

Abstract
The mdxCv3 mouse is a model for Duchenne muscular dystrophy (DMD). DMD is an X-linked disorder with defective expression of the protein dystrophin, and which is associated with a reduced b-wave and has other electroretinogram (ERG) abnormalities. To assess potential causes for the abnormalities, we recorded ERGs from pieces of isolated C57BL/6J and mdxCv3 mouse retinas, including measurements of transretinal and intraretinal potentials. The ERGs from the isolated mdxCv3 retina differ from those of control retinas in that they show reduced b-wave amplitudes and increased b-wave implicit times. Photovoltages obtained by recording across the photoreceptor outer segments of the retinas did not differ from normal, suggesting that the likely causes of the reduced b-wave are localized to the photoreceptor to ON-bipolar synapse. At a concentration of 50 μM, the glutamate analog DL-2-amino-4-phosphonobutyric acid (APB) blocks the b-wave component of the ERG, by binding to sites on the postsynaptic membrane. The On-bipolar cell contribution to the ERG was inferred by extracting the component that was blocked by APB. We found that this component was smaller in amplitude and had longer response latencies in the mdxCv3 mice, but was of similar overall time course. To assess the sensitivity of sites on the postsynaptic membrane to glutamate, the concentration of APB in the media was systematically varied, and the magnitude of blockage of the light response was quantified. We found that the mdxCv3 retina was 5-fold more sensitive to APB than control retinas. The ability of lower concentrations of APB to block the b-wave in mdxCv3 suggests that the ERG abnormalities may reflect alterations in either glutamate release, the glutamate postsynaptic binding sites, or in other proteins that modulate glutamate function in ON-bipolar cells.
Keywords: Isolated mdxCv3 mouse retina, Intraretinal ERG, Normal light transduction, Abnormal depolarizing bipolar cell activity, High sensitivity to APB
Introduction
Duchenne muscular dystrophy (DMD) is an X-linked disorder that is best known as a severe and progressive muscular dystrophy that leads to death by the third decade of life. It is due to defects in the expression of the protein dystrophin (Dp427) (Emery, 1993). The disorder is less well known for its non-muscle manifestations. These are variably expressed, and include such diverse features as cardiomyopathy, gastric dilatation, learning disorders, mental retardation, and defects in retinal electrophysiology resulting in an abnormal electroretinogram (ERG) (Tokarz et al., 1998; Pillers et al., 1999a). Specifically, patients with DMD, or its milder allelic variant Becker muscular dystrophy (BMD), have been shown to have a reduced amplitude b-wave in the scotopic ERG (Pillers et al., 1990; 1993; Cibis et al., 1993).
The DMD gene produces several gene products referred to as isoforms which have varying tissue distribution and which are named according to their size in kilodaltons (Ahn & Kunkel, 1993; Tokarz et al., 1998). Thus, the site of a mutation within the DMD gene affects the phenotype that an individual may manifest. The ERG abnormalities correlate with mutations affecting expression of one such isoform of dystrophin: Dp260 (D'Souza et al., 1995; Ino-ue et al., 1997: Pillers et al., 1999a). Dp260 is predominantly expressed in retina and is localized to the outer plexiform layer (D'Souza et al., 1995), a site typically associated with generation of the ERG b-wave. The mdxCv3 mouse model for DMD has been shown to be a good model for the ERG changes found in DMD/BMD patients (Pillers et al., 1995, 1999b). In response to moderately bright flashes, the scotopic b-wave of this mouse is markedly reduced resulting in a negative ERG. Unlike man, the mouse phenotype includes prolongation of the implicit times of the b-wave and oscillatory potentials.
The mdxCv3 mouse has a disruption of the normal expression of each of the known carboxyl terminus dystrophin isoforms (Cox et al., 1993). As is the case in man, the ERG changes in mouse models correlate with mutations that affect dystrophin isoform Dp260 (Pillers et al., 1999b). The mdxCv3 mouse also has reduced expression of α- and β-dystroglycan (Dalloz et al., 2001). The importance of isoform Dp260 and its interaction with members of the dystrophin-associated glycoprotein complex in the generation of the normal b-wave continues to be an important subject for study.
The amplitude and kinetics of the a-wave are often used as indicators of photoreceptor function. The a-wave data for the mdxCv3 have been compared to that of the control strain C57BL/6J and the early time course fitted to a transduction activation model (Pillers et al., 1999b). This approach suggests that the photoreceptors respond normally and that the reduced b-wave responses are due to changes at the rod to ON-bipolar cell synapse (Pillers et al., 1999b). Whether the defect is presynaptic or postsynaptic could not be determined.
Materials and methods
Pieces of dark-adapted retina from either mdxCv3 or C57BL/6J mice were superfused with an oxygenated Ringer solution at 36–37°C. The mdxCv3 mice were derived from a colony established at OHSU from breeding pairs supplied by Dr. Verne Chapman (now available from the Jackson Laboratories, Bar Harbor, ME, as the mdx3cv strain). The mdxCv3 mouse is a model for Duchenne muscular dystrophy and has a point mutation in intron 65 in a splice acceptor site of the dmd gene. The mutation results in dysfunctional dystrophin transcripts (Cox et al., 1993). The C57BL/6J were derived from a colony at OHSU established using mice from the Jackson Laboratories. Animal experiments conformed to the principles regarding the care and use of animals adopted by the American Physiological Society and the Society for Neuroscience.
Preparation of retinas:
Six- to ten-week-old mice weighing about 30 g were dark adapted for 12 h or more. Using night vision devices (Nitemate NAV-3, Litton Industries, Watertown, CT) and infrared illumination, the animal was euthanized by cervical dislocation. Both eyes were enucleated and an incision was made at the equator of the globe with a razor-blade. The anterior portion of the eye was removed by holding the globe with straight forceps along the outer edge of the cut and successively enlarging the incision with microscissors. The eyecup, hemisected slightly below the ora serrata, was then placed in a dish of Ringer solution at room temperature that had been bubbled with 95% O2 and 5% CO2. The retina was gently detached from the retinal pigment epithelium (RPE) using our earlier method (Green & Kapousta-Bruneau, 1999a). A small piece (approximately 2 × 2 mm) of retina was selected and placed in the recording chamber.
Perfusion system
The piece of the retina was placed receptor side up on a slightly smaller piece of Millipore filter (type SC). The filter and retina were positioned horizontally in the perfusion chamber between two 12-mm-diameter circular pieces of nylon mesh that had been glued to Teflon O-rings with cyanoacrylate adhesive (Crazy Glue, Borden, Columbus, OH). The volume of the fluid in the chamber was 0.5 ml. The fluid entered the open chamber (12-mm diameter) through a small orifice above the retina on one side of the open chamber and flowed over the retina and through the mesh to perfuse both sides of the retina. It was drained away by a small strip of tissue on the opposite side of the chamber. The tissue acted as a wick moving the fluid into a second chamber where it was sucked away by a vacuum pump (Hagen Elite, 801, Taiwan) into an accumulation reservoir. The bathing solution entering the chamber was gravity-fed from bottles above the recording chamber. To avoid bubble formation in the chamber, we found that it was necessary to preheat the perfusate in the bottles to 38–39°C (using aquarium heaters). In addition, using heated Nichrome wire wrapped Teflon tubing which was threaded under the chamber itself, the temperature inside the chamber was maintained at 36–37°C. To do this, a small thermistor probe (YSI, Model 73a, Yellow Springs, OH) was inserted into the fluid in the chamber and connected to an on–off controller (YSI, Model 554, Yellow Springs, OH) attached to a constant current power DC supply (Tektronix, PS282, Beaverton, OR) that heated the Nichrome wire.
The composition of the Ringer was NaCl (110 mM), KCl (5 mM), Na2HPO4 (0.8 mM), NaH2PO4 (0.1 mM), NaHCO3 (30 mM), MgSO4 (1 mM), CaCl2 (1.8 mM), glucose (22 mM), and glutamine (0.5 mM). The perfusate in the bottles was bubbled with 95% O2 and 5% CO2. The pH of the solution at 38oC was between 7.45–7.55 and required no adjustment. The retina was continuously perfused at 3 ml/min. After the tissue was placed in the chamber it took about 30 min to stabilize.
Recordings
Electrical potentials were amplified with a high-gain amplifier (CyberAmp 320, Axon Instruments, San Francisco, CA), using a bandwidth of 0–400 Hz. Responses were digitized and stored using an on-line computer data acquisition system (LabVIEW, National Instruments, Austin, TX). Each record was 2.4 s long and was sampled at a rate of 1000 points/s. Fifty prestimulus points were included with each record. All recordings were performed in a light-tight Faraday cage. Light stimuli were projected onto the retina through a Zeiss operating microscope from an optical stimulator mounted outside the cage. Recordings were made between an Ag–AgCl macroelectrode (Type E202, IVM, Healdsburg, CA) built in the bottom of the chamber and a glass capillary microelectrode filled with physiological saline. The microelectrodes were advanced using microdrives (Kopf Model 650, CA) mounted on an x, y, z manipulator (Narishige, Model MM3). The chamber was mounted on a vertical microjack (Model EL 80, Newport/Klinger, Garden City, NY) onto which horizontal slides had been mounted (Model 16021, Oriel, Stratford, CT). A dial micrometer mounted above the chamber registered the vertical position of the chamber and allowed its height to be varied in a controlled manner. The microelectrode entered the chamber from above and made angles of about 70 deg with respect to the retina. The tip openings were 1.5–5.0 μm in diameter. The resistances of the micropipettes were typically 5–25 M Ω.
Prior to placing the retina in the chamber, the light stimuli were focused on the surface of the fluid in the chamber. The microelectrode was carefully advanced until it touched the fluid surface in the center of the stimulus. The reading on the microdrive position counter at this point defined the location of the plane of focus. The microdrive was then withdrawn several millimeters and the retina was placed in the chamber. Typically there was 1.5–2.0 mm of fluid above the retina and consequently the chamber was raised so that the electrodes touched the fluid at this height. The plane of focus was then approximately at the tips of the rod outer segments. The microelectrode was advanced until it just touched the tips of the rod outer segments. This point was identified by the voltage shifts that occurred when the electrode entered the retina. Transretinal ERGs were recorded between the microelectrodes and an Ag–AgCl reference electrode located under the retina. To record the photovoltages, recordings were made across the rod outer segments. These were measured sequentially by first measuring an intensity series at the tips of the outer segments and then advancing the microelectrode 40 μm and repeating the intensity series. The two sets of records were subtracted from each other to obtain the photovoltages.
Stimulation
The timing of the 40-ms flash was under computer control. Data were collected using a fixed stimulus set in which the intensity increased progressively in 0.5 log steps. Low-intensity flashes were interposed between brighter flashes to assure that there was sufficient time between flashes for the prior sensitivity to be restored. Sixty seconds was generally sufficient for the response amplitude to be unaffected by earlier high-intensity flashes. The measured photon flux on the piece of retina in quanta/μm2/s was converted to quanta/rod/flash using the flash duration of 0.04 s, a quantum efficiency of bleaching of 0.5, a pigment density of 0.3, and a rod cross-sectional area of 2.3 μm2, as described previously (Green & Kapousta-Bruneau, 1999b, Sieving et al., 2001).
Chemicals
The ability of DL2-amino-4-phosphonobutyric acid (APB) to block the b-wave was assessed by adding various concentrations (0.625 to 50 μM) to the perfusate. The APB and all the other chemicals used in these studies were from Sigma (St. Louis, MO).
Results
Reduced and delayed b-wave
We wanted to establish whether the abnormalities seen in anesthetized mdxCv3 mice were also present in retinas isolated from the pigment epithelium and perfused with a rodent Ringer solution. Fig. 1 addresses this question and shows ERGs recorded from isolated pieces of mdxCv3 retina. Panel a shows typical ERGs evoked by a moderately intense flash (log I = −1.5). As previously reported for the ERG of intact and anesthetized mdxCv3 mice (Pillers et al., 1995, 1999b), the ERG from the isolated retina is predominantly negative. The b-wave amplitudes in panel b were measured from the trough of the a-wave to the peak of the b-wave. As reported for the intact animals, the ERGs from isolated mdxCv3 retinas had reduced scotopic b-wave amplitudes. To quantify these data, the function V/Vmax = I/(I + σ) was fit to the lower range of intensities (log I = 5.5−3.5). For the normal mice, the data were fit well by values Vmax and log σ of 0.222 and −4.79. The fit of this function was also good for mdxCv3 mice and values of Vmax and log σ were 0.139 and −4.22. Thus, relative to the normal controls, the amplitude of the mdxCv3 b-wave was reduced by about 40%, and the response–intensity curve was shifted to the right by half a log unit. Panel c shows that the b-wave implicit times (the time from the start of the flash to the peak of the b-wave) were increased by about 20 ms at all intensities in the mdxCv3 animals. These differences are very similar to those previously reported for intact mice (Pillers et al., 1995, 1999b).
Fig. 1.
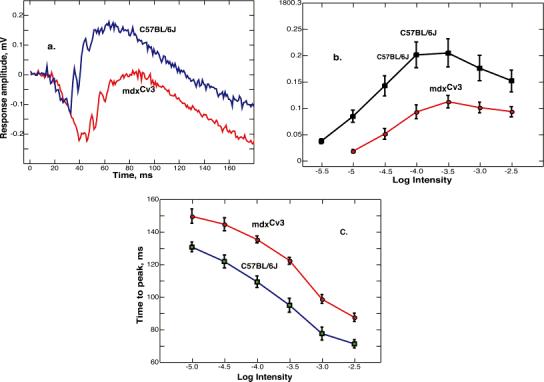
(a) Representative ERG responses to bright flashes of light. The tracings are single responses from a mdxCv3 and a C57BL/6J mouse (log I = -1.5). This intensity corresponds to a quantal flux of 35,000 quanta/ μm2/s incident on the piece of retina (see Materials and methods for other details). (b) B-wave intensity-response functions for mdxCv3 (n = 7) and C57BL/6J (n = 7) mice. The points are the averages ± the standard error of the mean. An intensity of -5.5 corresponds to about 0.1 R*/rod/flash. The b-wave intensity functions in mdxCv3 mice were 40 % smaller and shifted to the right on the intensity axis by about 0.5 log units. (c) The time from the start of the flash to the peak of the b-wave (implicit time) as a function of intensity for mdxCv3 (n = 7) and C57BL/6J (n = 7) mice. The points are the averages ± the standard error of the mean. There were significant differences between control and mdxCv3 mice in b-wave amplitudes and implicit times at all intensities.
Normal phototransduction
The reduced b-wave could result from either an abnormality in the mdxCv3 mouse rod outer segment transduction apparatus or in an abnormality at the rod-to-bipolar synapse. The isolated retina preparation allows this question to be directly addressed by measuring the rod outer segment photocurrents. That is, with electrodes across the outer segments one can record the changes in the extracellular potentials that result from reductions in the dark current evoked by light (Penn & Hagins, 1972; Green & Kapousta-Bruneau, 1999b; Sieving et al., 2001). Fig. 2 shows the results of photovoltage recordings from a group of four mdxCv3 (panel a) and four C57BL/6J (panel b) mice. For intensities from −4.0 to −2.0 both sets of responses grow in amplitude as intensity increases. At the highest two intensities (log I = −2.0 and −1.5), the response becomes longer in duration but not larger in amplitude. The brightest intensity corresponds to about 800 R*/rod/flash (see Materials and methods for further details of how the stimulus was calibrated). The virtually identical photoresponses from the mdxCv3 and normal mice provide strong evidence for phototransduction being normal in the mdxCv3 mouse.
Fig. 2.
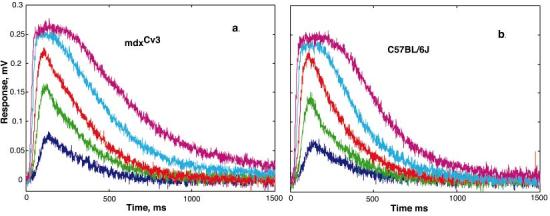
Photovoltages from mdxCv3 (n = 4) and C57BL/6J (n = 4) mice. The curves are the averages of a set of responses that were obtained at a range of intensities (log I = -3.5 to -1.5). An intensity of -1.4 log corresponds to a bleaching strength estimated to be about 1000 R*/rod/flash. In both mdxCv3 (panel a) and C57BL/6J (panel b), a stimulus intensity of about 25 R*/rod/flash produced a half-saturated response.
Blocking depolarizing bipolar cell activity
The b-wave has been linked to responses from depolarizing (ON) bipolar cells (Stockton & Slaughter, 1989; Gurevich & Slaughter, 1993; Tian & Slaughter, 1995; Robson & Frishman, 1995). Blocking ON- bipolar cells pharmacologically (given minimal contributions from more proximal neurons) allows one to extract the contributions of ON-bipolar cells to the ERG (Knapp & Schiller, 1984; Frishman et al., 1992; Gurevich & Slaughter, 1993; Robson & Frishman, 1995, 1996; Shiells & Falk, 1999; Green & Kapousta-Bruneau, 1999b; Jamison et al., 2001; McGillem & Dacheux, 2001; Kang Derwent & Linsenmeier, 2001). APB acts at the metabotropic glutamate receptors on ON-bipolar cells and blocks the b-wave.
The component of the ERG that is blocked by APB, which we refer to as P2, can be extracted from the total ERG by subtracting the ERG obtained after administering 50 μM of APB from the ERG recorded before administering APB. Figs. 3 (b & d) show that over a range of intensities P2 responses in mdxCv3 mice (whose b-wave is abnormal) have a similar time course to those of normal mice but are smaller. In Figs. 3 (a & c) the P2 responses are displayed on a shorter time scale. Here one can see that the onsets of the P2 responses in mdxCv3 are delayed by about 20 ms as compared to control mice. It is worth noting that if the normal P2 responses are delayed and scaled down in amplitude, they are a reasonably close fit to the mdxCv3 P2 responses. Thus, the kinetics of activation and recovery in the ON-bipolar cells in the mdxCv3 retina appear to be relatively normal. An increase in the synaptic delay without a change in the kinetics is consistent with a defect in synaptic transmission. This defect could be either presynaptic or postsynaptic.
Fig. 3.
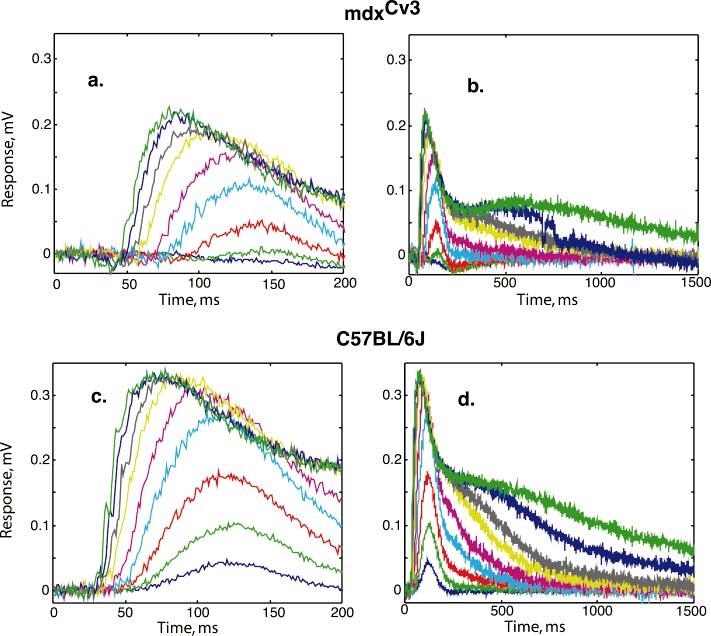
P2, the component blocked by APB, as a function of nine stimulus intensities (log I = -5.5 to -1.5). Panels a and b show the average P2's for mdxCv3 mice (n = 5) on two different time scales. Panels c and d show P2 for C57BL/6J mice (n = 5) . In panels a and c, the P2(t) responses are shown on a shorter time scale and in b and d on a longer time scale.
Figs. 4 (a & b) show another way in which the mdxCv3 is different from normal. At low intensities, there are negative response components to the P2 while the P2 from the C57BL/6J mice is always positive. Thus, in the mdxCv3, there appears to be a negative component that is blocked by APB. This postsynaptic negative component looks similar to and may have the same generator as the negative scotopic threshold response (STR) in the mouse (Saszik et al., 2002). It may also contribute to producing the predominantly negative ERG in the muscular dystrophy mouse. Part c of Fig. 4 plots the amplitude of P2 as a function of light intensity.
Fig. 4.
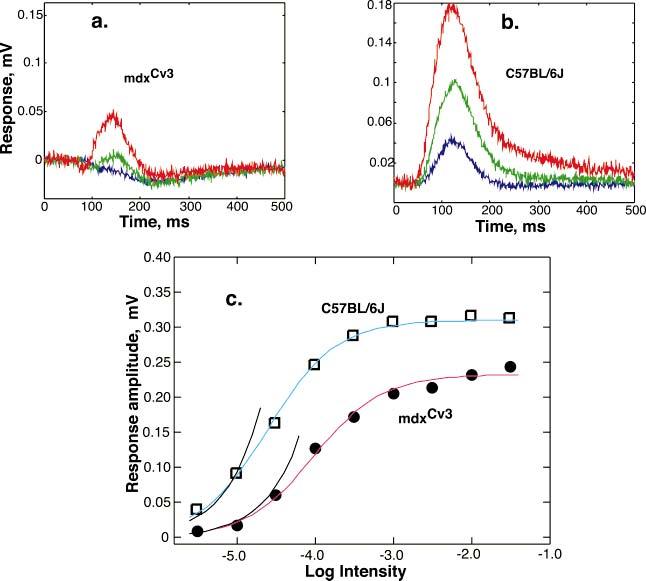
Panels a and b show P2(t) responses at low intensities (log I = -5.5 to 4.5) for mdxCv3 (n = 5) and C57BL/6J (n = 5) mice, respectively. These stimulus intensities correspond to R*/rod/flash of 0.1 to 1.0 . At the lowest intensities the amplitudes of the responses grew linearly with light intensity. Panel c shows the P2 intensity-response functions for mdxCv3 (n = 5) and C57BL/6J (n = 5) mice. The curves fit to the points are of the form V/Vmax = I/(I σ) with log sigma = [-4.01, -4.6 ] and Vmax = [0.2324, 0.31], respectively. The curved segment to the left of each curve shows a linear growth with flash intensity.
Sensitivity to APB
The ability of APB to block the b-wave was assessed by adding various concentrations of APB (0.625-5 μM) to the perfusate. Figs. 5 (a & b) show sample results from a mdxCv3 mouse and a normal mouse retina. We find that the mdxCv3 retina is more sensitive to APB than control retinas. For example, in Fig. 5a concentrations from 1.25 μM to 5 μM of APB were variously effective in blocking the b-wave in the mdxCv3 mouse. In contrast, the C57Bl/6J mouse in Fig. 5b required 5 μM to produce a measurable effect. A concentration of 10 μM was required to have the same effect in a normal mouse as a 1.25 μM concentration in the mdxCv3. Fig. 6 shows the results from 14 mice in which various concentrations of APB were applied to the retinas. Thus, we find that the b-wave is blocked by concentrations of APB that are five times lower than in controls.
Fig. 5.
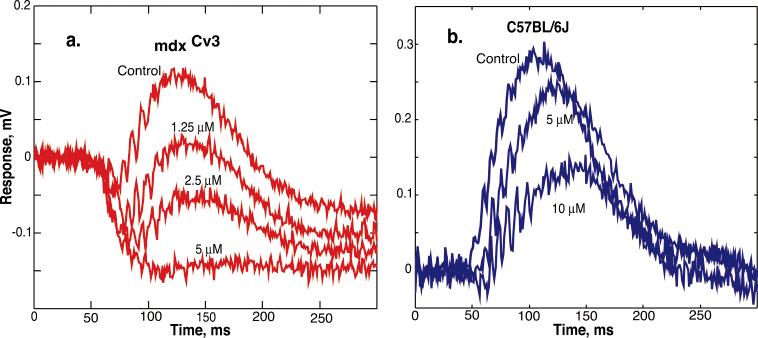
Representative ERG responses with various concentrations of APB in the perfusate. The tracings in panela are from a mdxCv3 mouse. The tracings in panel b are from a C57BL/6J mouse. The stimulus intensity in both was 3 R*/rod/flash.
Fig. 6.
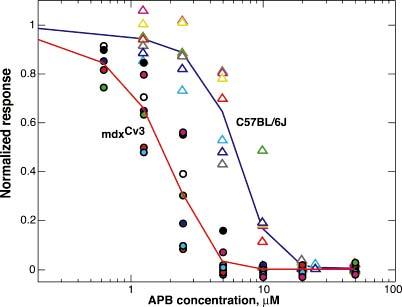
APB dose–response functions. The points plot the normalized b-wave amplitudes as a function of APB concentration for a flash of an intensity of 3 R*/rod/flash. The triangles are individual b-wave amplitudes from seven C57BL/6 J mice on which measurements were made at a variety of APB concentrations. A smooth curve is drawn through the averages. The mean and standard deviation of the response amplitudes before APB was administered were 165 ± 78 μV. The circles are individual b-wave amplitudes from seven mdxCv3 mice. The mean and standard deviation of the amplitudes before APB were 114 ± 45 μV. The mdxCv3 mice are more sensitive to APB.
Conclusions
While recording have been made from single cells in the isolated mouse retina (Balkema et al., 1981; Suzuki & Pinto, 1986; Stone & Pinto, 1993), we believe this to be the first report of ERG recordings which are similar in form to those recorded from the mouse retina in vivo (Fig. 1a). The isolated retina preparation offers several advantages over ERG recordings from the intact mouse. Electrodes could be inserted into the retina so that intraretinal records could be obtained (Fig. 2) and drugs can be applied in a controlled manner (Figs. 3, 4, 5, & 6).
The data in Fig. 2 provides evidence that the rod photocurrents in the mdxCv3 are the same as those of the normal mice. An earlier study in the intact mouse in which the leading edge of the a-wave was analyzed suggested that this might be the case (Pillers et al., 1999b). The responses in Fig. 2 provide strong evidence that the photoreceptor transduction apparatus is normal in mdxCv3 retinas. Consequently, the reduced b-wave (Fig. 1) and reduced P2 amplitudes (Figs. 3 & 4) must be due to an abnormality in function beyond the rod outer segments. This defect could be either in the presynaptic transmitter release mechanism or in the postsynaptic response to the changes in glutamate release.
The tracings shown in Figs. 3a and 3c, which we call P2, show the transretinal ERG components that are blocked by APB. Our previous studies have shown that the implicit time of the b-wave is delayed in the mdxCv3 mouse. The P2 tracings allow the early part of the component that is secondary to depolarizing bipolar cell activity to be examined. It takes 20 ms longer for the mdxCv3 responses to rise out of the noise of recording and yet the rising edge of the response to the same intensity is not markedly different in shape. Since the photoreceptor delay in mdxCv3 is not different from wild type (Pillers et al., 1999b and Fig. 2), we conclude that there is an increase in the synaptic delay at the rod-to-rod bipolar synapse.
Synaptic delays in the central nervous system (CNS) are typically of the order of several milliseconds (Sabatini & Regehr, 1999). This is not the case for the rod-to-rod bipolar synapse. Our studies on the isolated retinas of normal rats have shown that, over a wide range of intensities, P2 response latencies are 19 ms longer than those of photovoltage response latencies (Green & Kapousta-Bruneau, 1999b). Previous studies which have compared the light responses of depolarizing and hyperpolarizing bipolar cells (Ashmore & Falk, 1980; Kim & Miller, 1993) have found significantly longer response latency in depolarizing bipolar cells.
Shiells and Falk (1994) have measured the responses of hyperpolarizing and depolarizing bipolar cells to step increases in glutamate. They have shown an 11-ms delay in synaptic transfer at the rod-to-depolarizing bipolar synapse in the dog fish retina and no measurable delay in hyperpolarizing bipolar cells. This long delay is presumably a reflection of the processes intervening between the binding of transmitter to the postsynaptic membrane and the changes in ionic conductance at the plasma membrane of the rod bipolar cell.
The increased delay in mdxCv3 mice could be mediated presynaptically or postsynaptically. Synaptic delays of as long as 10 ms have been found under low [Ca2+]i at the calyx-type synapse in the rat (Bollmann et al., 2000). If the [Ca2+]i concentration in the rod terminals of mdxCv3 is abnormally low, this might account for the increase. The other possibility is that there are differences in the activation kinetics of the processes intervening between the binding of glutamate and the closing of cation channels in mdxCv3 rod bipolar cells.
APB selectively blocks the responses of depolarizing bipolar cells by binding to sites on the postsynaptic membrane. The sensitivity of the postsynaptic membrane to blockade by APB is determined by the concentration of indigenous glutamate, the affinity of the postsynaptic binding site to APB, and by the rates at which the APB comes off the sites. Thus, the increased sensitivity in mdxCv3 might be due to changes in any of the above.
Dystrophin has been reported to be localized in small extensions of rod and cone photoreceptor terminals in the mouse retina (Blank et al., 1999). Whether it is also localized in the postsynaptic cells is unclear. Evidence for a postsynaptic localization has only been detected in rabbit retina (Blank et al., 1999). However, it should be noted that the use of C-terminal antibodies to detect dystrophin by immunocytochemistry may detect several isoforms of dystrophin, and not specifically Dp260, which is the isoform associated with the b-wave abnormality (Pillers et al., 1999a;b). Studies localizing Dp260 do suggest that it is exclusively localized in photoreceptor cells (Claudepierre et al., 1999).
In the above list only the rate of glutamate release in the dark is controlled presynaptically. To address the question of how the rate of dark glutamate release affects APB sensitivity, we have used a model for the kinetics of enzymatic reactions in the presence of a competitive antagonist (Cheng & Prusoff, 1973). The form of the relationship between response, R, and glutamate concentration, [G], that the model yields is a dose–response curve:
| (1) |
where [A] is the antagonist concentration, Ka is a constant that depends on the relative rates at which glutamate binds and unbinds from the postsynaptic sites, and Kb is a constant that depends of the relative rates at which the antagonist binds, and unbinds from the postsynaptic sites. IC50, the concentration required for obtaining 50% of the maximum effect, is
| (2) |
The magnitude of the light evoked response is determined by the size of the postsynaptic change that results from a light-induced reduction in the dark glutamate release (i.e., the slope at the operating point). In our experiments on the dark-adapted retina, the sensitivity to the antagonist depends upon where on the glutamate dose–response curve one is operating. Within the conceptual framework above, the effect of having APB in the bath is to increase IC50. On linear response versus logarithmic glutamate coordinates, APB causes the dose–response curve to shift to the right. If the dark release of glutamate is small with respect to Ka, the light sensitivity will be reduced to half when the APB concentration is equal to Kb. On the other hand, if the dark release of glutamate is high enough to saturate the postsynaptic machinery in the wild-type retina, the above increase in APB will have little effect in reducing light sensitivity.
The model predicts that five times higher concentrations of APB will be required to reduce the light sensitivity by half, if 67% of sites on the postsynaptic membrane are closed in the dark in wild-type retina and 35 % are closed in the dark in the mdxCv3 retina. Thus, a reduction in dark release in mdxCv3 could move the operating point to a more linear portion of the curve. Fewer sites on the postsynaptic membrane would be occupied by glutamate and this would produce greater sensitivity to the agonist. It also could explain the reduction in the maximum amplitude of the response.
Saturation of the postsynaptic membrane could cause the light response at low light levels to be a nonlinear function of light intensity. Consistent with this, a recent report has suggested that rod–rod bipolar signal transfer in the mouse at very low light levels is nonlinear (Field & Rieke, 2002). The evidence for this comes from the observation that in mouse rod bipolar cells the responses at low levels increased with flash strength supralinearly (Field & Rieke, 2002). This does not seem to be the case with our P2 responses or those of Saszik et al. (2002) who made measurements for stimuli that ranged two orders of magnitude lower in intensity on intact C57BL/6 J mice and have shown that P2 rises linearly if measured near its peak (110 ms after a brief flash) up to saturation around 1 R*/rod/flash. Over the common range of intensities, their results and ours agree remarkably well.
Robson et al. (2003) have analyzed the rising portion of the P2 responses from the intact C57BL/6 J mice and found a change in waveform that provides evidence for a threshold nonlinearity. Their results are consistent with a threshold that is 25–30% of the amplitude of the single photon response. It is worth noting that the low intensity P2 records shown in Fig. 3, when plotted on double logarithmic coordinates (not shown), also exhibit this change in waveform at early times.
In summary, we find an abnormality in synaptic transmission from photoreceptors to depolarizing bipolar cells in the mdxCv3 retina that is associated with an increased synaptic delay and hypersensitivity to APB. The presence of dystrophin in the rod membrane suggests that the most likely underlying cause would be a presynaptic defect. Low [Ca2+]i, for example, might lead to lower dark release and slower light-evoked glutamate release. The lower dark release might eliminate saturation at the postsynaptic membrane and cause an increase in APB sensitivity.
On the other hand, a postsynaptic cause cannot be ruled out. The hypersensitivity to APB would be explained if dystrophin were playing a role in establishing the specificity and selectivity of the metabotropic glutamate receptor on the postsynaptic membrane of the rod bipolar cells. To account for the increased delay, dystrophin would have to be linked to the events intervening between the unbinding of glutamate from the postsynaptic membrane and opening of cationic channels in the depolarizing rod bipolar cells.
Acknowledgments
Haijun Guo assisted with the collection of the data shown in Fig. 2. We would like to thank Theodore Cohn, Catherine Morgans, Richard Neubig, and W. Rowland Taylor for helpful discussions. This work was supported by National Institute of Health Grants EY 10084 (D.M.P.), EY 07003-CORE, the Arline Silfberg Trust (D.M.P.), and a Research to Prevent Blindness Senior Investigator Award (D.G.G.).
References
- AHN AH, KUNKEL LM. The structural and functional diversity of dystrophin. Nature Genetics. 1993;3:283–291. doi: 10.1038/ng0493-283. [DOI] [PubMed] [Google Scholar]
- ASHMORE JF, FALK G. Responses of rod bipolar cells in the dark adapted retina of the dogfish, Scyliorhinus canicula. Journal of Physiology. 1980;300:115–150. doi: 10.1113/jphysiol.1980.sp013155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BALKEMA GW, PINTO LH, DRAGER UC, VANABLE JW. Characterization of abnormalities in the visual system of the mutant mouse pearl. Journal of Neuroscience. 1981;1:1320–1329. doi: 10.1523/JNEUROSCI.01-11-01320.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BLANK M, KOULEN P, BLAKE DJ, KROGER S. Dystrophin and beta-dystroglycan in photoreceptor terminals from normal and mdx3Cv mouse retinae. European Journal of Neuroscience. 1999;11:2121–2133. doi: 10.1046/j.1460-9568.1999.00636.x. [DOI] [PubMed] [Google Scholar]
- BOLLMANN JH, SAKMANN B, BORST JG. Calcium sensitivity of glutamate release in a calyx-type terminal. Science. 2000;289:953–957. doi: 10.1126/science.289.5481.953. [DOI] [PubMed] [Google Scholar]
- CHENG Y, PRUSOFF WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochemical Pharmacology. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- CIBIS GW, FITZGERALD KM, HARRIS DJ, ROTHBERG PG, RUPANI M. The effects of dystrophin gene mutations on the ERG in mice and humans. Investigative Ophthalmology and Visual Science. 1993;34:3646–3652. [PubMed] [Google Scholar]
- CLAUDEPIERRE T, RODIUS F, FRASSON M, FONTAINE V, PICAUD S, DREYFUS H, MORNET D, RENDON A. Differential distribution of dystrophins in rat retina. Investigative Ophthalmology and Visual Science. 1999;40:1520–1529. [PubMed] [Google Scholar]
- COX GA, PHELPS SF, CHAPMAN VM, CHAMBERLAIN JS. New mdx mutation disrupts expression of muscle and nonmuscle isoforms of dystrophin. Nature Genetics. 1993;4:87–93. doi: 10.1038/ng0593-87. [DOI] [PubMed] [Google Scholar]
- DALLOZ C, CLAUDEPIERRE T, RODIUS F, MORNET D, SAHEL J, RENDON A. Differential distribution of the members of the dystrophin glycoprotein complex in mouse retina: Effect of the mdx(3Cv) mutation. Molecular and Cellular Neuroscience. 2001;17:908–920. doi: 10.1006/mcne.2001.0978. [DOI] [PubMed] [Google Scholar]
- D'SOUZA VN, NGUYEN TM, MORRIS GE, KARGES W, PILLERS DM, RAY PN. A novel dystrophin isoform is required for normal retinal electrophysiology. Human Molecular Genetics. 1995;4:837–842. doi: 10.1093/hmg/4.5.837. [DOI] [PubMed] [Google Scholar]
- EMERY AEH. Duchenne Muscular Dystrophy. 2nd Oxford University Press; Oxford, UK: 1993. [Google Scholar]
- FIELD GD, RIEKE F. Nonlinear signal transfer from mouse rods to bipolar cells and implications for visual sensitivity. Neuron. 2002;34:773–785. doi: 10.1016/s0896-6273(02)00700-6. [DOI] [PubMed] [Google Scholar]
- FRISHMAN LJ, YAMAMOTO F, BOGUCKA J, STEINBERG RH. Light-evoked changes in [K+]o in proximal portion of light-adapted cat retina. Journal of Neurophysiology. 1992;67:1201–1212. doi: 10.1152/jn.1992.67.5.1201. [DOI] [PubMed] [Google Scholar]
- GREEN DG, KAPOUSTA-BRUNEAU NV. Electrophysiological properties of a new isolated retina preparation. Vision Research. 1999a;39:2165–2177. doi: 10.1016/s0042-6989(98)00318-6. [DOI] [PubMed] [Google Scholar]
- GREEN DG, KAPOUSTA-BRUNEAU NV. A dissection of the ERG from the isolated rat retina with microelectrodes and drugs. Visual Neuroscience. 1999b;16:727–741. doi: 10.1017/s0952523899164125. [DOI] [PubMed] [Google Scholar]
- GUREVICH L, SLAUGHTER MM. Comparison of the waveforms of the ON bipolar neuron and the b-wave of the electroretinogram. Vision Research. 1993;33:2431–2435. doi: 10.1016/0042-6989(93)90122-d. [DOI] [PubMed] [Google Scholar]
- INO-UE M, HONDA S, NISHIO H, MATSUO M, NAKAMURA H, YAMAMOTO M. Genotype and electroretinal heterogeneity in Duchenne muscular dystrophy. Experimental Eye Research. 1997;65:861–864. doi: 10.1006/exer.1997.0389. [DOI] [PubMed] [Google Scholar]
- JAMISON JA, BUSH RA, LEI B, SIEVING PA. Characterization of the rod photoresponse isolated from the dark-adapted primate ERG. Visual Neuroscience. 2001;18:445–455. doi: 10.1017/s0952523801183112. [DOI] [PubMed] [Google Scholar]
- KANG DERWENT JJ, LINSENMEIER RA. Intraretinal analysis of the a-wave of the electroretinogram (ERG) in dark-adapted intact cat retina. Visual Neuroscience. 2001;18:353–363. doi: 10.1017/s0952523801183021. [DOI] [PubMed] [Google Scholar]
- KIM HG, MILLER RF. Properties of synaptic transmission from photoreceptors to bipolar cells in the mudpuppy retina. Journal of Neurophysiology. 1993;69:352–360. doi: 10.1152/jn.1993.69.2.352. [DOI] [PubMed] [Google Scholar]
- KNAPP AG, SCHILLER PH. The contribution of on-bipolar cells to the electroretinogram of rabbits and monkeys. A study using 2-amino-4-phosphonobutyrate (APB) Vision Research. 1984;24:1841–1846. doi: 10.1016/0042-6989(84)90016-6. [DOI] [PubMed] [Google Scholar]
- MCGILLEM GS, DACHEUX RF. Rabbit cone bipolar cells: Correlation of their morphologies with whole-cell recordings. Visual Neuroscience. 2001;18:675–685. doi: 10.1017/s0952523801185019. [DOI] [PubMed] [Google Scholar]
- PENN RD, HAGINS WA. Kinetics of the photocurrent of retinal rods. Biophysical Journal. 1972;12:1073–1094. doi: 10.1016/S0006-3495(72)86145-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PILLERS DM, BULMAN DE, WELEBER RG, SIGESMUND DA, MUSARELLA MA, POWELL BR, MURPHEY WH, WESTALL C, PANTON C, BECKER LE. Dystrophin expression in the human retina is required for normal function as defined by electroretinography. Nature Genetics. 1993;4:82–86. doi: 10.1038/ng0593-82. [DOI] [PubMed] [Google Scholar]
- PILLERS DM, FITZGERALD KM, DUNCAN NM, RASH SM, WHITE RA, DWINNELL SJ, POWELL BR, SCHNUR RE, RAY PN, CIBIS GW, WELEBER RG. Duchenne/Becker muscular dystrophy: Correlation of phenotype by electroretinography with sites of dystrophin mutations. Human Genetics. 1999a;105:2–9. doi: 10.1007/s004399900111. [DOI] [PubMed] [Google Scholar]
- PILLERS DM, WELEBER RG, GREEN DG, RASH SM, DALLY GY, HOWARD PL, POWERS MR, HOOD DC, CHAPMAN VM, RAY PN, WOODWARD WR. Effects of dystrophin isoforms on signal transduction through neural retina: Genotype-phenotype analysis of duchenne muscular dystrophy mouse mutants. Molecular Genetics and Metabolism. 1999b;66:100–110. doi: 10.1006/mgme.1998.2784. [DOI] [PubMed] [Google Scholar]
- PILLERS DM, WELEBER RG, POWELL BR, HANNA CE, MAGENIS RE, BUIST NR. Åland Island eye disease (Forsius-Eriksson ocular albinism) and an Xp21 deletion in a patient with Duchenne muscular dystrophy, glycerol kinase deficiency, and congenital adrenal hypoplasia. American Journal of Medical Genetics. 1990;36:23–28. doi: 10.1002/ajmg.1320360106. [DOI] [PubMed] [Google Scholar]
- PILLERS DM, WELEBER RG, WOODWARD WR, GREEN DG, CHAPMAN VM, RAY PN. mdxCv3 mouse is a model for electroretinography of Duchenne/Becker muscular dystrophy. Investigative Ophthalmology and Visual Science. 1995;36:462–466. [PubMed] [Google Scholar]
- ROBSON JG, FRISHMAN LJ. Response linearity and kinetics of the cat retina: The bipolar cell component of the dark adapted electroretinogram. Visual Neuroscience. 1995;12:837–850. doi: 10.1017/s0952523800009408. [DOI] [PubMed] [Google Scholar]
- ROBSON JG, FRISHMAN LJ. Photoreceptor and bipolar cell contributions to the cat electroretinogram: A kinetic model for the early part of the flash response. Journal of the Optical Society of America A—Optic and Image Science. 1996;13:613–622. doi: 10.1364/josaa.13.000613. [DOI] [PubMed] [Google Scholar]
- ROBSON JG, MAEDA H, SASZIK SM, FRISHMAN LJ. In vivo studies of signaling in rod pathways of the mouse using the electroretinogram. Vision Research. 2004 doi: 10.1016/j.visres.2004.09.002. In press. [DOI] [PubMed] [Google Scholar]
- SABATINI BL, REGEHR WG. Timing of synaptic transmission. Annual Review Physiology. 1999;61:521–542. doi: 10.1146/annurev.physiol.61.1.521. [DOI] [PubMed] [Google Scholar]
- SASZIK SM, ROBSON JG, FRISHMAN LJ. The scotopic threshold response of the dark-adapted electroretinogram of the mouse. Journal of Physiology. 2002;543:899–916. doi: 10.1113/jphysiol.2002.019703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIELLS RA, FALK G. Responses of rod bipolar cells isolated from dogfish retinal slices to concentration-jumps of glutamate. Visual Neuroscience. 1994;11:1175–1183. doi: 10.1017/s0952523800006970. [DOI] [PubMed] [Google Scholar]
- SHIELLS RA, FALK G. Contribution of rod, on-bipolar, and horizontal cell light responses to the ERG of dogfish retina. Visual Neuroscience. 1999;16:503–511. doi: 10.1017/s0952523899163119. [DOI] [PubMed] [Google Scholar]
- SIEVING PA, FOWLER ML, BUSH RA, MACHIDA S, CALVERT PD, GREEN DG, MAKINO CL, MCHENRY CL. Constitutive “light” adaptation in rods from G90D rhodopsin: A mechanism for human congenital nightblindness without rod cell loss. Journal of Neuroscience. 2001;21:5449–5460. doi: 10.1523/JNEUROSCI.21-15-05449.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STOCKTON RA, SLAUGHTER MM. B-wave of the electroretinogram: A reflection of ON bipolar cell activity. Journal of General Physiology. 1989;93:101–122. doi: 10.1085/jgp.93.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STONE C, PINTO LH. Response properties of ganglion cells in the isolated mouse retina. Visual Neuroscience. 1993;10:31–39. doi: 10.1017/s0952523800003205. [DOI] [PubMed] [Google Scholar]
- SUZUKI H, PINTO LH. Response properties of horizontal cells in the isolated retina of wild-type and pearl mutant mice. Journal of Neuroscience. 1986;6:1122–1128. doi: 10.1523/JNEUROSCI.06-04-01122.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TIAN N, SLAUGHTER MM. Correlation of dynamic responses in the ON bipolar neuron and the b-wave of electroretinogram. Vision Research. 1995;35:1359–1364. doi: 10.1016/0042-6989(95)98715-l. [DOI] [PubMed] [Google Scholar]
- TOKARZ SA, DUNCAN NM, RASH SM, SADEGHI A, DEWAN AK, PILLERS DM. Redefinition of dystrophin isoform distribution in mouse tissue by RT-PCR implies role in nonmuscle manifestations of Duchenne muscular dystrophy. Molecular Genetics and Metabolism. 1998;65:272–281. doi: 10.1006/mgme.1998.2763. [DOI] [PubMed] [Google Scholar]