

Abstract

Carboxylic acids were converted in high yield to their 1,1-dimethylallyl (DMA) esters in two steps. Palladium-catalyzed deprotection of DMA esters was shown to be compatible with tert-butyl, benzyl and Fmoc protecting groups, and Fmoc deprotection could be carried out selectively in the presence of DMA esters. DMA esters were also shown to be resistant to nucleophilic attack, suggesting that they will serve as alternatives to tert-butyl esters when acidic deprotection conditions need to be avoided.
Allyl-based protecting groups have become a popular choice for the protection of carboxylic acids, especially in peptide and glycopeptide synthesis, owing primarily to the mildness and selectivity of deprotection using catalytic palladium(0).1 Allyl-based protection strategies for the synthesis of peptides are an attractive complement to Boc-and Fmoc-based strategies for both solid and solution phase peptide synthesis, particularly of sensitive glycol-, nucleo-, and sulfopeptides, providing another level of “orthogonality” in biopolymer synthesis.2
On resin cyclization of peptides is an important case where a combination of Fmoc and Allyl protection is needed to selectively free the required carboxyl group.3, In particular, the backbone amide linker (BAL) approach reported by Barany, Albericio and co-workers in 19984a offers a general method for preparing cyclic peptides on solid support and requires the use of a C-terminal allyl ester.4 However, a side-reaction noted in this and other applications of allyl esters is the unwanted nucelophilic attack on the allyl ester, in the case of the BAL linker, this resulted in the unwanted formation of diketopiperazines.5 Whereas the sterically hindered t-butyl ester is known to resist nucleophilic attack, the strongly acidic conditions needed to deprotect it often preclude its use for the preparation of acid-sensitive molecules. For instance, in the case of the BAL linker, the acidic conditions needed to deprotect a t-butyl ester would also result in cleavage of a substrate from the BAL linker. The logical solution is the use of a tertiary ester than can be deprtected under mild conditions. Herein, we report a general approach to the protection of carboxylic acids as 1,1,-dimethylallyl (DMA) esters, which combine the mild deprotection conditions of allyl esters with the resistance to nucleophilic attack of t-butyl esters.
Several previous syntheses of DMA esters have been reported6 but to date no general method for their synthesis from carboxylic acids has been reported. Direct synthesis of DMA esters through the esterification of 2-methyl-3-buten-2-ol proved impractical owing to the hindered nature of the tertiary alcohol. Likewise, trapping of various 1,1-dimethyl π-allyl metal complexes with carboxylate nucleophiles gave prenyl esters rather than DMA esters owing to the greater stability of the prenyl ester and kinetic preference for prenyl ester formation. Instead, the approach adopted for the synthesis of DMA esters involves the intermediacy of 1,1-dimethylpropargyl esters, which can be subsequently reduced to DMA esters. This approach was suggested to us by a copper-catalyzed synthesis of aryl propargyl ethers via nucleophilic attack on tertiary propargylic leaving groups.7
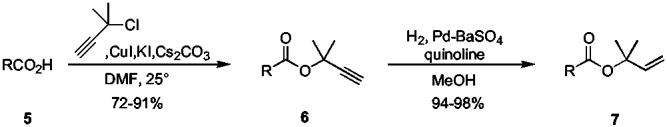
Treatment of 3-chloro-3-methyl-1-butyne (1) with benzoic acid in the presence of anhydrous copper(I) iodide did in fact afford the desired 1,1-dimethylpropargyl ester 3 in good yield (Scheme 1). Although initial results were obtained using K2CO3 as the base, use of Cs2CO3 as base was found to give superior results. Partial hydrogenation of the alkyne afforded the desired DMA ester 4 in excellent yield. Having thus established the viability of this approach, a series of N-protected amino acids were esterified using these conditions (Table 1). In all cases, the DMA esters were synthesized in good yield and with complete regiochemical control. Subsequent reduction by partial hydrogenation of the alkyne afforded the corresponding DMA esters in very high yields. The DMA esters of Fmoc-, Boc- and Cbz-protected amino acids were synthesized using these conditions.
Scheme 1.

Table 1.
Synthesis of DMA Esters from Protected Amino Acids
 |
| R | Yield of 6, % | Yield of 7, % |
|---|---|---|
| Fmoc-Val (5a) | 81 | 96 |
| Fmoc-Pro (5b) | 85 | 96 |
| Fmoc-Thr(Trt) (5c) | 91 | 94 |
| Fmoc-Abu (5d) | 76 | 97 |
| Fmoc-Tyr(t-Bu) (5e) | 90 | 98 |
| Fmoc-Orn(Boc) (5f) | 87 | 96 |
| Fmoc-Glu(t-Bu) (5g) | 82 | 97 |
| Fmoc-MeVal (5h) | 79 | 97 |
| Fmoc-Gly (5i) | 88 | 94 |
| Fmoc-Ala (5j) | 89 | 99 |
| Boc-Val (5k) | 72 | 96 |
| Cbz-Phe (5l) | 82 | 92 |
With various DMA esters in hand, it remained to be seen whether the DMA ester would prove to be orthogonal to the other protecting groups. In Table 2 it is shown that both the Fmoc group and the DMA ester can be removed selectively in the presence of the other. Moreover, DMA deprotection using catalytic Pd(0) and N-methylmorpholine proceeded in high yield and was compatible with t-butyl protection of various amino acid sidechains. While DMA esters could be readily deprotected in the presence of Fmoc, Boc and Cbz groups, neither Boc or Cbz could be selectively deprotected in the presence of a DMA ester. Thus, DMA esters should be considered orthogonal only with the Fmoc group.
Table 2.
Selective Deprotection of DMA Ester and Fmoc Group.
 |
| PG | AA | Yield of 5, % | Yield of 8, % |
|---|---|---|---|
| Fmoc | Val (7a) | 82 | 93 |
| Fmoc | Pro (7b) | 90 | 73 |
| Fmoc | Abu (7d) | 90 | 70 |
| Fmoc | Tyr(t-Bu) (7e) | 91 | 88 |
| Fmoc | Orn(Boc) (7f) | 83 | 86 |
| Fmoc | Glu(t-Bu) (7g) | 87 | 92 |
| Boc | Val (7k) | 84 | b |
| Cbz | Phe (7l) | 93 | b |
Deprotection conditions for Fmoc can be found in the Supporting Information; Boc deprotection was carried out using various concentrations of TFA in CH2Cl2 with various acid scavengers; Cbz deprotection was carried out by hydrogenolysis over various Pd catalysts.
Deprotection of Boc and Cbz lacked selectivity, resulting in loss of DMA ester.
It is especially noteworthy that, during deprotection of the γ-amino acid Fmoc-Abu (7d), the liberated primary amine 8d does not cyclize to form γ-butyrolactam 9 (Scheme 2). The fact that this normally facile reaction fails to occur under the reaction conditions suggests that, like t-butyl esters, DMA esters resist nucleophilic attack under all but the most forcing conditions. It is therefore suggested that DMA esters could successfully prevent unwanted diketopiperazine formation when using the BAL linker for peptide synthesis.
Scheme 2.

In conclusion, it has been demonstrated that carboxylic acids can be protected as their 1,1-dimethylallyl (DMA) esters in two steps. This methodology should prove quite general. Using this methodology, several DMA esters of different amino acids with various sidechain protecting groups were prepared and selectively deprotected. The DMA ester was shown to be orthogonal to the Fmoc group and compatible with t-butyl and benzyl-based protecting groups. The DMA ester should prove to be a useful alternative to the t-butyl ester for acid-sensitive substrates and provide protection against nucleophilic attack on the ester carbonyl.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (AI-50888) for support of this work.
Footnotes
Supporting Information Available. Experimental procedures and full characterization of compounds. This material is available free of charge on the Internet at http://pubs.acs.org.
References
- 1.Lipton MA. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E, editor. Vol. 2. John Wiley & Sons; New York: 2002. p. 1904. [Google Scholar]
- 2.Spivey AC, Woodhead SJ. Annu. Rep. Prog. Chem., Sect. B. 1998;94:77. [Google Scholar]
- 3.(a) Albericio F, Kates SA, Barany G, Sole NA, Johnson CR, Hudson D. Tet.Lett. 1993;34:1549. [Google Scholar]; (b) Tromelin A, Fulachier MH, Mourier G, Ménez A. Tet.Lett. 1992;33:5197. [Google Scholar]
- 4.(a) Barany G, Albericio F, Jensen KJ, Alsina J, Songster MF, Vagner J. Am. Chem. Soc. 1998;120:5441. [Google Scholar]; (b) Kurth MJ, Sammelson RE. Chem. Rev. 2001;101:137–202. doi: 10.1021/cr000086e. [DOI] [PubMed] [Google Scholar]
- 5.(a) Shute RE, Daniel H. J. Chem. Soc., Chem. Commun. 1987;15:1155. [Google Scholar]; (b) Fields GB, Noble RL. Int. J. Peptide Protein Res. 1990;35:161. doi: 10.1111/j.1399-3011.1990.tb00939.x. [DOI] [PubMed] [Google Scholar]
- 6.(a) Yamaguschi M, Okuma T, Scheloske M, Pardo DG. Tetrahedron Lett. 1994;35:1539. [Google Scholar]; (b) Saigo K, Usui M, Kikuchi K, Shimada E, Mukaiyama T. Bull. Chem. Soc. Jpn. 1977;7:1863. [Google Scholar]; (c) Piazza L, Paul K. Angew. Chem., Int. Ed. 2002;17:3263. [Google Scholar]; (d) Adock W, Cotton J, Trout NA. J. Org. Chem. 1995;21:7072. [Google Scholar]
- 7.Bell D, Davies MR, Green GR, Mann IS. Synthesis. 1995:707. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.