

Abstract
Background
Ventricular repolarization and contractile function are frequently abnormal in ventricular myocytes from human failing hearts as well as canine hearts with experimentally induced heart failure (HF). These abnormalities have been attributed to dysfunction involving various steps of the excitation-contraction coupling process, leading to impaired intracellular sodium and calcium homeostasis. We previously reported that the slow inactivating component of the Na+ current (late INa) is augmented in myocytes from failing hearts, and this appears to play a significant role in abnormal ventricular myocytes repolarization and function. We tested the effect of ranolazine, a novel drug being developed to treat angina, on 1) action potential duration (APD), 2) peak transient and late INa (INaT and INaL respectively), 3) early afterdepolarizations (EADs), and 4) twitch contraction (TC) including aftercontractions and contracture.
Methods:
Myocytes were isolated from the left ventricle of normal dogs and of dogs with chronic HF caused by multiple sequential intracoronary microembolizations. INaT and INaL were recorded using conventional whole-cell patch-clamp techniques. APs were recorded using the β-escin perforated patch-clamp configuration at frequencies of 0.25 and 0.5 Hz. TCs were recorded using an edge movement detector at stimulation frequencies ranging from 0.5 to 2.0 Hz.
Results
Ranolazine significantly (p < 0.05) and reversibly shortened the APD of myocytes stimulated at either 0.5 or 0.25 Hz in a concentration-dependent manner. At a stimulation frequency of 0.5 Hz, 5, 10 and 20 μM ranolazine shortened the APD90 (APD measured at 90% repolarization) from 516 ± 51 to 304 ± 22, 212 ± 34 and 160 ± 11 ms, respectively, and markedly decreased beat-to-beat variability of APD90, EADs and dispersion of APDs. Ranolazine preferentially blocked INaL relative to INaT in a state-dependent manner; with a ~ 38-fold greater potency against INaL to produce tonic block (IC50 = 6.5 μ M) than INaT (IC50 =294 μM). When we evaluated inactivated state blockade of INaL from the steady-state inactivation mid-potential shift using a theoretical model, ranolazine was found to bind more tightly to the inactivated state than the resting state of the sodium channel underlying INaL, with apparent dissociation constants Kdr=7.47μ M and Kdi=1.71μ M, respectively. TCs of myocytes stimulated at 0.5 Hz were characterized by an initial spike followed by a dome-like aftercontraction, which was observed in75% of myocytes from failing hearts and coincided with the long AP plateau and EADs. Ranolazine at 5, and 10 μM reversibly shortened duration of TCs and abolished the aftercontraction. When the rate of myocyte stimulation was increased from 1.0 to 2.0 Hz, there was a progressive increase in diastolic “tension”, i.e., contracture. Ranolazine at 5, and 10 μM reversibly prevented this frequency-dependent contracture.
Keywords: Heart failure, patch clamp, myocytes, action potential, contraction, late sodium current
INTRODUCTION
Ventricular arrhythmias are a frequent cause of sudden death in patients with heart failure (HF) (see review1). Despite the efforts of numerous investigators (see review 2), the underlying electrophysiological mechanisms responsible for these arrhythmias remain unclear. Increased dispersion of ventricular repolarization and early afterpolarizations (EADs) are major mechanisms involved in polymorphic ventricular tachycardia. 3, 4 Studies using different animal models of HF as well as heart tissue from HF patients revealed prolonged action potentials (APs), EADs, and increased spatial and temporal heterogeneity of ventricular repolarization. 5–8
A delicate balance between inward and outward currents maintains the cardiac AP plateau. Accordingly, AP prolongation in HF can be explained by either a decrease in repolarizing outward currents 5, 6 and/or an increase in depolarizing inward currents. We have shown that an increase in the slow-inactivating component of the sodium current (i.e., late INa, INaL) contributes to abnormal repolarization of ventricular myocytes from human and canine failing hearts. 7, 8 Recently Valdivia et al 9 found that INaL is significantly increased in ventricular myocytes from explanted failing human hearts as well as a canine pacing model of heart failure, confirming our observation.7, 8, 10 Thus, increased INaL may contribute to abnormal ventricular repolarization (AP prolongation and EADs)7, 8 and contractility 11 in HF, while inhibition of INaL should reverse these abnormalities. Therefore, INaL is a plausible target for drugs intended to reduce both arrhythmogenesis and mechanical dysfunction of failing ventricular myocytes. Indeed, one such antiarrhythmic agent, amiodarone, has been shown to selectively block INaL relative to the peak transient sodium current (INaT) in failing human myocardium. 12
Ranolazine, a novel drug being developed to treat angina 13, 14 has also been shown to have a number of cardiac electrophysiological effects, including inhibition of sea anemone toxin (ATX-II)-induced INaL and suppression of resulting ventricular arrhythmias in guinea pig and canine hearts. 15, 16 Thus, we determined the effects of ranolazine on INaT and INaL, AP duration, and twitch contractions of left ventricular myocytes isolated from dogs with HF to test the hypothesis that ranolazine reverses the HF-related abnormal repolarization and contractions by inhibiting augmented INaL.
METHODS
Canine HF Model and Myocyte Isolation
We developed a reproducible canine model of chronic HF that manifests marked and sustained depression of ventricular function, frequent ventricular ectopy and sudden death in ~13% of animals.17, 18 In the present study, healthy mongrel dogs weighing 24–31 kg underwent sequential coronary embolization to produce HF. This protocol was approved by the Institutional Animal Care and Use Committee of Henry Ford Hospital and conformed to the guiding principles of the Declaration of Helsinki.
Left ventricular mid-myocardial myocytes were enzymatically isolated from 26 dogs with HF (ejection fraction = 24 ± 1 %) and 5 normal dogs as described previously. 7 The yield of viable rod-shaped, Ca2+-tolerant myocytes varied from 50 to 80%.
Action Potential and Twitch Contraction Recordings
APs were recorded in current-clamp mode using a β-escin perforated patch- clamp configuration 11 with an Axopatch 200A patch-clamp amplifier (Axon Instruments, Foster City, CA), while pClamp 8.0 software (Axon) in conjunction with a PC computer was used to operate the current-clamp stimulation protocol. Myocytes were stimulated with current pulses lasting 0.2 ms duration. Contraction and relaxation were recorded using an edge detection system. Myocyte contraction was elicited using electrical field stimulation by applying pulses of 16 ms in duration, and a voltage 1.5- to 2–fold greater than the threshold 11. APs and twitch contractions were recorded at 35°C. Solutions used for AP and contraction recordings are summarized in Table 1.
Table 1.
Extracellular (Bath) and intracellular (Pipette) solutions used in the study
| Pipette solutions mM | Bath solutions mM | ||||
|---|---|---|---|---|---|
| INaT, INaL | AP Recordings, Perforated Patch | INAT | INAL | AP | |
| β-escin | 50μ M | ||||
| NaCl | 5 | 10 | 5 | 140 | 140 |
| KCl | 143 | 5.4 | 5.4 | ||
| CsCl | 133 | 133 | 5.4 | ||
| Glucose | 5 | 5 | |||
| CaCl2 | 1.8 | 1.8 | 1.8 | ||
| MgCl2 | 2 | 2 | 2 | 2 | |
| MgATP | 2 | ||||
| TEA | 20 | ||||
| Nifedipine | 0.002 | 0.002 | |||
| EGTA | 10 | ||||
| HEPES | 5 | 5 | 5 | 5 | 5 |
| pH | 7.3 | 7.3 | 7.3 | 7.3 | 7.3 |
| CsOH | KOH | CsOH | NaOH | NaOH | |
Sodium Current Recordings
INa was recorded using the conventional whole-cell patch-clamp technique (pClamp9, Axopatch 200A patch-clamp amplifier, Axon). The resistance of the borosilicate glass patch pipettes was 600–800 KΩ. The currents were low-pass filtered at 5 kHz, digitized at a sampling rate of 10 kHz and recorded at room temperature (22–24ºC). Voltage-clamp quality of the in each cell was controlled as described previously. 7, 8
INaL was elicited using 2- s membrane depolarizations from a holding potential of −140 mV applied at a pacing frequency of 0.1 Hz. “Zero”current was determined either after TTX (25 μM) application or by completely inactivating INaL using a 2-s depolarizing pre-pulse to –40mV, and was subtracted from the current traces. 7
The amplitude of INaL was determined from the average current measured 200–220 ms after the onset of a 2-s depolarization to −30 mV. This time was chosen to avoid contamination of late INa with INaT, which is completely inactivated within 200 ms of the beginning of the depolarizing pulse. 19 INaT was measured at a symmetrical Na+ concentration of 5 mM (Table 1). Inhibition of INaL and INaT was measured 2–6 minutes after adding ranolazine to the bath. Different cells were used to determine the effects of various concentrations of ranolazine. Holding potential was always –140 mV in order to ensure maximal availability of sodium channels.
Data Analysis
The ranolazine concentration-response curve for tonic block, describing the portion of blocked INaL or INaT (B) was determined by a conventional one-to-one binding model:
| (1) |
Tonic block was assessed after a 10-s resting period of at negative potential (Vh = −140mV).Steady-state inactivation (SSI) of INaL was measured using a double-pulse protocol. An 8-s prepulse of was followed by a 2-s or 40-ms testing pulse to −30 mV for INaL and INaT, respectively. Relative INaL or INaT was plotted against the pre-pulse voltage (Vp) and fitted to a Boltzmann function (SSI):
| (2) |
where V½ and k are the mid-point potential and slope factor, respectively. Interaction of the drug with the open and inactivated states of the channel was evaluated as suggested by Bean et al 20 by fitting experimental points of the drug-induced steady-state inactivation mid-point potential shifts to the equation:
| (3) |
where ΔV½ is the drug-induced shift of the mid-point potential of the SSI-voltage relation (Equation 2), k is the SSI slope factor, and Kdr and Kdi are apparent dissociation constants for the resting and inactivated states, respectively. The time course of INaL decay was fit to a single exponential model starting 200 ms after the onset of membrane depolarization:
| (4) |
where τ is the time constant, and Io and Is are the amplitude and steady-state component, respectively. INaL was fitted within the time interval from 0.2 s to 2 s after the onset of membrane depolarization. Chemicals
Enzymes: collagenase type II (291 U/mg) was purchased from Worthington (Freehold, NJ) and all other chemicals were purchased from Sigma (St. Louis, MO). Ranolazine was supplied by CV Therapeutics (Palo Alto, CA).
Statistical Analysis
Multiple comparisons were made using one way analysis of variance (ANOVA) followed by Bonferroni’s post-hoc test; taking p < 0.05 as significant. Deviations from the mean are reported in terms of the standard error, except where noted as standard deviation.
The quality of the models representing one-to-one binding (Figs. 4,5) and shifts in steady-state inactivation (SSI) caused by ranolazine (Fig. 6) were evaluated by F-test (StatMost 2.5 Software, DataMost Corp., Salt Lake City, UT) testing the null hypothesis that variability of the experimental data and values predicted by the model are the same. The F-value was determined as the ratio of the larger over the smaller variance and compared with tabulated F-values for the respective degrees of freedom and a confidence level of 0.95.
Figure 4.
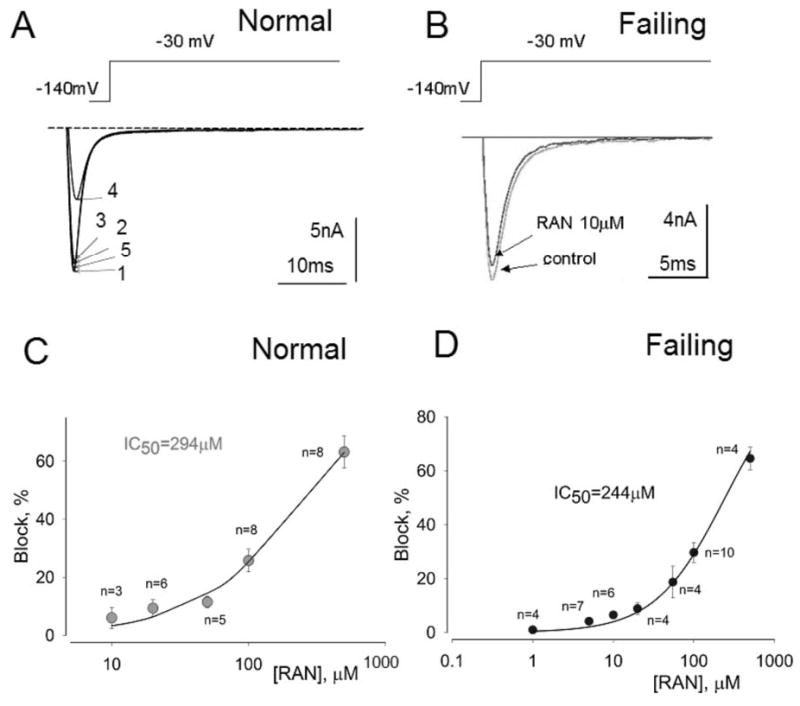
Ranolazine (RAN) weakly inhibits peak transient sodium current.
Effect of ranolazine on peak INa recorded from left ventricular myocytes isolated from normal (A, C) and failing canine hearts (B,D). Panel A: Superimposed current traces recorded in a non failing myocyte in the absence of drugs (1), and in the presence of 10 (2), 20 (3) and 500 μM ranolazine (4) and after drug washout (5). Panel B: Superimposed current traces recorded without (control) and with 10 μM ranolazine recorded in failing myocytes. Panels C and D: Concentration-response relationships for inhibition of peak INa by ranolazine in normal (C) and failing hearts (D). Points and error bars represent mean and standard error of peak sodium currents obtained in 13 myocytes from 2 normal (non-failing) hearts (C) and 14 myocytes from 4 failing hearts (D). Data points are fitted to a one-to-one binding model (solid line; Equation 1, Methods). The IC50 value is the concentration of ranolazine that caused 50% inhibition of peak INa.
Figure 5.
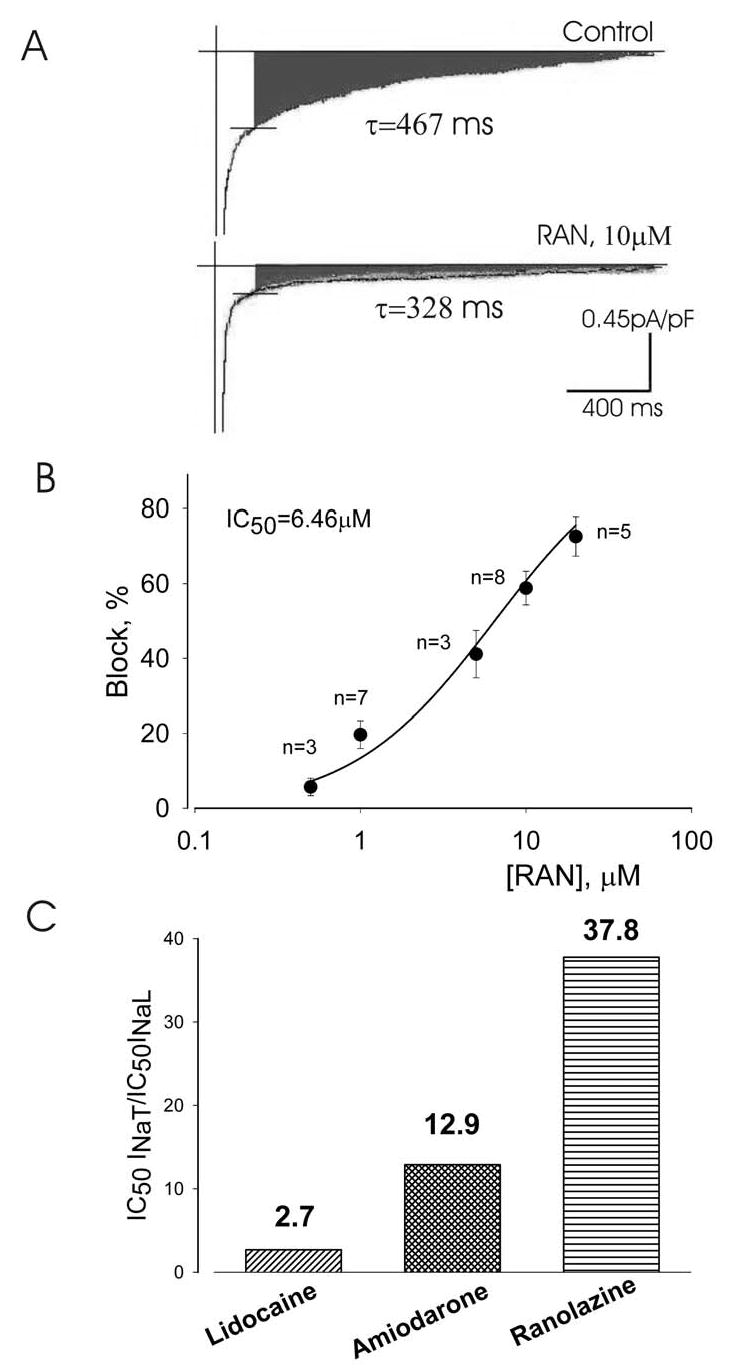
Inhibition by ranolazine (RAN) of the late INa of left ventricular myocytes isolated from canine failing hearts.
Panel A: Traces of late INa recorded without (control), and with 10 μM ranolazine. The peak transient inward sodium current is truncated. The decay time constant for late INa (τ values, single exponential fit; Equation 4, Methods) is given next to the current traces. Panel B: Concentration-response relationship for the inhibition of late INa by ranolazine. Points represent from 6 myocytes from 2 hearts. Data points are fit to a one-to-one binding model (Equation 1, Methods). The amplitude of late INaL was determined as the average current within 200–220 ms after the onset of a 2-s depolarization. Data are mean ± SEM. Panel C: Potencies of lidocaine, amiodarone 12 and ranolazine to inhibit peak and late INa representing the drug concentrations necessary to cause 50% block of peak and late INa.
Figure 6.
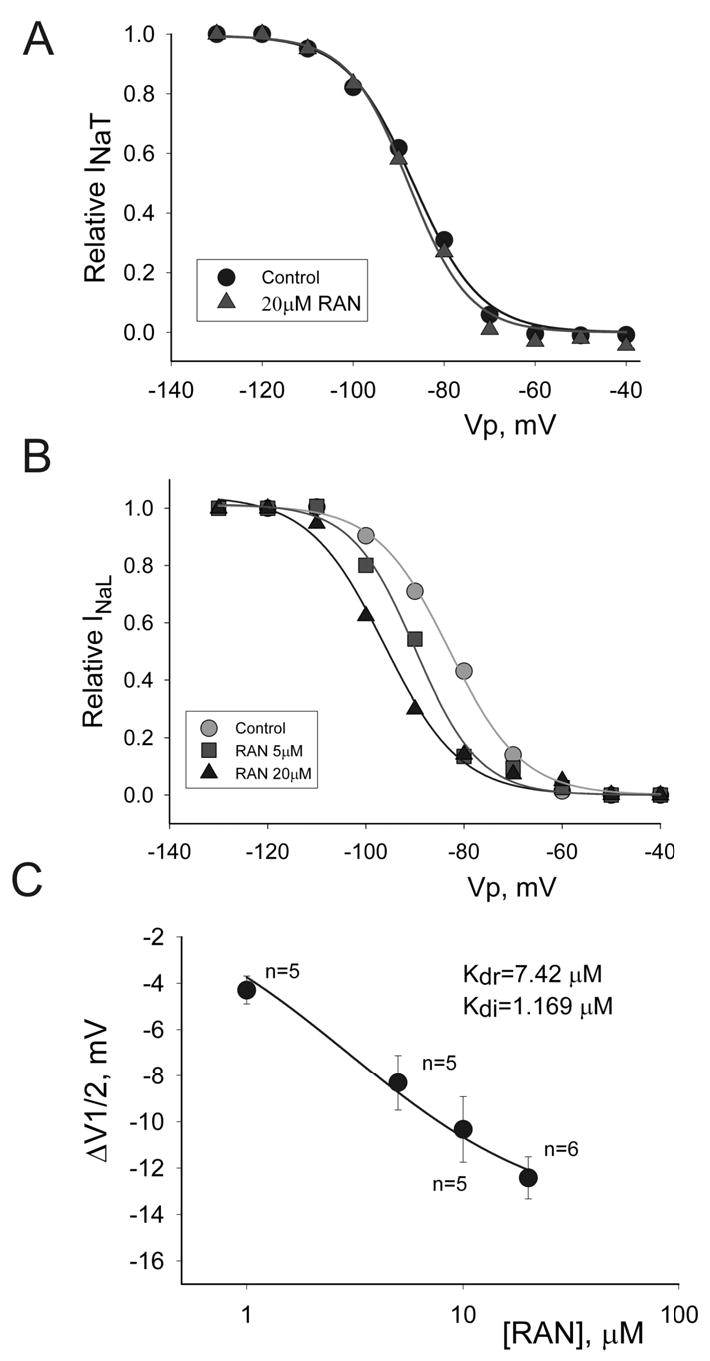
Concentration-dependent effect of ranolazine (RAN) on steady-state inactivation (SSI) of peak transient INa (INaT) and late INa (INaL) recorded in left ventricular myocytes isolated from canine failing hearts. Panel A: SSI-voltage relationship for peak INa in controls (•), and in the presence of 20 μM of ranolazine (▴), which failed to shift the SSI curve. Solid lines in Panels A and B represent data point fit to the Boltzmann function (Equation 2, Methods). All data were obtained in the same cell. Panel B: SSI-voltage relationship for late INa in controls (•) and in the presence of 5 (▪), and 10 μM ranolazine (▴), showing the leftward shift of the SSI mid-potential caused by ranolazine. All data were obtained in the same cell. Panel C: SSI mid potential shift (ΔV½) caused by ranolazine in 11 ventricular myocytes from 5 failing hearts. Solid line represents a data fit to equation 3 (Methods) to obtain dissociation constants of ranolazine for the resting (Kdr) and inactivated (Kdi) states of INaL as shown in the graph. The slope factor k in equation 3 was taken as 7.6mV, the evaluated mean for the cells presented in the panel C. Data are means ± SEM.
RESULTS
Effects of Ranolazine on Action Potentials of Ventricular Myocytes from Failing Hearts
The effects of 5, 10 and 20 μM ranolazine on AP duration, beat-to-beat variability of APD and EADs are described below and summarized in Figure 1.
Figure 1.
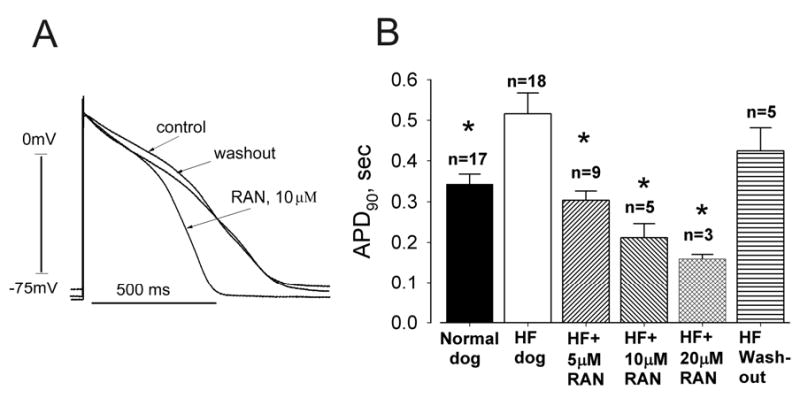
Ranolazine (RAN) reversibly shortens action potential (AP) duration (APD) of ventricular myocytes isolated from canine failing hearts (HF).
Panel A: Representative APs of single left ventricular cardiomyocytes. Superimposed APs were recorded at a pacing rate of 0.5 Hz in the absence of drug (control), in the presence of ranolazine (10 μM) and after drug washout.
Panel B: Effect of ranolazine on APD at 90% repolarization (APD90) of ventricular myocytes stimulated at a rate of 0.5 Hz. Ranolazine at concentrations of 5, 10 and 20 μM significantly and reversibly shortened APD90. Data were pooled from 3 non-failing and 6 failing hearts. All APs longer than 2 s were excluded.
n= number of myocytes; * p < 0.05 vs. HF (ANOVA followed by Bonferroni’s post-hoc test against HF).
APDs were significantly longer in ventricular myocytes from failing hearts (hereafter referred to as failing myocytes) than fnon-failing hearts (Fig. 1). At a pacing rate of 0.5 Hz (30 beats/min) ranolazine at 10 μM reversibly shortened APD of failing myocytes (Fig. 1A). While at 5, 10 and 20 μM it shortened APD90 (Fig. 1B) in a reversible and concentration-dependent manner.
The effect of ranolazine on beat-to-beat variability of APD and heterogeneity (dispersion) of APDs of failing myocytes was determined at 0.25 Hz (15 beats/min). Abnormally long APs, beat-to-beat APD variability and EADs were often observed in failing myocytes (Fig. 2A). Ranolazine at10 μM effectively shortened APD, abolished beat-to-beat variability, and suppressed EADs (Fig. 2B) and these effects were reversible upon ranolazine washout (Fig. 2C). As shown in Figure 3, ranolazine also markedly and reversibly reduced APD heterogeneity of myocytes stimulated at the rate of 0.25 Hz. Histograms of the distribution of 282 APDs from 5 myocytes isolated from 3 canine hearts (Fig. 3A) revealed that APDs of failing myocytes varied widely from as short as 208 ms to as long as 3.9 s. Ranolazine at5 μM markedly reduced this heterogeneity (Fig. 3B), and this effect was partially reversed upon ranolazine washout (Fig. 3C). It also significantly and reversibly reduced the variability consecutive APDs (Fig. 3D).
Figure 2.
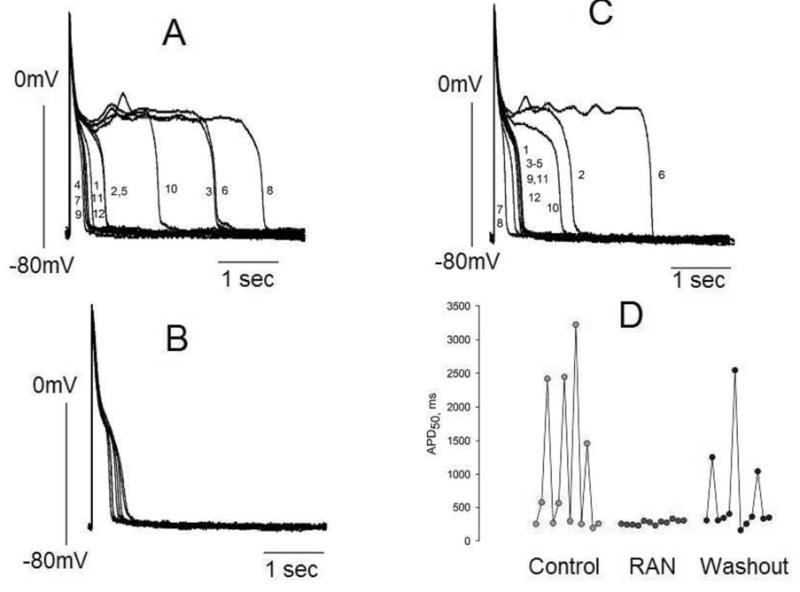
Ranolazine (RAN) reduces APD variability in left ventricular myocytes isolated from canine failing hearts. Twelve consecutive APs recorded at a pacing rate of 0.25 Hz are superimposed. Panel A: APs in the absence of drugs (control). Panel B: APs recorded in the presence of 10 μM of ranolazine. Panel C: APs recorded 3–4 minutes after drug washout. Panel D: Summary of the data shown in panels A-C. Each data point represents the APD50 from a series of APs recorded consecutively at a pacing rate of 0.25 Hz. The numbers next to the AP tracings in panels A and C are consecutive APs recorded during a period of ~ 45 seconds.
Figure 3.
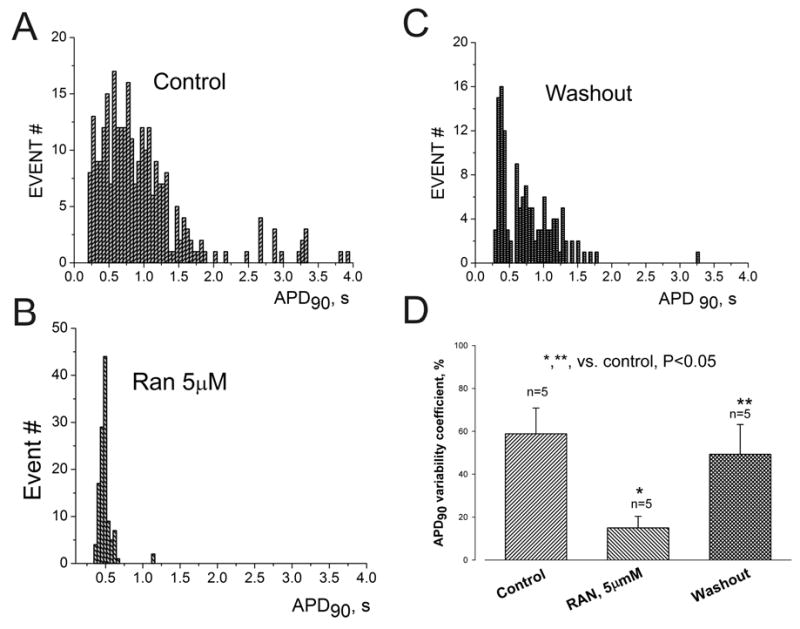
Ranolazine (RAN) reversibly reduces APD dispersion in left ventricular myocytes isolated from canine failing hearts. Histograms of APD distribution measured at 90% repolarization (APD90).APs were recorded in the absence of drugs (control) (A), in the presence of 5 μM ranolazine (B), and after drug washout (C). Bin size = 0.05 s for all histograms. D. APD variability measured as the coefficient of variability, CV = (SD/mean APD90) x 100%. Ranolazine (5 μM) reversibly reduced ADP90 variability in failing myocytes. Data were obtained in 5 ventricular myocytes from 3 failing dog hearts. P < 0.05 control vs. RAN and RAN vs. washout (ANOVA).
Effects of Ranolazine on Peak and Late Na+ Current
We determined the potency of ranolazine to cause 50% inhibition (IC50) of INaT and INaL during low-frequency stimulation (0.1 Hz, tonic block). Ranolazine weakly reduced the magnitude of INaT from either normal (Fig. 4A,C) or failing myocytes (Fig. 4B,D) with a potency of 294 and 244 μM, respectively. In comparison, the potency of ranolazine to inhibit INaL was much greater (6.46 μM) (Fig. 5). Therefore, ranolazine was approximately 38-fold more potent in inhibiting late INa than peak INa (Fig. 5C). Compared to lidocaine (potency ratio 2.7; Maltsev and Undrovinas, unpublished) and amiodarone (potency ratio 12.9) 12, ranolazine appears to be a more selective INaL inhibitor (Fig. 5C). For example, 10 μM ranolazine inhibited INaL by ~ 55% but had no or minimal (5%) inhibition of INaT (compare Fig. 4B,C with Fig.5A). Ranolazine at5 and 10 μM slightly accelerated the time-course of INaL decay as evaluated by a single exponential fit to equation 4 (see τ values in Fig. 5A).
We also determined the effects of ranolazine on peak transient and late INa at various membrane holding voltages and a low simulation frequency (0.1 Hz), i.e., SSI curve. At a rather high concentration (20 μM), ranolazine did not cause a significant shift in INaT SSI (Fig. 6A). In contrast, besides a strong resting state block (tonic block at very negative voltages, i.e., −140 mV) and low stimulation rate (Fig. 5), ranolazine also caused a marked voltage-dependent block of the INaL (Fig. 6B,C). Ranolazine caused a significant concentration-dependent leftward shift of the voltage-dependence of INaL SSI (Fig. 6C). The average shift of the midpoint potential V½ for at least 4 different concentrations of ranolazine ranged from 1 to 20 mV as determined by fitting the data points to a Boltzmann function (Equation 2, solid lines Fig. 6B) before and 6–10 min after adding ranolazine, respectively.
Based on the theoretical approach suggested by Bean et al 20 and the voltage shifts of the SSI curve (Fig 6C), the interaction of ranolazine with both the resting (R) and inactivated states (I) of the Na+ channel was evaluated, and is illustrated in the following diagram:

During the SSI assessment protocol, the sodium channel moves from the resting to the inactivated state. Ranolazine (D) binds to the resting state (RD), producing tonic block. SSI shift occurs when ranolazine binds to the inactivated state (RI). The data points were fit to Equation 3 (solid line in Fig. 6C), and the dissociation constants for the resting and inactivated states (Kdr and Kdi, respectively) were calculated (Fig. 6C). The resting-state block determined by fitting of the concentration-response curve (IC50 = 6.46 μM) (Fig. 5B) compared rather well with Bean’s model (Kdr = 7.42 μ M). These data validate both the experimental and theoretical approaches used to assess drug affinity for the different sodium channel states associated with late INa. Ranolazine appears to be ~ 6 times more potent in blocking the inactivating than the resting state (Kdi = 1.17 μ M vs. Kdr = 7.42 μM). According to equation 3 the maximal predicted SSI shift of INaL induced by ranolazine can be evaluated as (ΔV½)[RAN]=∞=kln(Kdi/Kdr)= −14mV. The shift produced by 20-μM ranolazine was −12 ± 0.9 mV (see Fig. 6C), close to the maximal predicted effect for INaL. At the same time, there was no indication of drug binding to the INaT inactivated state (Fig 6A) at this concentration.
Effects of Ranolazine on Contractility
Twitch contractions (cell shortening) of a single canine failing ventricular myocyte is often characterized by a phasic-tonic contraction (i.e., spike-and-dome-like shape) and/or early aftercontractions. 8 In comparison to the phasic contraction observed in normal myocytes, this abnormal shape has also been observed in multicellular preparations isolated from failing human hearts.21, 22 We determined the effects of ranolazine on abnormal relaxation of failing canine LV myocytes and found that at 10 μM it significantly suppressed the tonic (i.e., dome-like) component of the twitch contraction, thereby restoring normal relaxation, and this effect was reversed upon ranolazine washout (Fig. 7).
Figure 7.
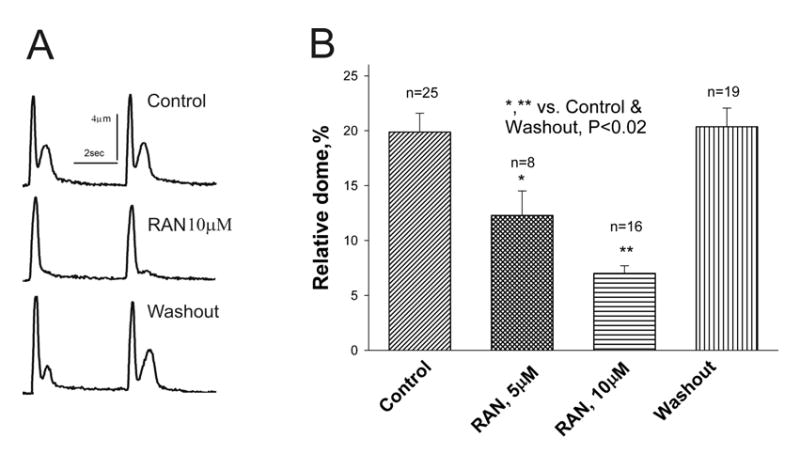
Ranolazine inhibition of abnormal relaxation of twitch contractions representing single ventricular myocytes from failing dog hearts. Panel A: Individual twitch shortenings recorded in control conditions (no drug, top), in the presence of ranolazine (10 μM, middle), and after drug washout (bottom). Panel B: Data from myocytes of 6 dogs with HF. Twitch contraction of a failing myocyte is characterized by a spike-dome shape. Ranolazine normalized the shape of the twitch contraction by abolishing the dome (tonic) component. *p < 0.001, **p < 0.02 vs. control (ANOVA).
Because exacerbated rate-dependent increases in resting tension (i.e., contracture) are a common feature of HF, the effect of ranolazine on the magnitude of the contracture measured at the end of a 1 min stimulation at different rates (1.0, 1.5 and 2.0 Hz) was also determined. As expected, contracture, , increased in a rate-dependent manner; ranolazine at 5 and 10 μM not only reduced the magnitude of the contracture at the basal stimulation rate of 1.0 Hz, but also blunted the increase in resting tension as the stimulation frequency increased to 1.5 and 2.0 Hz, and this effect was reversible upon ranolazine washout (Fig. 8).
Figure 8.
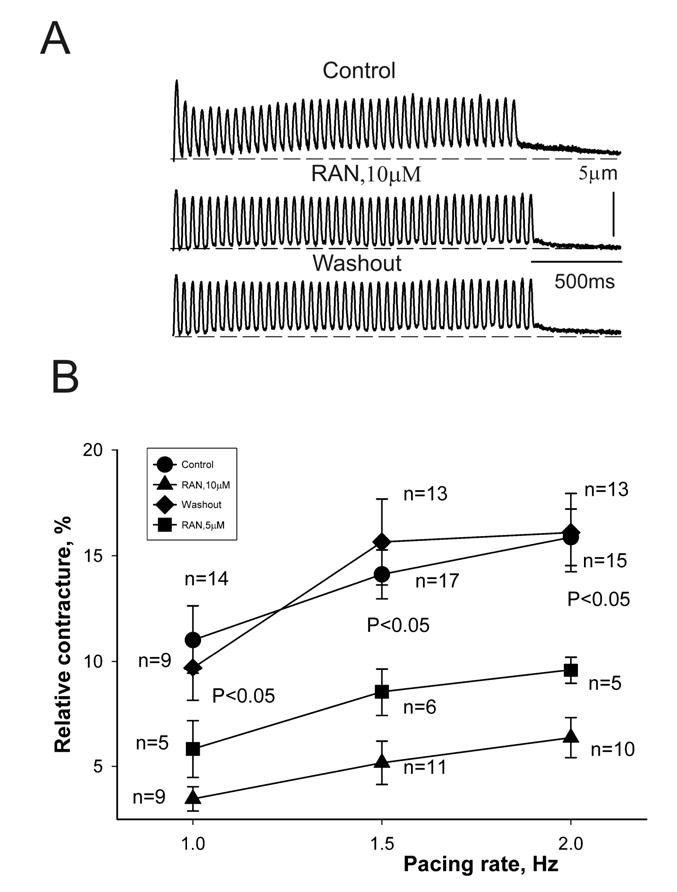
Ranolazine inhibition of frequency-dependent contracture (i.e., increase in diastolic tension) in failing myocytes. Panel A: Twitch contractions recorded at a pacing rate of 1.5 Hz under control conditions (top), in the presence of 10 μM ranolazine (middle), and after drug washout (bottom). Panel B: Data from myocytes of 7 dogs with HF at stimulation rates of 1.0, 1.5 and 2.0 Hz. Ranolazine at concentrations of 5 (▪) and 10 μM (▴) reversibly reduced the frequency-dependent contracture. p < 0.05 vs. control (•) and washout (♦) (ANOVA).
DISCUSSION
The two major findings of this study are: 1) ranolazine is approximately 38-fold more potent in inhibiting late (sustained) than peak (transient) INa; and 2) ranolazine at concentrations ≤10 μM (i.e., within the clinically relevant therapeutic range) reverses abnormalities of repolarization (prolonged APD, beat-to-beat variability and dispersion of APD and EADs), and contractile dysfunction (early aftercontractions, and contracture) of ventricular myocytes from failing canine hearts.
Inhibition of INaL by ranolazine is state-dependent. Ranolazine causes tonic (resting state) block of INaL in failing canine myocytes with an estimated potency of 6.46 μM, in keeping with similar findings in normal guinea pig ventricular myocytes treated with the Na+ channel modifier sea anemone toxin ATX-II 15, 23, as well as normal canine LV mid-myocardial cells 16. Ranolazine inhibits sodium channels responsible for INaL in the inactivated state with a potency of 1.17 μ M, greater than the resting state. This can be important at depolarized membrane potentials such as resting potential of about –80 mV, and AP plateau (−20 to 0 mV) where the probability of sodium channels inactivation is significantly greater, and the drug potency to block INaL may be closer to 1 μ M. Because INaT and INaL were measured at a different external Na+ (5 and 140 mM, respectively), one should expect some reduction of ranolazine-induced INaT block for, because increases in extracellular [Na+]o could attenuate the blocking action of class I drugs 24; thus, INaT would be even less susceptible to ranolazine at physiological Na+ concentrations. The lack of inactivated state of the sodium channels in the ATX-II-treated myocytes used as an LQT-3 model 15, 23 does not allow the evaluation of the ranolazine binding to the inactivated state of the sodium channel.
Can the electrophysiological and contractile effects of ranolazine reported in the present study be explained solely by the inhibition of INaL? It has been shown that ranolazine inhibits rapid inward rectifying current, IKr, with a potency of 11.2 μ M, and weakly interacted with ICaL or with the current generated by Na+/Ca2+ exchanger with potencies of 296 and 91 μ M, respectively 16. Thus, it is very likely that INaL is the major target for ranolazine to prevent repolarization and contraction abnormalities in ventricular myocytes from failing hearts.
The present study is the first to show that ranolazine weakly (IC50 = 244 to 294 μM) reduces INaT in both normal and failing myocytes. These two sodium currents, INaT and INaL, have different physiological importance, contributing to separate phases of the action potential, and their targeting is expected to have distinct consequences. INaT is responsible for AP upstroke, excitation and impulse propagation, and has been found to be down regulated in failing hearts. 9, 25, 26 Accordingly, further blockade of INaT, which is characteristic of class I anti-arrhythmic drugs, may greatly increase risk in HF patients. On the other hand, augmented INaL plays a significant role in repolarization and electromechanical coupling abnormalities in HF 1, and its blockade can be beneficial to suppress ventricular arrhythmias related to abnormal repolarization and contraction. Based on this, differential blockade of INaL relative to INaT may be an attractive feature for new anti-arrhythmic agents that may also reduce mechanical dysfunction. This differential potency of ranolazine to inhibit peak and late INa in the failing heart is greater than either amiodarone 12 or lidocaine (Fig. 5C). Thus, ranolazine appears to be a selective inhibitor of INaL, and single-channel studies are necessary to further evaluate the mechanism(s) of state- and mode-dependent 27 ranolazine block of the sodium channel responsible for INaL in failing myocytes.
Inhibition of late INa by ranolazine can explain its ability to reverse both the electrical and mechanical abnormalities of LV myocytes from failing canine hearts. In failing myocytes INaL is increased 8, 9 and ranolazine, by inhibiting INaL, should suppress EADs. Indeed, reduction of beat-to-beat variability of APDs and dispersion of APDs from failing ventricular myocytes by ranolazine are similar to the findings that STX and TTX abolished the APD variability observed in failing human and dog myocytes 7,8. Both the electrophysiological and mechanical abnormalities of failing myocytes were attenuated by ranolazine. Consistent with the observation that ranolazine suppresses EADs, it blocks dome-like early aftercontractions. The shortening of APDs and suppression of EADs explains how ranolazine can abolish early aftercontractions and improve myocyte relaxation. The fact that resting tension increases in together with the rate of stimulation is a common feature of failing myocytes from both animal 28 and human hearts. 29, 30 The mechanism proposed to explain this frequency-dependent rise in diastolic tension is an increase in intracellular [Na+]i leading to the [Ca++]i overload associated with rapid depolarization. 30, 31 By inhibiting late INa ranolazine should curtail the rise in [Na+]i and thereby reduce intracellular Ca++ overload.
In summary, in ventricular myocytes of dogs with chronic heart failure, ranolazine preferentially blocked INaL relative to INaT in a state-dependent manner, being ~ 38-fold more potent at producing tonic block of INaL than INaT. Furthermore, ranolazine bound more tightly to the inactivated than the resting state of the sodium channel responsible for INaL, with the apparent dissociation constants of Kdr =7.47 μ M and Kdi =1.71 μ M, respectively, yet without interacting with INaT. Thus, ranolazine is expected to be a more potent INaL blocker following membrane depolarization. Ranolazine can also reduce dispersion of APDs and improve contractile function. These findings raise the possibility that in the failing heart, ranolazine may suppress ventricular arrhythmias associated with repolarization abnormalities, and improve LV contractility. The data also provide additional evidence for participation of the late sodium current in repolarization and contractile abnormalities of the failing heart. Future studies are needed to establish the validity of inhibiting late INa as a target for treatment of heart failure.
Acknowledgments
The study was supported in part by grants from the National Heart Lung and Blood Institute [HL-53819, HL074328-01 (Undrovinas) and PO1 HL074237-02 (Sabbah)], a grant in-aid (0350472Z) from the American Heart Association (Undrovinas), and a grant from CV Therapeutics (Undrovinas and Sabbah)
References
- 1.Tomaselli GF, Zipes DP. What causes sudden death in heart failure? Circ Res. 2004;95:754–763. doi: 10.1161/01.RES.0000145047.14691.db. [DOI] [PubMed] [Google Scholar]
- 2.Tomaselli GF, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270–283. doi: 10.1016/s0008-6363(99)00017-6. [DOI] [PubMed] [Google Scholar]
- 3.Galinier M, Vialette JC, Fourcade J, Cabrol P, Dongay B, Massabuau P, Boveda S, Doazan JP, Fauvel JM, Bounhoure JP. QT interval dispersion as a predictor of arrhythmic events in congestive heart failure. Importance of aetiology. Eur Heart J. 1998;19:1054–62. doi: 10.1053/euhj.1997.0865. [DOI] [PubMed] [Google Scholar]
- 4.Boccalandro F, Velasco A, Thomas C, Richards B, Radovancevic B. Relations among heart failure severity, left ventricular loading conditions, and repolarization length in advanced heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol. 2003;92:544–7. doi: 10.1016/s0002-9149(03)00722-7. [DOI] [PubMed] [Google Scholar]
- 5.Beuckelmann DJ, Nabauer M, Erdmann E. Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circulation Research. 1993;73:379–85. doi: 10.1161/01.res.73.2.379. [DOI] [PubMed] [Google Scholar]
- 6.Kaab S, Nuss HB, Chiamvimonvat N, O'Rourke B, Pak PH, Kass DA, Marban E, Tomaselli GF. Ionic mechanism of action potential prolongation in ventricular myocytes from dogs with pacing-induced heart failure. Circulation Research. 1996;78:262–73. doi: 10.1161/01.res.78.2.262. [DOI] [PubMed] [Google Scholar]
- 7.Maltsev VA, Sabbah HN, Higgins RSD, Silverman N, Lesch M, Undrovinas AI. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation. 1998;98:2545–2552. doi: 10.1161/01.cir.98.23.2545. [DOI] [PubMed] [Google Scholar]
- 8.Undrovinas AI, Maltsev VA, Sabbah HN. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: Role of sustained inward current. Cell Mol Life Sci. 1999;55:494–505. doi: 10.1007/s000180050306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, Kamp TJ, Makielski JC. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. 2005;38:475–83. doi: 10.1016/j.yjmcc.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 10.Maltsev VA, Lesch M, Undrovinas A. A non-inactivating inward current in cardiomyocytes of dogs with chronic heart failure. Circulation. 1995;92:I-504. (Abstract). [Google Scholar]
- 11.Maltsev VA, Sabbah HN, Tanimura M, Lesch M, Goldstein S, Undrovinas AI. Relationship between action potential, contraction-relaxation pattern, and intracellular Ca2+ transient in cardiomyocytes of dogs with chronic heart failure. Cellular and Molecular Life Sciences. 1998;54 :597–605. doi: 10.1007/s000180050187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maltsev VA, Sabbah HN, Undrovinas AI. Late sodium current is a novel target for amiodarone: Studies in failing human myocardium. J Mol Cell Cardiol. 2001;33:923–932. doi: 10.1006/jmcc.2001.1355. [DOI] [PubMed] [Google Scholar]
- 13.Chaitman BR, Skettino SL, Parker JO, Hanley P, Meluzin J, Kuch J, Pepine CJ, Wang W, Nelson JJ, Hebert DA, Wolff AA. Anti-ischemic effects and long-term survival during ranolazine monotherapy in patients with chronic severe angina. J Am Coll Cardiol. 2004;43:1375–82. doi: 10.1016/j.jacc.2003.11.045. [DOI] [PubMed] [Google Scholar]
- 14.Chaitman BR, Pepine CJ, Parker JO, Skopal J, Chumakova G, Kuch J, Wang W, Skettino SL, Wolff AA. Effects of ranolazine with atenolol, amlodipine, or diltiazem on exercise tolerance and angina frequency in patients with severe chronic angina: a randomized controlled trial. Jama. 2004;291:309–16. doi: 10.1001/jama.291.3.309. [DOI] [PubMed] [Google Scholar]
- 15.Song Y, Shryock JC, Wu L, Belardinelli L. Antagonism by ranolazine of the pro-arrhythmic effects of increasing late INa in guinea pig ventricular myocytes. J Cardiovasc Pharmacol. 2004;44 :192–9. doi: 10.1097/00005344-200408000-00008. [DOI] [PubMed] [Google Scholar]
- 16.Antzelevitch C, Belardinelli L, Zygmunt AC, Burashnikov A, Di Diego JM, Fish JM, Cordeiro JM, Thomas G. Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation. 2004;110:904–10. doi: 10.1161/01.CIR.0000139333.83620.5D. Epub 2004 Aug 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sabbah HN, Stein PD, Kono T, Gheorghiade M, Levine TB, Jafri S, Hawkins ET, Goldstein S. A canine model of chronic heart failure produced by multiple sequential coronary microembolizations. American Journal of Physiology. 1991;260:H1379–84. doi: 10.1152/ajpheart.1991.260.4.H1379. [DOI] [PubMed] [Google Scholar]
- 18.Sabbah HN, Goldberg AD, Schoels W, Kono T, Webb C, Brachmann J, Goldstein S. Spontaneous and inducible ventricular arrhythmias in a canine model of chronic heart failure: relation to haemodynamics and sympathoadrenergic activation. European Heart Journal. 1992;13 :1562–72. doi: 10.1093/oxfordjournals.eurheartj.a060102. [DOI] [PubMed] [Google Scholar]
- 19.Undrovinas AI, Maltsev VA, Kyle JW, Silverman NA, Sabbah HN. Gating of the late Na+ channel in normal and failing human myocardium. J Mol Cell Cardiol. 2002;34:1477–1489. doi: 10.1006/jmcc.2002.2100. [DOI] [PubMed] [Google Scholar]
- 20.Bean BP, Cohen CJ, Tsien RW. Lidocaine block of cardiac sodium channels. Journal of General Physiology. 1983;81:613–42. doi: 10.1085/jgp.81.5.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gwathmey JK, Copelas L, MacKinnon R, Schoen FJ, Feldman MD, Grossman W, Morgan JP. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circulation Research. 1987;61:70–6. doi: 10.1161/01.res.61.1.70. [DOI] [PubMed] [Google Scholar]
- 22.Dipla K, Mattiello JA, Margulies KB, Jeevanandam V, Houser SR. The sarcoplasmic reticulum and the Na+/Ca2+ exchanger both contribute to the Ca2+ transient of failing human ventricular myocytes. Circ Res. 1999;84:435–44. doi: 10.1161/01.res.84.4.435. [DOI] [PubMed] [Google Scholar]
- 23.Wu L, Shryock JC, Song Y, Li Y, Antzelevitch C, Belardinelli L. Antiarrhythmic effects of ranolazine in a guinea pig in vitro model of long-QT syndrome. J Pharmacol Exp Ther. 2004;310:599–605. doi: 10.1124/jpet.104.066100. Epub 2004 Mar 18. [DOI] [PubMed] [Google Scholar]
- 24.Barber MJ, Wendt DJ, Starmer CF, Grant AO. Blockade of cardiac sodium channels. Competition between the permeant ion and antiarrhythmic drugs. J Clin Invest. 1992;90:368–81. doi: 10.1172/JCI115871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zicha S, Maltsev VA, Nattel S, Sabbah HN, Undrovinas AI. Post-transcriptional alterations in the expression of cardiac Na(+) channel subunits in chronic heart failure. J Mol Cell Cardiol. 2004;37:91–100. doi: 10.1016/j.yjmcc.2004.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maltsev VA, Sabbah HN, Undrovinas AI. Down-regulation of sodium current in chronic heart failure: effects of long-term therapy with carvedilol. Cell Mol Life Sci. 2002;59:1561–8. doi: 10.1007/s00018-002-8529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maltsev VA, Undrovinas AI. A multi-modal composition of the late Na+ current in human ventricular cardiomyocytes. Cardiovasc Res 2005; in press. [DOI] [PMC free article] [PubMed]
- 28.Wang Z, Lam CF, Mukherjee R, Hebbar L, Wang Y, Spinale FG. Relationship between external load and isolated myocyte contractile function with CHF in pigs. Am J Physiol. 1997;273:H183–91. doi: 10.1152/ajpheart.1997.273.1.H183. [DOI] [PubMed] [Google Scholar]
- 29.Davies CH, Davia K, Bennett JG, Pepper JR, Poole-Wilson PA, Harding SE. Reduced contraction and altered frequency response of isolated ventricular myocytes from patients with heart failure. Circulation. 1995;92:2540–9. doi: 10.1161/01.cir.92.9.2540. [DOI] [PubMed] [Google Scholar]
- 30.Pieske B, Maier LS, Piacentino V, 3rd, Weisser J, Hasenfuss G, Houser S. Rate dependence of [Na+]i and contractility in nonfailing and failing human myocardium. Circulation. 2002;106:447–53. doi: 10.1161/01.cir.0000023042.50192.f4. [DOI] [PubMed] [Google Scholar]
- 31.Baartscheer A, Schumacher CA, Belterman CN, Coronel R, Fiolet JW. [Na+]i and the driving force of the Na+/Ca2+-exchanger in heart failure. Cardiovasc Res. 2003;57:986–95. doi: 10.1016/s0008-6363(02)00848-9. [DOI] [PubMed] [Google Scholar]