

Abstract
Signals generated by the extracellular matrix (ECM) promote cell survival. We have shown that chondrocytes detached from their native ECM and plated without serum at low density on poly-l-lysine undergo significant cell death that is associated with the production of reactive oxygen species (ROS). No cell death or ROS production was observed when cells were plated on fibronectin under the same conditions. Cell death on poly-l-lysine could be completely inhibited with the addition of either antioxidants or inhibitors of specific protein kinase C (PKC) isoforms including PKC-βI. PKC-βI was noted to translocate from the cytosol to the particulate membrane after plating on poly-l-lysine, and this translocation was inhibited by the addition of an antioxidant. Time-course analyses implicated endogenous ROS production as a secondary messenger leading to PKC-βI activation and subsequent chondrocyte cell death. Cell survival on poly-l-lysine was significantly improved in the presence of oligomycin or DIDS, suggesting that ROS production occurred via complex V of the electron transport chain of the mitochondria and that ROS were released to the cytosol via voltage-dependent anion channels. Together, these results represent a novel mechanism by which ROS can initiate cell death through the activation of PKC-βI.
Keywords: articular cartilage, osteoarthritis, cell signaling, fibronectin
cell contact with the extracellular matrix (ECM) promotes survival in many cell types, but the exact mechanism is not completely understood. Studies have shown apoptotic cell death in epithelial (9) and endothelial (27) cells after detachment from their native ECM. The cell death that occurs when cells lose matrix contact has been termed “anoikis” (Greek for “homelessness”) (9) and is thought to safeguard an organism from the growth of cells at sites where they do not belong, the latter being a hallmark of malignancy (10). Matrix survival signals can be generated through binding of ECM proteins to members of the integrin family of cell adhesion receptors, including the α5β1 fibronectin receptor (46). In most cell types, matrix signaling from integrins works in concert with signals generated by growth factors to promote survival signaling through the phosphatidylinositol (PI) 3-kinase/Akt pathway, although in some cell types MAPK pathways also are required (11).
Articular cartilage is a unique tissue that contains one cell type, the chondrocyte, which is surrounded by a very abundant ECM that provides the tissue with viscoelastic properties necessary for maintenance of normal joint function (31). Chondrocytes are responsible for production and maintenance of the cartilage over the lifetime of an individual. A better understanding of mechanisms responsible for regulating chondrocyte survival is important, especially because undifferentiated precursor cells are not locally available to replace cells lost by cell death and because increased cell death is noted in arthritic cartilage (reviewed in Refs. 1, 19).
Unlike epithelial and endothelial cells, which die when they are detached from their matrix, chondrocytes readily survive in suspension culture in the absence of exogenous growth factors provided the cell density is sufficient (3, 15, 24). Chondrocytes appear to receive survival signals from both the ECM as well as from autocrine production of growth factors, including insulin-like growth factor-I (IGF-I). In previous work, we have noted significant chondrocyte death in vitro after inhibition of the α5β1-integrin by using integrin blocking antibodies (36) or by inhibition of IGF-I signaling with antibodies to the IGF-I receptor (24). In vivo, significant apoptosis has been observed in chondrocytes in transgenic mice lacking collagen II (43), the principal component of articular cartilage, and reduced cellularity was noted in the cartilage of α1-integrin knockout mice (45).
The objective of the present study was to further investigate the cell signaling mechanisms that regulate human articular chondrocyte survival, specifically pathways potentially relevant to growth factor and matrix survival signaling. For these studies, chondrocytes were tested in a cell culture system that in previous work (36) was found to modulate chondrocyte survival in a manner that depended on growth factor and matrix survival signals. In that study, ~60% of chondrocytes removed from their native ECM by proteolytic matrix digestion died in an overnight culture when plated at low density (to reduce autocrine growth factor signaling) in serum-free conditions on poly-l-lysine. Attachment to poly-l-lysine would not provide the necessary matrix survival signals to a chondrocyte because this attachment is charge mediated rather than receptor mediated, like the attachment to fibronectin. We considered this a potential model for chondrocyte anoikis because anoikis includes death that occurs after cell attachment to a matrix that is distinct from the native ECM of the respective cell (10). In support of this model, the cells that did not die on poly-l-lysine (40% of total cells) were found to be producing fibronectin and died with inhibition of fibronectin binding to the α5β1-integrin (36).
In the present study, a large number of signaling inhibitors were screened for their ability to modulate survival of chondrocytes under similar culture conditions. Interestingly, various antioxidants were sufficient to increase survival to close to 100% on poly-l-lysine, as were a combination of specific protein kinase C (PKC) inhibitors. Further experiments were performed indicating that reactive oxygen species (ROS)-dependent activation of PKC-βI resulted in chondrocyte death on poly-l-lysine, whereas PKC-δ was required for promotion of survival on poly-l-lysine or fibronectin.
MATERIALS AND METHODS
Materials
Cell culture medium, antibiotics, fibronectin, and FBS were obtained from GIBCO-BRL (Gaithersburg, MD). Pronase was obtained from Calbiochem (San Diego, CA), and collagenase-P was obtained from Boehringer-Mannheim. Poly-l-lysine (100–150 kDa) and 4,5-dihydroxy-1,3-benzene-disulfonic acid (Tiron) were purchased from Sigma (St. Louis, MO). The various inhibitors listed in Tables 1 and 2 were purchased from Sigma, Calbiochem, and Biomol (Plymouth Meeting, PA). PKC antibodies were obtained from Transduction Laboratories (BD Biosciences, San Jose, CA).
Table 1.
Effects of ROS and mitochondrial specific pharmacological inhibitors on chondrocyte survival when cells were plated on either poly-l-lysine or fibronectin
| Compound | Function* | Effect on Poly- l-Lysine | Effect on Fibronectin |
|---|---|---|---|
| Deferoxamine | Chelates iron | No effect | No effect |
| DPI | Flavin-dependent enzymes, NADPH oxidase | No effect | No effect |
| Oxypurinol | Xanthine oxidase | No effect | No effect |
| 3-Amino-1,2,4-triazole | Catalase | No effect | No effect |
| l-NIL | iNOS | No effect | No effect |
| l-NMMA | cNOS, eNOS, iNOS | No effect | No effect |
| Ketoconazole | Cytochrome P-450, 5-lipoxygenase | Toxic (≥10 μM) | Toxic (≥100 μM) |
| ETYA | 5-, 12-, 15-lipoxygenase | Toxic (≥10 μM) | Toxic (≥100 μM) |
| Indomethacin | COX-1, COX-2 | No effect | No effect |
| Bongkrekic acid | Mitochondrial permeability pore | No effect | No effect |
| Ruthenium red | Mitochondrial Ca2+ channels | Toxic (≥2.5 μM) | Toxic (≥10 μM) |
| Rotenone | Complex I of mitochondrial electron transport chain | Toxic (≥25 μM) | No effect |
| TTFA | Complex II of mitochondrial electron transport chain | No effect | No effect |
| Myxathiazole | Complex III of electron transport chain | Toxic (≥2.5 μM) | Toxic (≥50 μM) |
| MCCCP | Uncouples mitochondrial electron transport chain | Toxic (≥1 μM) | Toxic (≥10 μM) |
| Oligomycin | ATP synthase (complex V of mitochondrial electron transport chain | Protective | No effect |
| DIDS | Voltage-dependent ion channels | Protective | No effect |
Unless otherwise indicated, the compounds listed function as chemical inhibitors. Cells were preincubated with inhibitors in dose-response experiments ranging from nanomolar to micromolar concentrations for 15 min, followed by culture on fibronectin- or poly-l-lysine-coated coverslips for 24 h. DPI, diphenyleneiodonium; l-NIL, N6-(1-iminoethyl)-l-lysine; l-NMMA, N-monomethyl-l-arginine; ETYA, eicosatetrosynoic acid; TTFA, thenoyltrifluoroacetone; mCCCP, carbonyl cyanide m-chlorophenylhydrazone; iNOS, inducible nitric oxide synthase (NOS); eNOS, endothelial NOS; nNOS, neuronal NOS; COX, cyclooxygenase.
Table 2.
Effects of pharmacological signaling inhibitors on chondrocyte survival when cells were plated on either poly-l-lysine or fibronectin
| Compounds | Functions | Effect on Poly-l-Lysine | Effect on Fibronectin |
|---|---|---|---|
| U0126 | MEK1/2 | No effect | No effect |
| PD-98059 | MEK1/2 | No effect | No effect |
| LY-294002 | PI 3-kinase | No effect | No effect |
| SB-203580 | p38 | No effect | No effect |
| SP-600125 | JNK | No effect | No effect |
| Lavendustin C | c-Src, CaM II | Toxic (≥10 μM) | Toxic (≥25 μM) |
| IQD | PARP | No effect | No effect |
| SN-50 | NF-κB | Toxic (≥12.5 μM) | Toxic (≥50 μM) |
| PP2 | Lyk, Fyn, Hck, EGFR, ZAP-70 | No effect | No effect |
| Perillic acid | p21ras, small G proteins | No effect | No effect |
| Pifithrin-α | p53 | No effect | No effect |
| Cyclosporin A | PP2B | Toxic (≥125 μM) | No effect |
| Roscovitine | Cyclin A/B/E, Cyclins 1/2/5 Erk1/2 | No effect | No effect |
| Ionomycin | Ca2+ ionophore | Toxic (≥0.5 μM) | Toxic (≥5 μM) |
| BAPTA-AM | Chelates intracellular divalent cations (Ca2+) | Toxic (≥0.5 μM) | Toxic (≥4 μM) |
| RCH-80267 | All DAG lipases | No effect | No effect |
| MAFP | Phospholipase A | Toxic (≥2 μM) | Toxic (≥10 μM) |
| Propanolol | Phospholipase D | No effect | No effect |
| Thapsigargin | Endoplasmic reticular Ca2+-ATPase | Toxic (≥0.01 μM) | Toxic (≥1μM) |
| Nifedipine | L-type Ca2+ channels | No effect | No effect |
| Methoxyverapamil | Broad Ca2+ channels | No effect | No effect |
| Cypermethrin | Calcineurin, PTP-2B | No effect | No effect |
| ALLN | Calpain I/II, Cathepsins B/L, Proteosome | No effect | No effect |
| H-89 | PKA, CaM II, Casein kinase I, Myosin light chain kinase | Toxic (≥6.25 μM) | Toxic (≥100 μM) |
Unless otherwise indicated, the compounds listed function as chemical inhibitors. Cells were preincubated with inhibitors in dose-response experiments ranging from nanomolar to micromolar concentrations for 15 min, followed by culture on fibronectin- or poly-l-lysine-coated coverslips for 24 h. EGFR, epidermal growth factor receptor; DAG, diacyl glycerol; MAFP, methyl arachidonyl fluorophosphate; ALLN, N-acetyl-Leu-Leu-norleucinal.
Tissue acquisition and chondrocyte cell culture
Human ankle articular cartilage was obtained within 72 h of death through the Gift of Hope Organ and Tissue Donor Network (Elmhurst, IL) in accordance with institutional protocol and review board approval. Only human donors with no known history of joint disease were used for this study. Full-thickness cartilage was removed from all surfaces of both the left and right tali and pooled for chondrocyte isolation. Cartilage slices were digested in DMEM-Ham’s F-12 (1:1) culture medium in the presence of gentamicin, penicillin, streptomycin, and amphotericin B with sequential treatments of 0.2% Pronase for 1 h in serum-free medium and then overnight with 0.025% bacterial collagenase in medium supplemented with 5% FBS in a humidified atmosphere of 5% CO2 at 37°C with continuous agitation. Cells were counted using a hemacytometer, and initial survival before plating, determined by Trypan blue exclusion, was >90%. Cells were plated at low density (3.75 × 104 cells/cm2) on poly-l-lysine- or fibronectin-coated coverslips as previously described (36). For inhibitor studies, the cells were added to wells containing the specified inhibitor in the medium and then incubated at room temperature for 15 min before placement in the incubator. Inhibitors were tested in dose-ranging experiments from nanomolar to micromolar concentrations. Unless otherwise noted, the cells were incubated for 24 h before analysis of cell death.
Determination of cell death
Cell death was quantified using the LIVE/DEAD cell survival assay (Molecular Probes, Eugene, OR) with the use of ethidium bromide homodimer 1 to stain dead cells and calcein-AM to visualize live cells as previously described (7, 36). Percent survival was determined by performing a total cell count in triplicate (n = 3) with the number of cells counted always exceeding 100 for each data point.
Cell death also was analyzed using a combination of Trypan blue staining to detect dead cells and a fluorescence-based terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL) assay (Promega, Madison, WI) to detect DNA cleavage. For this, cells were treated sequentially with the following steps (in chronological order): 1) stained with Trypan blue for 1 min, 2) fixed with 4% paraformaldehyde, 3) permeabilized with 1% Triton X-100, and 4) treated with a reaction mixture containing fluorescein-labeled dUTP with a TdT enzyme. Cells were incubated with propidium iodide as a fluorescent counterstain, and coverslips were mounted onto microscope slides. Dead cells stained with Trypan blue were first visualized using phase-contrast light microscopy on an Olympus BX 60 fluorescent microscope, and then the same cells were visualized using fluorescence microscopy to determine whether they were TUNEL positive or negative. DNA fragmentation also was measured using an ELISA (Boehringer Mannheim) for histone-associated DNA fragments as previously described (7).
Quantification of intracellular ROS production
ROS production was detected using 5-(6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester (CM-H2DCFDA) from Molecular Probes. This is a cell-permeable dye that is retained in cells by the action of intracellular esterases on the acetyl ester group. The nonfluorescent dihydro form of fluorescein is readily oxidized to the highly fluorescent parent dye by intracellular ROS and thus serves as a fluorogenic probe for detecting oxidative activity. The dye was solubilized in DMSO to make a concentrated solution and was then added to cells plated in DMEM-Ham’s F-12 medium without phenol red in 96-well plates that had been precoated with either fibronectin or poly-l-lysine. Relative fluorescent intensities were quantified on a fluorescent microplate reader at the indicated times.
Isolation of cytosolic and particulate membrane fractions and immunoblot analysis
After cells were plated on either fibronectin- or poly-l-lysine-coated coverslips for the indicated time periods, cells were washed once for 3 min with ice-cold phosphate-buffered saline (PBS) containing 1 mM diisopropyl fluorophosphate. Cells were scraped and briefly sonicated in a small volume of a hypotonic lysis buffer (1 ml buffer per 1 × 107 cells) containing 50 mM Tris·HCl (pH 7.5), 2 mM 4-(2-aminoethyl)-benzenesulfonyl fluoride, 1 mM EDTA, 1 mM diisopropyl fluorophosphate, 14 μM E-64, 130 μM bestatin, 1 mM leupeptin, and 0.3 μM aprotinin. Cell extracts were then gently placed on top of a 15% sucrose cushion (4 ml) and centrifuged at 75,000 g for 45 min. The clear supernatant at the top of the sucrose was collected and used as the cytosolic fraction as previously described (5). After careful removal of the remaining sucrose, the pellet at the bottom of the centrifuge tube was solubilized with brief sonication in the above lysis buffer (0.5 ml buffer/pellet) with the addition of 150 mM NaCl and 1% Triton X-100 (final concentration). This lysate was transferred to a microfuge tube and centrifuged at 14,000 g for 10 min to remove insoluble cellular debris. The clear supernatant was collected and used as the particulate membrane fraction (proteins from plasma membrane and intracellular organelles).
Protein was measured in cytosolic and membrane fractions using the BCA Protein Assay (Pierce, Rockford, IL). Samples with equal amounts of total protein were separated using SDS-PAGE with 10% acrylamide gels and transferred to nitrocellulose for immunoblot analysis. Immunoreactivity was determined with enhanced chemiluminescence (ECL; Amersham).
ATP quantification
A luciferase-based ATP assay kit (Calbiochem) was used to determine the intracellular ATP concentrations. Briefly, freshly isolated chondrocytes were plated on either poly-l-lysine- or fibronectin-coated 12-well plates, and ATP levels were measured at the indicated time points by following the manufacturer’s instructions. Bioluminescence was quantified in a Sirius luminometer (Berthold Detection Systems, Oak Ridge, TN).
Caspase activity
A fluorescent caspase detection kit (Calbiochem) utilizing the broad-spectrum caspase inhibitor Z-Val-Ala-Asp-fluoromethyl ketone (Z-VAD-fmk) conjugated to quenched FITC was used to determine general caspase activity at the indicated time points in freshly isolated chondrocytes plated on either poly-l-lysine- or fibronectin-coated 12-well plates. Relative fluorescence intensities were quantified on a fluorescent microplate reader.
Caspase activity also was analyzed using a colorimetric assay (Sigma) utilizing the hydrolysis of the peptide substrate Z-Asp-Glu-Val-Asp-fluoromethyl ketone (Z-DEVD-fmk) conjugated to p-nitroaniline, which displays an increased specificity for caspase-3. Relative absorbance was quantified on a microplate reader at 405 nm.
Statistical analysis
Data were analyzed using one-way ANOVA to detect a difference in group means with a post hoc Bonferroni correction by using the Windows-based Statistical Package for the Social Sciences software (SPSS, Chicago, IL). A P value <0.05 was considered significant.
RESULTS
Chondrocyte death observed on poly-l-lysine requires endogenous production of ROS
Consistent with our previous publication (36), overnight incubation of freshly isolated chondrocytes in serum-free medium on poly-l-lysine reduced cell survival to <40% (Fig. 1A), whereas almost 100% survival was noted in cells plated on fibronectin (Fig. 2B). We sought to determine whether the cell death observed on poly-l-lysine was a function of endogenous ROS production because of previous findings that chondrocytes cultured in low-density suspension required antioxidants for survival (15, 40). We found that Tiron (125 μM), a scavenger of O2−·(30), and Mn(III) tetrakis(4-benzoic acid)porphyrin (125 μM), a superoxide dismutase mimetic that reduces production of both O2−·and ONOO− (39), or N-acetyl-l-cysteine (40 mM), an antioxidant and a substrate for synthesis of reduced glutathione, could completely prevent the cell death displayed by chondrocytes plated on poly-l-lysine (Fig. 1). As expected, cell death on poly-l-lysine was not seen in the presence of 10% serum. Death of all cells was noted when cells were placed in medium without cystine, a condition that we have previously found to significantly deplete intracellular stores of glutathione and thereby increase cell death mediated by ROS (6).
Fig. 1.
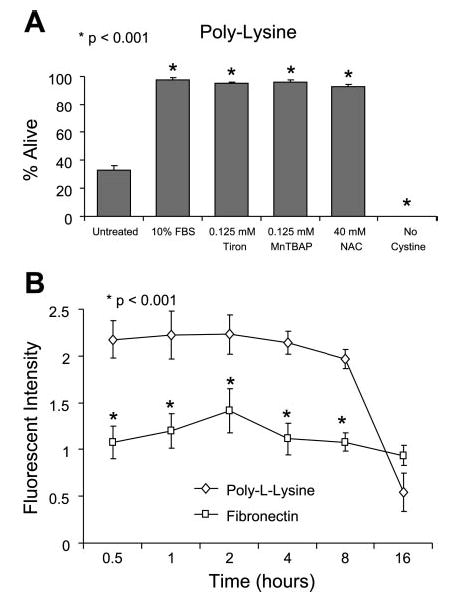
Reactive oxygen species (ROS) and chondrocyte survival on poly-l-lysine. A: cells were plated at low density in 24-well plates at 5 × 105 cells/ml (0.5 ml/well) in serum-free DMEM-Ham’s F-12 on poly-l-lysine-coated coverslips in the presence of various antioxidants for 24 h before cell survival was quantified using the LIVE/DEAD cell survival assay. Results represent means ± SE of 3 separate experiments using primary chondrocytes derived from different donors. *P < 0.001 vs. untreated control. MnTBAP, Mn(III) tetrakis(4-benzoic acid)porphyrin; NAC, N-acetyl-l-cysteine. B: cells were plated at low density in 96-well plates coated with either fibronectin or poly-l-lysine at 5 × 105 cells/ml (0.2 ml/well) in serum-free DMEM-Ham’s F-12 without phenol red. ROS were quantified with the ROS-sensitive dye 5-(6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester (CM-H2DCFDA) at a final concentration of 5 μM. Results represent means ± SE of 3 separate experiments using primary chondrocytes derived from different donors. *P < 0.001 vs. untreated control.
Fig. 2.
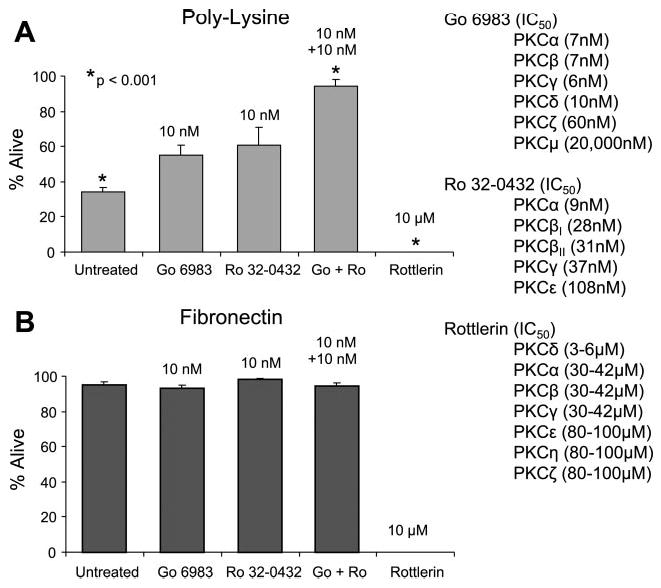
Effect of PKC inhibitors on chondrocyte survival on poly-l-lysine. Freshly isolated chondrocytes were plated on coverslips coated with either poly-l-lysine (A) or fibronectin (B) at 5 × 105 cells/ml (0.5 ml/well) for 24 h under serum-free conditions in the presence or absence of the PKC inhibitors Gö-6983, Ro 32–0432, and rotterlein. Survival was determined using the LIVE/DEAD cell survival assay. The IC50 of the inhibitors for each respective PKC isoform is shown at right. Results represent means ± SE of 3 separate experiments using primary chondrocytes derived from different donors. *P < 0.001 vs. untreated control.
Because antioxidants could prevent chondrocyte death on poly-l-lysine, experiments were performed to quantify intracellular ROS production using the ROS-sensitive dye CM-H2DCFDA. Measurements were started at 30 min after plating because this was a time point well before cell death was detected. Cells plated on poly-l-lysine produced approximately twice as much ROS compared with cells plated on fibronectin (Fig. 1B). The fluorescent signal produced by cells on poly-l-lysine dropped below the signal produced on fibronectin at the 16-h time point, because >50% of chondrocytes were dead by that time point (Fig. 1B). These results demonstrate that culturing freshly isolated chondrocytes on poly-l-lysine in serum-free conditions can serve as a model system for evaluating the mechanism of cell death stimulated by endogenous ROS production.
Inhibitors of mitochondrial electron transport chain and voltage-dependent anion channels promote chondrocyte survival on poly-l-lysine
Notwithstanding Fenton-derived and spontaneous autooxidation reactions, ROS are produced by enzymes that are capable of one-electron transfers to O2. This includes NADPH oxidase, xanthine oxidase, cytochrome P-450, and various enzyme complexes that comprise the mitochondrial electron transport chain (reviewed in Ref. 8). A pharmacological approach was used to rapidly assess potential sources of ROS in chondrocytes plated on poly-l-lysine (Table 1). Inhibitors of NADPH oxidase, xanthine oxidase, or catalase did not affect survival. Likewise, inhibitors of NO production did not affect survival, indicating that NO was not required for the observed cell death on poly-l-lysine.
Among this set of inhibitors tested, only oligomycin, which inhibits the terminal complex of the electron transport chain, and DIDS, which blocks voltage-dependent anion channels, had the ability to significantly promote chondrocyte survival on poly-l-lysine (Table 1). Chondrocyte survival increased from 36 ± 4 to 60 ± 5% with 1 μM oligomycin and to 72 ± 5% with 50 μM DIDS. A previous study by Han et al. (13) demonstrated release of ROS from mitochondria to the cytosol via voltage-dependent anion channels, suggesting that inhibition of mitochondrial ROS release was promoting chondrocyte survival. The survival-promoting capacity of oligomycin was less than that of DIDS, probably because of the inhibitory effects of oligomycin on mitochondrial ATP synthase at higher concentrations. We also tested the electron transfer inhibitor myxothiazol and found that it increased cell death on poly-l-lysine at concentrations ≥2.5 μM and, at ≥50 μM, increased cell death on fibronectin consistent with previous work, demonstrating that myxothiazol can be a potent stimulator of mitochondrial ROS production (44).
Chondrocyte death observed on poly-l-lysine requires activation of PKC
To further delineate the cell signaling mechanisms regulating chondrocyte survival under these conditions, we screened a large series of cell signaling regulators at various concentrations, from nanomolar to high micromolar values (Table 2). No effect on survival with cells plated on poly-l-lysine or fibronectin was noted with MAPK inhibitors to MEK, p38, or JNK or with a PI 3-kinase inhibitor. A significant increase in cell death was noted with lavendustin C (c-Src and CaM II inhibitor), SN-50 (NF-κB inhibitor), cyclosporine A (protein phosphatase 2B inhibitor), regulators of intracellular calcium levels (ionomycin, BAPTA-AM, and ruthenium red), methyl arachidonylfluorophosphate (phospholipase A inhibitor), thapsigargin, and H-89 (PKA, CaM II, casein kinase I, and myosin light chain kinase inhibitor).
In the initial inhibitor screen, compounds that could inhibit certain isoforms of PKC were found to modulate cell survival on poly-l-lysine or fibronectin (Fig. 2). Specifically, both of the staurosporine derivatives Gö-6983 and Ro 32–0432 could individually promote survival on poly-l-lysine by ~50% but only at the 10 nM concentration. Concentrations below 10 nM had no effect, and concentrations above 10 nM became toxic (data not shown), probably because of inhibition of other PKC isoforms at higher concentrations. Interestingly, treatment with the combination of both inhibitors at 10 nM each completely prevented the cell death observed on poly-l-lysine. As can be determined from the IC50 values of the inhibitors shown in Fig. 2, these findings suggest that activity of one or more isoforms of PKC is required for chondrocyte death on poly-l-lysine, and other PKC isoforms may promote survival. Neither Gö-6983 nor Ro 32–0432 displayed any toxicity with cells plated on fibronectin at the concentrations tested (Fig. 2B). In contrast, an inhibitor of PKC-δ induced 100% cell death at a concentration similar to the IC50 of PKC-δ (10 μM) with cells plated on either poly-l-lysine or fibronectin.
PKC-βI activation on poly-l-lysine is inhibited with addition of antioxidant
The results described above suggested an association between ROS production and PKC activity in the regulation of chondrocyte survival. To determine whether specific PKC isoforms were activated in chondrocytes plated on poly-l-lysine in an ROS-dependent manner, we used immunoblot analysis to detect the translocation of 10 PKC isoforms from the cytosol to the particulate membrane fraction as a measure of PKC activation. The contribution of ROS was determined by culturing cells in the presence of the O2−· scavenger Tiron, which had been found to promote survival. Cytoplasmic and particulate membrane fractionations of total cell lysates that were obtained from time-course experiments indicated a slight decrease in the membrane fraction of PKC-α at 30 min in the presence of Tiron but without a significant change in the cytosolic fraction (Fig. 3A). However, PKC-βI immunoblots of lysates from cells plated on poly-l-lysine showed that Tiron increased the amount of PKC-βI retained in the cytosol at 30 min and 1 h and also inhibited the translocation of PKC-βI to the membrane, which appeared at ~1 h and was maximal by 6 h (Fig. 3A).
Fig. 3.
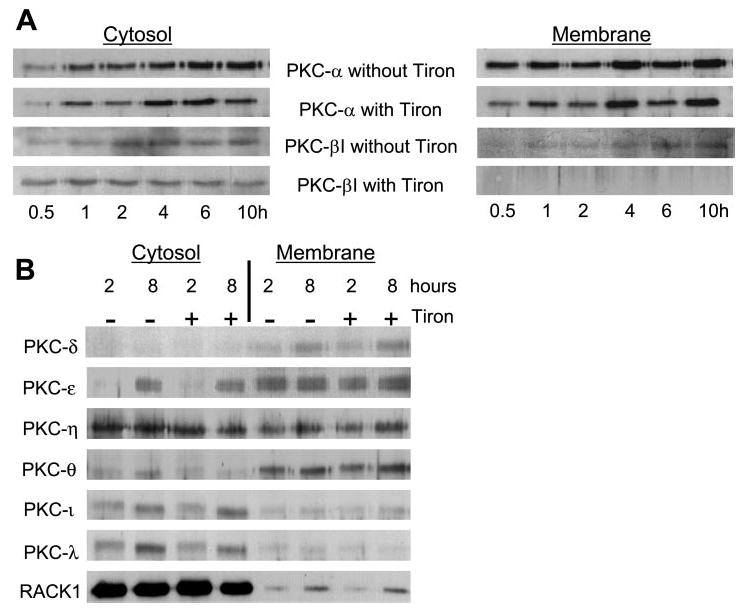
PKC isoform translocation in chondrocytes plated on poly-l-lysine in the absence and presence of an antioxidant. Immunoblot analysis was used to detect the translocation of 10 PKC isoforms from the cytosol to the particulate membrane when freshly isolated chondrocytes were plated on poly-l-lysine in serum-free DMEM-Ham’s F-12 in the presence and absence of the antioxidant Tiron (125 μM). Cell lysates were prepared after culture for the indicated time points, and the particulate membrane was separated from the cytosol by ultracentrifugation. A: translocation of PKC-α and -βI. B: translocation of PKC-δ, -ε, -η, -θ, -ι, and -λ. No evidence of PKC-βII or PKC-γ was found in human articular chondrocytes by using immunoblot analysis (data not shown). Results are representative of at least 3 separate experiments that gave similar results.
Tiron had no apparent effect on the translocation of PKC-δ, -ε, -η, -θ, -ι, -λ, or RACK1, a receptor for activated C-kinase (Fig. 3B). There appeared to be an increase in PKC-δ in the membrane fraction and an increase in PKC-ε in the cytosol at 8 h compared with 2 h that was not affected by Tiron. We could not detect PKC-βII or PKC-γ in human articular chondrocytes using immunoblot analysis (data not shown). Combining these results with the effects of the inhibitors shown in Fig. 2 strongly suggests that PKC-βI is activated in response to ROS and is required for a signal that mediates cell death in chondrocytes, whereas PKC-δ activity may be necessary for survival.
Chondrocyte death observed on poly-l-lysine is not caspase dependent
Cell death on poly-l-lysine began at 4 h and was complete by 24 h as determined using Trypan blue staining (Fig. 4A). TUNEL-based immunocytochemistry was used to determine whether the cell death observed on poly-l-lysine was associated with DNA fragmentation. This method was combined with Trypan blue to differentiate between apoptotic and necrotic cell death, as previously described (34). Cells positive to Trypan blue and TUNEL staining were considered to be apoptotic, whereas cells positive to Trypan blue but negative to TUNEL staining were considered to be necrotic. The early cell death displayed by freshly isolated chondrocytes plated on poly-l-lysine, which at 0.5 h is only 10% of the cells (Fig. 4A), was mostly associated with apoptosis (Fig. 4B). As the percentage of dead cells increased on poly-l-lysine as a function of time, the percentage of Trypan blue-positive, TUNEL-positive cells (apoptotic) declined, whereas the percentage of Trypan blue-positive, TUNEL-negative cells (necrotic) increased (Fig. 4B). These results suggest that the majority of cell death occurring in response to endogenous ROS production in chondrocytes plated on poly-l-lysine was due to necrosis, whereas a small population of cells underwent apoptosis. No TUNEL reactivity was seen in cells plated on fibronectin (data not shown). The lack of apoptotic death on poly-l-lysine was further confirmed by using an assay for fragmented DNA as an indication of apoptotic death (Fig. 4C).
Fig. 4.
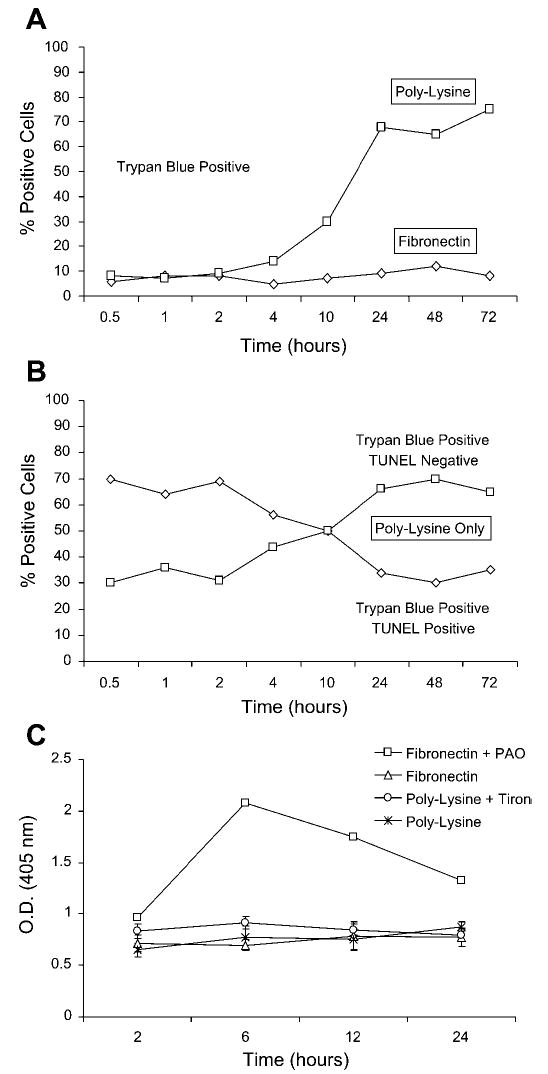
Quantitation of apoptotic and necrotic death of chondrocytes plated on poly-l-lysine. Freshly isolated chondrocytes were plated on poly-l-lysine or fibronectin as described for Fig. 1. At the indicated time points, the cells were sequentially stained with Trypan blue, fixed with 4% paraformaldehyde, and, finally, TdT-mediated dUTP nick end labeling (TUNEL) stained as described in materials and methods. Cells that were both Trypan blue and TUNEL positive were considered to be apoptotic, whereas cells that were Trypan blue positive and TUNEL negative were considered to be necrotic. At least 100 cells were counted for each time point, and the results are reported as the percentage that showed positive staining. A: cells staining with Trypan blue after plating on either poly-l-lysine or fibronectin. B: cells quantified with both Trypan blue and TUNEL staining after plating on poly-l-lysine. C: cells were plated on poly-l-lysine with or without 125 μM Tiron (to promote survival) or on fibronectin with or without 100 nM phenylarsine oxide (PAO; to induce apoptosis). DNA fragmentation was measured at the indicated time points using an ELISA for histone-associated DNA fragments. OD, optical density.
Caspases are widely accepted as downstream effector molecules in the progression of apoptosis with the ability to activate nuclear DNases, thereby inducing DNA fragmentation (38). In preliminary experiments, caspase inhibitors were tested in dose-response experiments that ranged from nanomolar to micromolar concentrations (data not shown). Neither the broad-spectrum caspase inhibitor Z-VAD-fmk nor the caspase-3-specific inhibitor Z-DEVD-fmk, nor both together, promoted survival of chondrocytes plated on poly-l-lysine (Fig. 5A). As expected, no change in survival was noted in cells plated on fibronectin (Fig. 5B).
Fig. 5.
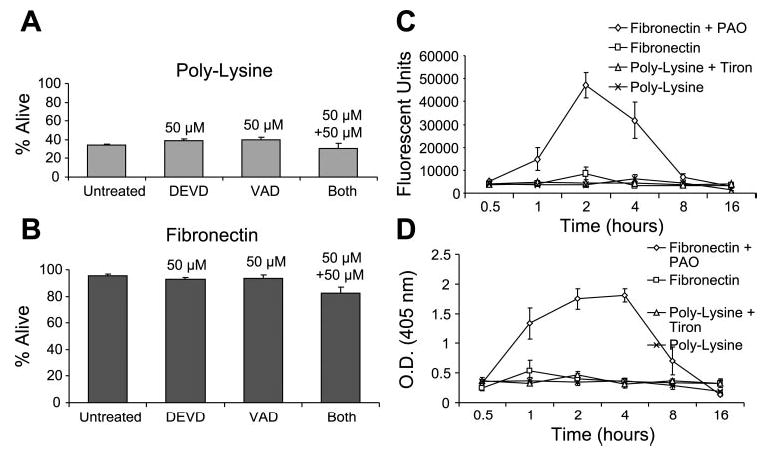
Chondrocyte death on poly-l-lysine does not require caspase activity. Freshly isolated chondrocytes were plated on either poly-l-lysine (A) or fibronectin (B) for 24 h under serum-free conditions, as described in Fig. 1, in the absence or presence of a broad caspase inhibitor (VAD) and/or a caspase-3-specific inhibitor (DEVD). Survival was determined using the LIVE/DEAD cell survival assay. Data presented in this section represent typical results from the dose-response experiments. Cells also were plated on poly-l-lysine with or without Tiron (to promote survival) or on fibronectin with or without PAO (to induce apoptosis), and caspase activity was measured using an assay for general caspase activity (C) or caspase-3 activity (D). Results represent means ± SE of 3 separate experiments.
To further confirm the lack of caspase involvement in chondrocyte death on poly-l-lysine, we measured caspase activity in freshly isolated chondrocytes plated on either poly-l-lysine or fibronectin. No significant caspase activity was observed in chondrocytes plated on either poly-l-lysine or fibronectin with the use of general (Fig. 5C) and caspase-3-specific assays (Fig. 5D). Phenylarsine oxide was included with cells plated on fibronectin as a positive control and was found to stimulate caspase activity in both assays as expected.
Chondrocyte death observed on poly-l-lysine is associated with decreased intracellular ATP levels
Because caspase activity is ATP dependent, and because it has been suggested that low intracellular levels of ATP can switch the cell death modality from apoptosis to necrosis (25), it was of interest to quantify intracellular ATP in chondrocytes plated on either poly-l-lysine or fibronectin. Cells plated on poly-l-lysine were found to have a significant decrease in intracellular ATP concentration at and beyond a 2-h time point relative to cells on fibronectin (Fig. 6). The 2-h time point was reached just before the majority of the cell death on poly-l-lysine was noted and when the percentage of TUNEL-positive cells started to decline (see Fig. 4). This would be consistent with the hypothesis that ATP depletion contributed to a switch from apoptotic to necrotic cell death.
Fig. 6.
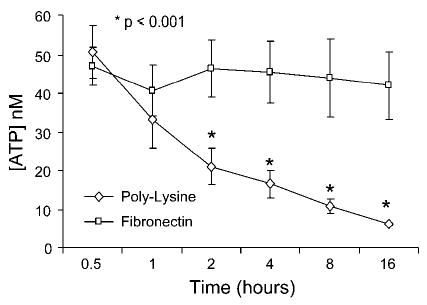
ATP levels in chondrocytes plated on poly-l-lysine or fibronectin. Freshly isolated chondrocytes were plated on either poly-l-lysine- or fibronectin-coated 12-well plates, and at the indicated time points, ATP levels were measured using a luciferase-based assay. Results represent means ± SE of 3 separate experiments. *P < 0.001 vs. fibronectin.
DISCUSSION
Death of chondrocytes due to the endogenous production of excessive amounts of ROS was found in the present study after freshly isolated chondrocytes were plated on poly-l-lysine under low-density, serum-free conditions. These culture conditions provided a model system for the study of chondrocyte survival signaling when normal matrix contacts have been lost and autocrine survival signaling is reduced. Such conditions may occur in vivo during advanced stages of arthritis, where significant loss of matrix and cells has been noted (reviewed in Ref. 1). In addition, this model system provides data relevant to chondrocyte death initiated by endogenous ROS production, which has been reported to occur after mechanical trauma (21). The inhibitor results suggested a novel mechanism of cell death signaling that linked ROS-dependent activation of PKC-βI to chondrocyte cell death (Fig. 7).
Fig. 7.
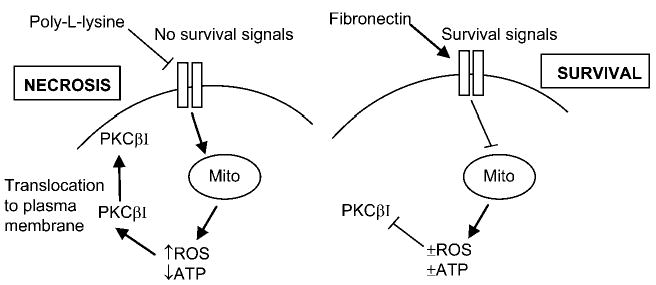
Theoretical model for pathways resulting in chondrocyte death when matrix and growth factor survival signals are lacking in cells plated under serum-free conditions on poly-l-lysine. Mito, mitochondria.
Previous studies have demonstrated the protective function of antioxidants when chondrocytes are cultured at low density in serum-free suspension cultures (15, 40). Antioxidants also have been found to protect chondrocytes from trauma-induced cell death (21), and dietary antioxidants have been shown to retard the development of spontaneous osteoarthritis in STR/1N mice (20). Although it has been recognized that ROS can play a significant role in cartilage loss and chondrocyte death during the development of arthritis (14), potential signaling mechanisms responsible for endogenous ROS mediated death in chondrocytes have not been explored.
Chondrocyte death on poly-l-lysine was completely inhibited with the addition of either an antioxidant or inhibitors of specific isoforms of PKC. Time-course studies suggest that the generation of ROS precedes the activation of PKC-βI. PKC family members can have both proapoptotic and antiapoptotic effects on eukaryotic cells that depend on the PKC isoform and the cell type studied. For example, PKC-α, -β, and -δ have been shown to induce cell death (22, 32, 37), whereas PKC-θ and -ε have been shown to promote survival (2). Although PKC-δ activity has been associated with cell death in some cell types (26, 37), our finding of a prosurvival role in chondrocytes is similar to the role of PKC-δ in TNF-α-mediated survival of neutrophils (17). Also similar to our findings, PKC-βI activation has been associated with cell death in U-937 cells (35).
There also is evidence that supports a link between ROS production and PKC activation. It has been shown that oxidants can directly activate several isoforms of PKC (reviewed in Ref. 12). This represents a mechanism by which ROS can participate as secondary messengers in cell death-related signal transduction. In addition to the ability of ROS to directly activate PKC, activated PKC also has been shown to induce the production of endogenous ROS (4, 18) and thus may participate in a positive feedback loop. These observations strongly suggest that PKC is highly intimated with ROS-dependent signal transduction that can result in either increased survival or cell death, depending on the specific isoform involved.
The cell death modality of chondrocytes plated on poly-l-lysine appeared to be caspase-independent, because an increase in caspase activity could not be detected and standard caspase inhibitors did not promote survival. Caspase-independent cell death has been previously described as a physiologically relevant form of cell death (29, 33). Recent studies in chondrocytes suggest that caspase-independent cell death occurs in situ during the development of osteoarthritis in the STR/ort mouse (28), and a study of chondrocytes treated in vitro with peroxynitrite found caspase-independent cell death that was mediated by calpains (42). However, we could not inhibit the cell death in our model system with a calpain inhibitor (N-acetyl-Leu-Leu-norleucinal), suggesting a different mechanism. Because NO inhibition also did not promote survival, the ROS-mediated cell death in our system did not require generation of peroxynitrite formed by the reaction of NO with superoxide.
Our results are analogous to previous studies showing that epithelial cells (9) and endothelial cells (27) experimentally detached from their native ECM undergo anoikis. The present study also is in agreement with a report that links the endogenous production of ROS to anoikis (23). However, previous studies describing anoikis have found the cell death to be associated with apoptosis. In the present study, only the very early time points after chondrocytes were plated on poly-l-lysine showed cells with positive TUNEL staining as a marker for apoptosis, whereas the majority of cell death that occurred after ~4 h was TUNEL negative, suggesting that necrosis was the likely mechanism. A switch from apoptotic to necrotic cell death may have occurred secondary to ATP depletion, which we observed after 2 h of culture on poly-l-lysine. Apoptosis requires energy, and previous work has documented that ATP depletion can result in necrosis under conditions that would otherwise result in apoptosis (25). Necrosis also may have resulted from direct membrane damage secondary to excessive production of endogenous ROS.
Studies using synovial fibroblasts have demonstrated that signals generated downstream from the α5β1-integrin can stimulate mitochondrial ROS production, which in turn is required for collagenase-1 expression (16, 41). In contrast, we found that the lack of integrin signals in chondrocytes plated on poly-l-lysine generated ROS, resulting in cell death, whereas α5β1-mediated attachment to fibronectin did not (Fig. 7). A key difference between the studies is the manner in which the α5β1 signals were generated. In the previous work, the increase in mitochondrial ROS generation was observed after adherent synovial fibroblasts were treated with an anti-α5-integrin antibody, which resulted in redistribution of the actin cytoskeleton and subsequent cell rounding due to a disruption in the normal cellular interaction with fibronectin (16, 41). That study differs from the present study of chondrocytes plated on fibronectin or poly-l-lysine. The cells plated in serum-free medium on poly-l-lysine model a situation where growth factor and integrin survival signals are lacking, whereas cells plated on fibronectin are receiving integrin survival signals from intact fibronectin. Similar to our results, Werner and Werb (41) found that the α5-integrin signals generated when cells bind to intact fibronectin did not result in mitochondrial depolarization or ROS production. The ROS production noted with synovial fibroblasts treated with α5-integrin blocking antibodies was inhibited by rotenone, which blocks complex I of the mitochondrial electron transport chain, whereas in the present study, survival on poly-l-lysine was not improved with rotenone but was promoted by oligomycin, which blocks complex V, and by DIDS, which blocks voltage-dependent anion channels. These results are consistent with a mitochondrial source for ROS in both studies, albeit at different points in the electron transport chain, and results obtained with DIDS suggest a potential mechanism for the release of mitochondrial ROS through voltage-dependent anion channels, as previously reported using isolated rat heart mitochondria (13).
There are important limitations to the present study. Although the chondrocyte death observed after plating on poly-l-lysine provided a model system for studying signaling pathways and cell death related to endogenous ROS production, it is not completely clear whether the same pathways would function to mediate chondrocyte death in vivo. A role for ROS in chondrocyte death in vivo has been suggested but not conclusively proven (14). Further work needs to be done in an animal model to substantiate the role of ROS and PKC signaling in vivo.
An additional limitation is the use of chemical inhibitors to modulate the activity of signaling pathways. Chemical inhibitors may not always be specific, particularly when used at concentrations above their IC50 values. For this reason, we performed dose-response experiments and only made conclusions about results within the range of concentrations that should provide optimal specificity. We were not able to confirm results with molecular studies, such as transfecting chondrocytes with a construct that would result in PKC-βI inhibition. This was because transfection required initial culture under conditions that promoted survival to transfect the cells, and once the cells had been cultured under these conditions, we found they do not die when replated on poly-l-lysine.
Together, our results demonstrate a link between the ROS-dependent activation of PKC-βI and chondrocyte cell death when normal matrix and growth factor survival signals are lost. Further studies are needed to delineate additional signaling events involved in this process, including how PKC activity regulates survival. In vitro analysis of chondrocytes derived from transgenic mice lacking PKC-βI could further define the role of this signaling kinase in chondrocyte anoikis. In opposition to PKC-βI, our findings suggest that PKC-δ activity may be important for promotion of chondrocyte survival.
Acknowledgments
We gratefully acknowledge the Gift of Hope Organ and Tissue Donor Network and the donor families for providing tissue and the assistance of Dr. Arkady Margulis for collecting donor tissues.
Footnotes
GRANTS
This work was supported by National Institutes of Health Grants AR-49003 and AG-16697.
References
- 1.Aigner T, Kim HA, Roach HI. Apoptosis in osteoarthritis. Rheum Dis Clin North Am. 2004;30:639–653, xi. doi: 10.1016/j.rdc.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 2.Bertolotto C, Maulon L, Filippa N, Baier G, Auberger P. Protein kinase C theta and epsilon promote T-cell survival by a rsk-dependent phosphorylation and inactivation of BAD. J Biol Chem. 2000;275:37246–37250. doi: 10.1074/jbc.M007732200. [DOI] [PubMed] [Google Scholar]
- 3.Bruckner P, Horler I, Mendler M, Houze Y, Winterhalter KH, Eich-Bender SG, Spycher MA. Induction and prevention of chondrocyte hypertrophy in culture. J Cell Biol. 1989;109:2537–2545. doi: 10.1083/jcb.109.5.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Datta R, Yoshinaga K, Kaneki M, Pandey P, Kufe D. Phorbol ester-induced generation of reactive oxygen species is protein kinase Cβ-dependent and required for SAPK activation. J Biol Chem. 2000;275:41000–41003. doi: 10.1074/jbc.M009322200. [DOI] [PubMed] [Google Scholar]
- 5.Dekker LV, Leitges M, Altschuler G, Mistry N, McDermott A, Roes J, Segal AW. Protein kinase C-β contributes to NADPH oxidase activation in neutrophils. Biochem J. 2000;347:285–289. [PMC free article] [PubMed] [Google Scholar]
- 6.Del Carlo M, Jr, Loeser RF. Increased oxidative stress with aging reduces chondrocyte survival: correlation with intracellular glutathione levels. Arthritis Rheum. 2003;48:3419–3430. doi: 10.1002/art.11338. [DOI] [PubMed] [Google Scholar]
- 7.Del Carlo M, Jr, Loeser RF. Nitric oxide-mediated chondrocyte cell death requires the generation of additional reactive oxygen species. Arthritis Rheum. 2002;46:394–403. doi: 10.1002/art.10056. [DOI] [PubMed] [Google Scholar]
- 8.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 9.Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;124:619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frisch SM, Ruoslahti E. Integrins and anoikis. Curr Opin Cell Biol. 1997;9:701–706. doi: 10.1016/s0955-0674(97)80124-x. [DOI] [PubMed] [Google Scholar]
- 11.Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- 12.Gopalakrishna R, Jaken S. Protein kinase C signaling and oxidative stress. Free Radic Biol Med. 2000;28:1349–1361. doi: 10.1016/s0891-5849(00)00221-5. [DOI] [PubMed] [Google Scholar]
- 13.Han D, Antunes F, Canali R, Rettori D, Cadenas E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J Biol Chem. 2003;278:5557–5563. doi: 10.1074/jbc.M210269200. [DOI] [PubMed] [Google Scholar]
- 14.Henrotin YE, Bruckner P, Pujol JP. The role of reactive oxygen species in homeostasis and degradation of cartilage. Osteoarthritis Cartilage. 2003;11:747–755. doi: 10.1016/s1063-4584(03)00150-x. [DOI] [PubMed] [Google Scholar]
- 15.Ishizaki Y, Burne JF, Raff MC. Autocrine signals enable chondrocytes to survive in culture. J Cell Biol. 1994;126:1069–1077. doi: 10.1083/jcb.126.4.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kheradmand F, Werner E, Tremble P, Symons M, Werb Z. Role of Rac1 and oxygen radicals in collagenase-1 expression induced by cell shape change. Science. 1998;280:898–902. doi: 10.1126/science.280.5365.898. [DOI] [PubMed] [Google Scholar]
- 17.Kilpatrick LE, Lee JY, Haines KM, Campbell DE, Sullivan KE, Korchak HM. A role for PKC-δ and PI 3-kinase in TNF-α-mediated antiapoptotic signaling in the human neutrophil. Am J Physiol Cell Physiol. 2002;283:C48–C57. doi: 10.1152/ajpcell.00385.2001. [DOI] [PubMed] [Google Scholar]
- 18.Korchak HM, Rossi MW, Kilpatrick LE. Selective role for β-protein kinase C in signaling for O2− generation but not degranulation or adherence in differentiated HL60 cells. J Biol Chem. 1998;273:27292–27299. doi: 10.1074/jbc.273.42.27292. [DOI] [PubMed] [Google Scholar]
- 19.Kuhn K, D’Lima DD, Hashimoto S, Lotz M. Cell death in cartilage. Osteoarthritis Cartilage. 2004;12:1–16. doi: 10.1016/j.joca.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 20.Kurz B, Jost B, Schunke M. Dietary vitamins and selenium diminish the development of mechanically induced osteoarthritis and increase the expression of antioxidative enzymes in the knee joint of STR/1N mice. Osteoarthritis Cartilage. 2002;10:119–126. doi: 10.1053/joca.2001.0489. [DOI] [PubMed] [Google Scholar]
- 21.Kurz B, Lemke A, Kehn M, Domm C, Patwari P, Frank EH, Grodzinsky AJ, Schunke M. Influence of tissue maturation and antioxidants on the apoptotic response of articular cartilage after injurious compression. Arthritis Rheum. 2004;50:123–130. doi: 10.1002/art.11438. [DOI] [PubMed] [Google Scholar]
- 22.Laouar A, Glesne D, Huberman E. Protein kinase C-β, fibronectin, α5β1-integrin, and tumor necrosis factor-α are required for phorbol di-ester-induced apoptosis in human myeloid leukemia cells. Mol Carcinog. 2001;32:195–205. doi: 10.1002/mc.10012. [DOI] [PubMed] [Google Scholar]
- 23.Li AE, Ito H, Rovira II, Kim KS, Takeda K, Yu ZY, Ferrans VJ, Finkel T. A role for reactive oxygen species in endothelial cell anoikis. Circ Res. 1999;85:304–310. doi: 10.1161/01.res.85.4.304. [DOI] [PubMed] [Google Scholar]
- 24.Loeser RF, Shanker G. Autocrine stimulation by insulin-like growth factor 1 and insulin-like growth factor 2 mediates chondrocyte survival in vitro. Arthritis Rheum. 2000;43:1552–1559. doi: 10.1002/1529-0131(200007)43:7<1552::AID-ANR20>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 25.Lyamzaev KG, Izyumov DS, Avetisyan AV, Yang F, Pletjushkina OY, Chernyak BV. Inhibition of mitochondrial bioenergetics: the effects on structure of mitochondria in the cell and on apoptosis. Acta Biochim Pol. 2004;51:553–562. [PubMed] [Google Scholar]
- 26.Majumder PK, Pandey P, Sun X, Cheng K, Datta R, Saxena S, Kharbanda S, Kufe D. Mitochondrial translocation of protein kinase C delta in phorbol ester-induced cytochrome c release and apoptosis. J Biol Chem. 2000;275:21793–21796. doi: 10.1074/jbc.C000048200. [DOI] [PubMed] [Google Scholar]
- 27.Meredith JE, Jr, Fazeli B, Schwartz MA. The extracellular matrix as a cell survival factor. Mol Biol Cell. 1993;4:953–961. doi: 10.1091/mbc.4.9.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mistry D, Oue Y, Chambers MG, Kayser MV, Mason RM. Chondrocyte death during murine osteoarthritis. Osteoarthritis Cartilage. 2004;12:131–141. doi: 10.1016/j.joca.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 29.Monney L, Otter I, Olivier R, Ozer HL, Haas AL, Omura S, Borner C. Defects in the ubiquitin pathway induce caspase-independent apoptosis blocked by Bcl-2. J Biol Chem. 1998;273:6121–6131. doi: 10.1074/jbc.273.11.6121. [DOI] [PubMed] [Google Scholar]
- 30.Moreno-Manzano V, Ishikawa Y, Lucio-Cazana J, Kitamura M. Selective involvement of superoxide anion, but not downstream compounds hydrogen peroxide and peroxynitrite, in tumor necrosis factor-α-induced apoptosis of rat mesangial cells. J Biol Chem. 2000;275:12684–12691. doi: 10.1074/jbc.275.17.12684. [DOI] [PubMed] [Google Scholar]
- 31.Muir H. The chondrocyte, architect of cartilage. Biomechanics, structure, function and molecular biology of cartilage matrix macromolecules. Bioessays. 1995;17:1039–1048. doi: 10.1002/bies.950171208. [DOI] [PubMed] [Google Scholar]
- 32.Okuda H, Adachi M, Miyazawa M, Hinoda Y, Imai K. Protein kinase Cα promotes apoptotic cell death in gastric cancer cells depending upon loss of anchorage. Oncogene. 1999;18:5604–5609. doi: 10.1038/sj.onc.1202946. [DOI] [PubMed] [Google Scholar]
- 33.Okuno S, Shimizu S, Ito T, Nomura M, Hamada E, Tsujimoto Y, Matsuda H. Bcl-2 prevents caspase-independent cell death. J Biol Chem. 1998;273:34272–34277. doi: 10.1074/jbc.273.51.34272. [DOI] [PubMed] [Google Scholar]
- 34.Perry SW, Epstein LG, Gelbard HA. Simultaneous in situ detection of apoptosis and necrosis in monolayer cultures by TUNEL and trypan blue staining. Biotechniques. 1997;22:1102–1106. doi: 10.2144/97226st01. [DOI] [PubMed] [Google Scholar]
- 35.Pongracz J, Deacon EM, Johnson GD, Burnett D, Lord JM. Doppa induces cell death but not differentiation of U937 cells: evidence for the involvement of PKC-β1 in the regulation of apoptosis. Leuk Res. 1996;20:319–326. doi: 10.1016/0145-2126(95)00074-7. [DOI] [PubMed] [Google Scholar]
- 36.Pulai JI, Del Carlo M, Jr, Loeser RF. The α5β1 integrin provides matrix survival signals for normal and osteoarthritic human articular chondrocytes in vitro. Arthritis Rheum. 2002;46:1528–1535. doi: 10.1002/art.10334. [DOI] [PubMed] [Google Scholar]
- 37.Reyland ME, Barzen KA, Anderson SM, Quissell DO, Matassa AA. Activation of PKC is sufficient to induce an apoptotic program in salivary gland acinar cells. Cell Death Differ. 2000;7:1200–1209. doi: 10.1038/sj.cdd.4400744. [DOI] [PubMed] [Google Scholar]
- 38.Salvesen GS, Dixit VM. Caspases: intracellular signaling by proteolysis. Cell. 1997;91:443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- 39.Szabo C, Day BJ, Salzman AL. Evaluation of the relative contribution of nitric oxide and peroxynitrite to the suppression of mitochondrial respiration in immunostimulated macrophages using a manganese mesoporphyrin superoxide dismutase mimetic and peroxynitrite scavenger. FEBS Lett. 1996;381:82–86. doi: 10.1016/0014-5793(96)00087-7. [DOI] [PubMed] [Google Scholar]
- 40.Tschan T, Hoerler I, Houze Y, Winterhalter KH, Richter C, Bruckner P. Resting chondrocytes in culture survive without growth factors, but are sensitive to toxic oxygen metabolites. J Cell Biol. 1990;111:257–260. doi: 10.1083/jcb.111.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Werner E, Werb Z. Integrins engage mitochondrial function for signal transduction by a mechanism dependent on Rho GTPases. J Cell Biol. 2002;15:15. doi: 10.1083/jcb.200111028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Whiteman M, Armstrong JS, Cheung NS, Siau JL, Rose P, Schantz JT, Jones DP, Halliwell B. Peroxynitrite mediates calcium-dependent mitochondrial dysfunction and cell death via activation of calpains. FASEB J. 2004;18:1395–1397. doi: 10.1096/fj.03-1096fje. [DOI] [PubMed] [Google Scholar]
- 43.Yang C, Li SW, Helminen HJ, Khillan JS, Bao Y, Prockop DJ. Apoptosis of chondrocytes in transgenic mice lacking collagen II. Exp Cell Res. 1997;235:370–373. doi: 10.1006/excr.1997.3692. [DOI] [PubMed] [Google Scholar]
- 44.Young TA, Cunningham CC, Bailey SM. Reactive oxygen species production by the mitochondrial respiratory chain in isolated rat hepatocytes and liver mitochondria: studies using myxothiazol. Arch Biochem Biophys. 2002;405:65–72. doi: 10.1016/s0003-9861(02)00338-7. [DOI] [PubMed] [Google Scholar]
- 45.Zemmyo M, Meharra EJ, Kuhn K, Creighton-Achermann L, Lotz M. Accelerated, aging-dependent development of osteoarthritis in α1 integrin-deficient mice. Arthritis Rheum. 2003;48:2873–2880. doi: 10.1002/art.11246. [DOI] [PubMed] [Google Scholar]
- 46.Zhang Z, Vuori K, Reed JC, Ruoslahti E. The α5β1 integrin supports survival of cells on fibronectin and up-regulates Bcl-2 expression. Proc Natl Acad Sci USA. 1995;92:6161–6165. doi: 10.1073/pnas.92.13.6161. [DOI] [PMC free article] [PubMed] [Google Scholar]