which dams and their litters were immediately transferred to environmental chambers and exposed to normobaric hypoxia (11% O2) or normoxia (21% O2) as described previously (31). Litter size was normalized to 10–11 pups per litter by cross-fostering.
Lactating dams were maintained with their litters for 7 d in a hypoxic or normoxic environment. Pups (n = 9 [two litters] per treatment) were weighed and then given either isotonic saline vehicle (5 μL/g) or dexamethasone phosphate (Sigma, St. Louis, MO) subcutaneously in a tapering regimen as follows: PD3 (0.5 mg/kg), PD4 (0.25 mg/kg), PD5 (0.125 mg/kg), and PD6 (0.05 mg/kg) (11). Between 0800 and 1000 on PD7, dams were removed from the chambers, and the pups were quickly decapitated and trunk blood was collected as a pool of two pups. Serum was obtained after the blood clotted and was centrifuged.
Serum adiponectin and resistin concentrations were measured by ELISA (B-Bridge International, Sunnyvale, CA). All samples were measured within one assay. The intraassay coefficients of variation were 4.8–6.6% for adiponectin and 4.9–5.2% for resistin. Data were analyzed by two-way analysis of variance and Duncan’s multiple range test (p < 0.05).
Acknowledgments
Thanks to Peter Homar and Barbara Jankowski for their excellent technical assistance. This work was supported by NIH/NIDDK grant DK54685.
References
- 1.Frankel L, Stevenson DK. Compreh Therap. 1987;13:14–19. [PubMed] [Google Scholar]
- 2.Friedman AH, Fahey JT. Semin Perinatol. 1993;17:106–121. [PubMed] [Google Scholar]
- 3.Low JA, Froese AB, Galbraith RS, Smith JT, Sauerberi EE, Derrick EJ. Acta Paediatr. 1993;82:433–437. doi: 10.1111/j.1651-2227.1993.tb12717.x. [DOI] [PubMed] [Google Scholar]
- 4.Thomas T, Marshall JM. J Physiol. 1995;487:513–525. doi: 10.1113/jphysiol.1995.sp020896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perlman JM. Semin Perinatol. 2004;28:415–424. doi: 10.1053/j.semperi.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 6.Low JA. J Obstet Gynaecol Res. 2004;30:276–286. doi: 10.1111/j.1447-0756.2004.00194.x. [DOI] [PubMed] [Google Scholar]
- 7.Raff H, Bruder ED, Jankowski BM, Colman J. Horm Metab Res. 2001;33:151–155. doi: 10.1055/s-2001-14929. [DOI] [PubMed] [Google Scholar]
- 8.Raff H, Bruder ED, Jankowski B, Oaks MK, Colman RJ. Endocrine. 2001;16:137–141. doi: 10.1385/ENDO:16:2:139. [DOI] [PubMed] [Google Scholar]
- 9.Raff H, Bruder ED, Jankowski BM. Endocrine. 1999;11:37–39. doi: 10.1385/ENDO:11:1:37. [DOI] [PubMed] [Google Scholar]
- 10.Dekelbab BH, Sperling MA. Diabet/Metabol Res Rev. 2004;20:189–195. doi: 10.1002/dmrr.448. [DOI] [PubMed] [Google Scholar]
- 11.Flagel SB, Vasquez DM, Watson SJ, Neal CR. Am J Physiol Integrat Comp Physiol. 2002;282:R55–R63. doi: 10.1152/ajpregu.2002.282.1.R55. [DOI] [PubMed] [Google Scholar]
- 12.Bruder ED, Lee PC, Raff H. Endocrinology. 2004;145:5364–5372. doi: 10.1210/en.2004-0582. [DOI] [PubMed] [Google Scholar]
- 13.Stark AR, Carlo WA, Tyson JE, The National Institute of Child Health Human Development Neonatal Research Network N Engl J Med. 2001;344:95–101. doi: 10.1056/NEJM200101113440203. [DOI] [PubMed] [Google Scholar]
- 14.Bruder ED, Lee PC, Raff H. J Appl Physiol. 2005;98:981–990. doi: 10.1152/japplphysiol.01029.2004. [DOI] [PubMed] [Google Scholar]
- 15.Bruder ED, Jacobson L, Raff H. J Endocrinol. 2005;185:477–484. doi: 10.1677/joe.1.06159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kadowaki T, Yamauchi T. Endocr Rev. 2005;26:439–451. doi: 10.1210/er.2005-0005. [DOI] [PubMed] [Google Scholar]
- 17.Yu YH, Ginsberg HN. Circ Res. 2005;96:1042–1052. doi: 10.1161/01.RES.0000165803.47776.38. [DOI] [PubMed] [Google Scholar]
- 18.Banerjee RR, Lazar MA. J Mol Med. 2003;81:218–226. doi: 10.1007/s00109-003-0428-9. [DOI] [PubMed] [Google Scholar]
- 19.Lihn AS, Pedersen SB, Richelsen B. Obesity Rev. 2005;6:13–21. doi: 10.1111/j.1467-789X.2005.00159.x. [DOI] [PubMed] [Google Scholar]
- 20.Ukkola O. Eur J Endocrinol. 2002;147:571–574. doi: 10.1530/eje.0.1470571. [DOI] [PubMed] [Google Scholar]
- 21.Kanantie E, Hytinantti T, Hovi P, Andersson S. J Clin Endocrinol Metab. 2004;89:4031–4036. doi: 10.1210/jc.2004-0018. [DOI] [PubMed] [Google Scholar]
- 22.Kotani Y, Yokota I, Kitamura S, Matsuda J, Naito E, Kuroda Y. Clin Endocrinol. 2004;61:418–423. doi: 10.1111/j.1365-2265.2004.02041.x. [DOI] [PubMed] [Google Scholar]
- 23.Iniquez G, Sotoa N, Avila A, et al. J Clin Endocrinol Metab. 2004;89:5500–5503. doi: 10.1210/jc.2004-0792. [DOI] [PubMed] [Google Scholar]
- 24.Gregoire FM. Exp Biol Med. 2001;226:997–1002. doi: 10.1177/153537020122601106. [DOI] [PubMed] [Google Scholar]
- 25.Lopez-Bermejo A, Casano-Sancho P, Fernandez-Real JM, et al. Clin Endocrinol. 2004;61:339–346. doi: 10.1111/j.1365-2265.2004.02102.x. [DOI] [PubMed] [Google Scholar]
- 26.Pannacciulli N, Vettor R, Milan G, et al. J Clin Endocrinol Metab. 2003;88:1748–1752. doi: 10.1210/jc.2002-021215. [DOI] [PubMed] [Google Scholar]
- 27.Monteleone P, Fabrazzo M, Martiadis V, et al. J Clin Endocrinol Metab. 2003;88:5387–5391. doi: 10.1210/jc.2003-030956. [DOI] [PubMed] [Google Scholar]
- 28.Verhaeghe J, van Bree R, van Herck E, Coopmans W. J Clin Endocrinol Metab. 2005;90:3449–3453. doi: 10.1210/jc.2004-2512. [DOI] [PubMed] [Google Scholar]
- 29.Bruder ED, Lee PC, Raff H. Am J Physiol Endocrinol Metab. 2004;288:E314–E320. doi: 10.1152/ajpendo.00362.2004. [DOI] [PubMed] [Google Scholar]
- 30.Raff H. Endocrine. 2002;17:157–160. doi: 10.1385/ENDO:17:3:157. [DOI] [PubMed] [Google Scholar]
- 31.Raff H, Jankowski BM, Bruder ED, Engeland WC, Oaks MK. Endocrinology. 1999;140:3147–3153. doi: 10.1210/endo.140.7.6794. [DOI] [PubMed] [Google Scholar]