

Abstract
The photoluminescence-following electron-transfer (PFET) technique, developed in our laboratory, is a sensitive chromatographic detection method for oxidizable analytes. Because the oxidations are homogeneous, the technique avoids the problem of electrode fouling. A liquid-phase oxidant reacts with the electrochemically active analytes after separation, becoming capable of photoluminescence. Laser-induced photoluminescence is measured to quantitate the analytes. Thus, the electrochemical properties of the oxidant determine the detection selectivity, and the spectroscopic properties define the sensitivity. The properties of tris(2,2′-bipyridine)osmium (1) were investigated for use as the liquid-phase oxidant in the PFET system. The redox potential of the complex is less positive than that of tris(2,2′-bipyridine)ruthenium (2); thus, on-line generation of 13+ by reaction with PbO2, and selective oxidation of catechols by 13+, was possible. The mild oxidizing power of 13+ led to a lower background signal (compared to 23+) when mixed with acidic mobile phases. Photoluminescence from 12+ was much weaker than that from 22+; nonetheless, the system achieved subnanomolar detection limits for dopamine, 3-methoxytyramine, and serotonin. Dopamine and 3-methoxytyramine in rat brain striatal dialysates were determined before and after the injection of nomifensine. The pH of the mobile phase can govern the detection selectivity, since oxidation of most organics is accompanied by proton transfer. Reaction of 1 with catechols showed pH-dependent sensitivity resulting from pH-dependent reaction rate changes. Since the reaction rate is also temperature dependent, increased temperature at the mixer resulted in higher sensitivity. However, the noise level also increased at elevated temperature; thus, the detection limit did not improve.
High-performance liquid chromatography with electrochemical detection (HPLC-EC) is a sensitive and selective technique for the quantitative determination of small biomolecules.1 It has been widely used to determine peptides,2 proteins,3,4 and neurotransmitters.5 One advantage of HPLC-EC is its sensitivity and selectivity. A limitation of HPLC-EC comes from the fact that the reaction always occurs at the electrode surface. Substances in biological samples compromise the electrode surface. Attention to the surface has been a major issue in HPLC-EC.
There are several ways to create an optical signal from an analyte’s electrochemical activity. Chemiluminescence detection is an alternative means of obtaining a quantitative signal from a redox reaction. Oxidizable drugs,6–10 amines,11,12 and other species13–15 yield signals. A chemical derivatization step before the chemiluminescent reaction permits determination of neurotransmitters and related species such as catecholamines and hydroxyindoles.16–18
Similarly, electrogenerated chemiluminescence (ECL) has been successful in detection schemes, for example, for ascorbate,19,20 amines21–23 and biopolymers.24,25 The mechanism of the production of ECL in schemes based on ruthenium complexes is now understood.26,27 This mechanistic understanding shines light on the strong dependence of the ECL signal on the analyte properties. A clever way to use ECL that avoids this problem is to require analytes and an ECL-active solution to compete for current.28 This microfluidic-based approach essentially replaces the current measurement in an electrochemical detector with ECL. While simpler from an instrumentation point of view than electrochemical detection, it suffers from poor sensitivity.
The photoluminescence-following electron-transfer (PFET) technique, developed in our laboratory,29,30 is another useful detection scheme for generating an optical signal from electrochemically active molecules. A fluid stream containing a homogeneous oxidant is mixed with chromatographic eluent in a diffusion-controlled mixer.31–33 The oxidant is subsequently reduced to produce photoluminescence upon optical excitation, which is measured quantitatively and recorded. The chemistry involved is much simpler than in the techniques of chemiluminescent detection or ECL described above. PFET does not include derivatization steps, only an electron-transfer reaction. Thus, the optically detected species in PFET is always the same: namely, the reduced form of the oxidant.
To date, PFET applications to peptides29 and explosives30 have used Ru(bpy)33+/2+ (bpy ) 2,2′-bipyridine) as the reagent/photoluminescor. While having certain advantages, it is by no means perfect. It has a rather long lifetime and, thus, is susceptible to quenching.34 This could lead to inaccuracy when using it as the basis for PFET. It is also an aggressive oxidant. The selectivity advantage of electrochemical detection is most obvious at modest potentials. The analogous situation must hold for PFET. Electrochemical detection has the potentiostat; PFET does not. It is imperative to demonstrate alternatives to the aggressive Ru(bpy)33+ as a reagent.
Tris(2,2′-bipyridine)osmium (1) may be a valuable PFET reagent. It has a fast electron-transfer exchange rate and is stable in oxidation states (III) and (II). As a result, it has been used as a mediator35–38 for many electrochemical studies. It happens to be ECL active, as well.39 Like many similar bipyridine or phenanthroline metal complexes, 1 changes its spectroscopic properties according to its oxidation state: 12+ is photoluminescent, while the 13+ complex is not. Interestingly, 12+ has a similar absorbance spectrum to 22+, so that the same laser line can be used to excite both complexes. Also favoring its application to PFET is it short lifetime,40 which will minimize quenching interferences. Most importantly, its redox potential is ~0.4 V less positive than that of the analogous ruthenium complex. Therefore, this complex seems to fulfill a need for a less-aggressive reagent than 22+. Unfortunately, 12+ is a very poor luminophore. Furthermore, its luminescence occurs in the far red region of the electronic spectrum, necessitating the use of a photocathode with a low work function. Photomultipliers based on low work-function photocathodes, of course, suffer from higher dark current than their higher work-function counterparts. Thus, it is not clear at the outset that 13+/2+ will be an effective PFET reagent. The purpose of this paper is to determine the utility of 13+/2+ as a PFET reagent. A companion paper demonstrates the application of the quantitative analysis of dopamine in rat brain microdialysate.
EXPERIMENTAL SECTION
Reagents
All commercial chemicals were used as received without further purification, except where noted. Reagent and sources were as follows: trifluoroacetic acid (TFA) from Acros (Geel, Belgium); monochloroacetic acid from Mallinckrodt (St. Louis, MO); acetonitrile, 2-propanol, and sodium acetate from EMD (Gibbstown, NJ); 1-propanol, lead dioxide, sodium hydroxide, glacial acetic acid, and disodium EDTA from J. T. Baker (Phillipsburg, NJ); tris(2,2′-bipyridine)ruthenium dichloride hexahydrate, glycine, and sodium perchlorate from Aldrich (Milwaukee, WI); chloral hydrate, nomifensine, dopamine (DA) hydrochloride, 3,4-dihydroxyphenylacetic acid (DOPAC), serotonin (5HT) hydrochloride, 3-methoxytyramine (3MT) hydrochloride, 5-hydroxy-indole-3-acetic acid (5HIAA), S-(–)-carbidopa, and sodium 1-octanesulfonate from Sigma (St. Louis, MO). Maleic acid is from Fisher (Fair Lawn, NJ) and is recrystallized once from water. Sodium perchlorate was recrystallized from methanol once to remove chloride. All aqueous solutions were prepared with deionized water (18.2 MΩ resistivity) from a Millipore Milli-Q Synthesis A10 system (Billerica, MA).
Preparation of Metal Polypyridyl Complexes and Solutions
1 was prepared and recrystallized in our laboratory according to previously reported procedures.41,42 Crystals of 1 were dissolved in acetonitrile to make 1.0 mM stock solutions. Aliquots of the stock solution were diluted in an acidic electrolyte solution (0.2% TFA and 0.1 M NaClO4 in acetonitrile or water) to prepare the PFET reagent solution. The resulting solution was used after passing a disposable 0.45 μm poly(tetrafluoroethylene)/ polypropene (PTFE/PP) syringe filter (Chrom Tech, Apple Valley, MN).
Absorbance and Photoluminescence Measurements
Absorbances were measured using an Agilent 8453 UV–visible spectrometer (Agilent Technologies, Wilmington, DE), and corresponding photoluminescence was measured using a fluorometer (Jobin-Yvon, Edison, NJ). Typically, 20 μM complexes in the acidic electrolyte solutions are mixed with 0.1% TFA in water (50:50) to mimic the postcolumn reaction in a chromatographic system. The final concentration of the complex is, thus, 10 μM. To check the reactivity with a solid oxidant, PbO2, complexes were mixed with PbO2 particles and stirred. The resulting slurry was passed through a syringe filter to retain the PbO2 particles, and absorbance and photoluminescence were measured.
Cyclic Voltammetry
All cyclic voltammograms were obtained using an AIS electrochemical analyzer (Model DLK-100A, AIS, Flemington, NJ) with a glassy carbon working electrode and a Ag/AgCl (3 M NaCl) reference electrode. All analytes were prepared in the acidic electrolyte solution (in acetonitrile)–water mixture (50:50) as described before. The analyte concentrations were 1 mM. Each cyclic voltammogram was obtained at the scan rate of 50 mV/s.
Stability of 13+
10 μM 12+ in the acidic electrolyte solution (acetonitrile) was mixed with PbO2 particles and then transferred into quartz cuvettes. An aliquot was prepared by taking the supernatant (without filtration), and another was taken by passing it through the disposable syringe filter (with filtration). UV–vis absorption spectra were measured for 3000 s with an interval of 10 s. Absorbances at 480 nm were corrected for simple baseline shifts by subtracting the absorbance at 800 nm. Corrected absorbances at 480 nm were compared to the absorbance of 12+ at 480 nm to calculate the amount of reconversion from 13+ to 12+.
The pH-dependent reversion of 13+ was measured in a flow-injection system. 13+ was prepared on-line by passing a solution of 12+ through a packed bed of PbO2, which was mixed with buffers at various pHs. Mixtures of monochloroacetic acid and acetic acid (combined concentration of 100 mM) made pH 2.00, 2.30, and 3.00 buffers. The pH of this mixture was adjusted with concentrated sodium hydroxide solution. For pH 4.00 and 5.00 buffers, 100 mM sodium acetate solution was titrated with glacial acetic acid. 100 mM maleate solutions were titrated with concentrated sodium hydroxide solution to adjust to pH 6.00 and 7.00. A 100 mM glycine solution with concentrated sodium hydroxide was used to make a pH 9.00 buffer.
Second-Order Reaction Rate Constant Measurements
The procedure to measure the second-order reaction rate constants of 13+ with solutes has been reported.33 Briefly, clear filtrate of 13+ was mixed with reactant solutions of the same volume in the fluorometer at room temperature. The reactant solutions contained either dopamine or DOPAC at pH ranging from 3.0 to 5.0. Solution pHs are buffered with monochloroacetic acid/acetic acid (pH 3.0 and 3.5) or with acetic acid (pH 4.0 to 5.0). The mixture was optically excited at 470 nm, and the photoluminescence was monitored at 722 nm for 60 s with an integration time of 0.01 s. Traces of signal intensity versus time were converted to product–time traces and fit into a second-order reaction kinetics model with the reaction rate constant as a variable. The Solver function in the Microsoft Excel spreadsheet found the optimum value of the variable, which gave the minimum sum of squared deviations.
Chromatographic System
Two HPLC pumps (models 590 and 600/626S, Waters, Milford, MA) or a syringe pump (model 100DM, ISCO, Lincoln, NE) with simple tees as flow splits delivered solutions at ~1 μL/min. The flow rate from each source was frequently checked by measuring the volume of solutions delivered in a specific time, to ensure the correct flow rate was maintained during experiments.
Homemade capillary columns were packed by previously described techniques43 using 100-μm i.d., 360-μm o.d. fused-silica capillaries as the column blank. The column was slurry packed with reversed-phase particles at 3000–4000 psi using a consta-Metric III metering pump (LDC Analytical, Riveria Beach, FL)32 and connected directly to an Upchurch loop microinjector (Oak Harbor, WA), which introduced 0.5 μL of each sample into the column.
A capillary Taylor reactor (CTR) was used to mix the chromatographic eluents and the postcolumn PFET solution. They were prepared according to the previously reported procedure.32,33,44 The ends of two 18-μm tungsten wires (Goodfellow, Devon, PA) were each threaded into two 50-μm fused-silica capillaries (Polymicro Technologies, Phoenix, AZ). The other ends were both threaded into another 50-μm fused-silica capillary to form a Y-shaped device. The third capillary was transparently coated. The junction between three capillaries was placed in a piece of dual shrink/melt tubing (Small Parts, Miami Lakes, FL). After sufficient heating, the device was allowed to cool and the tungsten wires were removed to create fluid conduits of 18-μm diameter between 50-μm capillaries. It is important to gently tug the wire at the initial stages of the cooling process, or else the wire will stick. The reaction length was set at 8 cm from the confluence according to the previous study.33
Optical Detector
Figure 1 is the diagram of the fluorescence detector. A laser beam from a 30-mW variable-power blue-line argon ion laser (model 2201-30BL, Cyonics/Uniphase, San Jose, CA) passed through a 488-nm band-pass filter and was focused onto an optically transparent capillary. Photoluminescence from 12+ was measured using epifluorescence: a microscopic objective lens (Plan Neofluar 20x, NA 0.5, Carl Zeiss, Thornwood, NY) focused the optical emission from the capillary and a combination of a dichroic mirror (cutoff at 500 nm) and optical filters (a 600-nm long-pass filter and a 750-nm band-pass filter, angle tuned) allowed the photoluminescence into an IR-sensitive photomultiplier tube (R374, Hamamatsu, Bridgewater, NJ). A Keithley 6485 picoammeter (Cleveland, OH) converted photocurrent from the photomultiplier tube to a dc voltage signal. An IBM-compatible computer with a PeakSimple chromatographic data system (SRI Instruments, Torrance, CA) collected the dc signal after a 0.4-Hz eight-pole low-pass filter (Wavetek 852 dual filter, San Diego, CA).
Figure 1.
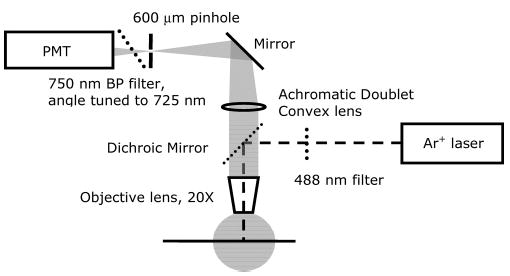
Optical parts of the detection system.
Chromatographic Conditions
Separation of catechols and 5-hydroxyindoleamines was achieved using an acidic mobile phase. Aqueous buffer containing 100 mM sodium acetate, 0.15 mM disodium EDTA, and 0.15 mM sodium 1-octanesulfonate (pH was adjusted to 4.7 with glacial acetic acid) was mixed with methanol (90:10, aqueous/methanol v/v) and passed through a nylon filter with 0.45 μm pores (Osmonics, Minnetonka, MN). The column was slurry packed to 9.5 cm with 3-μm AlltimaC18 (Alltech, Deerfield, IL) reversed-phase material.
Stock solutions of 1.0 mM catechols and standards were prepared in 0.1 M acetic acid and stored frozen. The frozen stock solutions were thawed before each use and were diluted to desired concentrations in degassed solutions. Typically, successive 10-fold dilutions were made using 0.1 M acetic acid, except for in the final dilution, where artificial cerebrospinal fluid (aCSF) was used to mimic the sample matrix of the microdialysis samples. The aCSF contained 145.0 mM NaCl, 2.7 mM KCl, 1.0 mM MgCl2, 1.2 mM CaCl2, 0.45 mM NaH2PO4, and 1.55 mM Na2HPO4 at pH 7.4.
Chromatographic Conditions for Temperature-Controlled Reaction Kinetics Experiments
An acidic mobile phase was used to attenuate the reaction kinetics in the CTR. An aqueous buffer containing 85 mM monochloroacetic acid, 15 mM sodium acetate, 0.15 mM disodium EDTA, and 10.0 mM SOS (pH was adjusted to 2.4 with concentrated sodium hydroxide solution) was mixed with acetonitrile (89:11, aqueous/acetonitrile v/v) and passed through a nylon filter. The column was slurry packed to 7.6 cm with 2.6-μm XTerra MS-C18 (Waters, Milford, MA) reversed-phase particles. Injection samples contained dopamine and serotonin, each at 100 nM, and 0.11 M acetic acid in aCSF.
Temperature Effect on Detection Limit
A homemade heating block controlled the temperature of the CTR. A Minco proportional–integral–derivative (PID) controller (Model CT15122, Minneapolis, MN) drove two square-shaped silicone rubber heaters (SRFG-202/10, 5 × 5 cm2, 2.5 W/in2, Omega, Stamford, CT). An aluminum block directly on top of the heaters transferred heat to the confluence end of the CTR. Between the CTR and the aluminum block were silicone rubber sheets (0.10 in. thickness, McMaster-Carr, Cleveland, OH) to enhance the thermal contact. The heating assembly is placed inside of insulator blocks made from calcium silicate (McMaster-Carr). The insulator assembly helped to stabilize the temperature of the heating block. An elastomer-coated temperature sensor (S665PDZ24A, Minco) in the insulator assembly monitored the actual temperature, and this information was used as a feedback to the PID controller.
For each new temperature setting, enough thermal equilibration time was given. Once the temperature became stable, a blank injection with aCSF was made to determine baseline noise level. The baseline signals of 1–4 min were analyzed with Microsoft Excel’s Regression function. After a linear regression was made for a given data set, the standard deviation of Y-residual values was taken as the noise level. Thus, the noise figure contains both high-frequency noise and low-frequency noise (drift), but not linear drift.
Animal and Surgical Procedures
All procedures involving animals were conducted with approval of the Institutional Animal Care and Use Committee of the University of Pittsburgh. Male Sprague–Dawley rats (250–375 g, Hilltop, Scottdale, PA) were anesthetized with chloral hydrate (initial dose of 300 mg/kg intraperitoneal (i.p.) with additional doses of 50 mg/kg i.p. as needed to maintain anesthesia) and wrapped in a homeothermic blanket (EKEG Electronics,Vancouver, BC, Canada). The rats were placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA) with the incisor bar set at 5 mm above the interaural line,45 and appropriately placed holes were drilled through the skull.
Microdialysis
Vertical concentric microdialysis probes (220 μm o.d., 4 mm long) were constructed with hollow-fiber dialysis membrane (Spectra-Por RC Hollow Fiber, molecular weight cutoff: 13 000, 160-μm i.d., Spectrum Laboratories Inc., Rancho Dominguez, CA) and fused-silica outlet lines (150-μm o.d., 75-μm i.d., Polymicro Technologies, Phoenix, AZ). The microdialysis probe was implanted into the brain over a period of 30 min to the following coordinates: 2.5 mm anterior to bregma, 2.5 mm lateral from midline, and 7.0 mm below dura. The probe was perfused at 0.586 μL/min. Nomifensine (20 mg/kg, i.p.) was prepared in phosphate-buffered saline (PBS) and administrated. All brain dialysates were collected in vials containing 10% (v/v) 0.1 M acetic acid.
RESULTS AND DISCUSSION
Spectroscopic Properties of Complexes
1 has a molar absorptivity, ε (cm−1 M−1), of 11 900 in 0.15% TFA, 0.05 M NaClO4 in 50:50 (v:v) AN/W, at λ = λmax = 483 nm. This is similar to the literature values of 11 100 cm−1 M1 and 478 nm in AN and 12 000 cm−1 M−1 and 480 nm in water.34 In comparison, 2 has a molar absorptivity of 14 200 cm−1 M−1 and λmax = 451 in the same electrolyte. These are very similar to the values in water reported earlier.34 The photoluminescent emission of 1 is somewhat solvatochromatic. Juris et al.34 report values for λmaxem of 715 nm in water and 743 nm in AN. We find a value of 722 nm in the electrolyte described previously. We did not determine photoluminescent quantum yields. Juris et al.34 report a value of 0.046 for 2 and 0.005 for 1. Figure 2 shows excitation and emission spectra for compounds 1 and 2. It is clear that there is considerable sacrifice in luminescent intensity accompanying the change from 2 to 1. Fortunately, the excitation maximum for 1 is closer to the laser line used (488 nm) than is the excitation maximum for 2.
Figure 2.
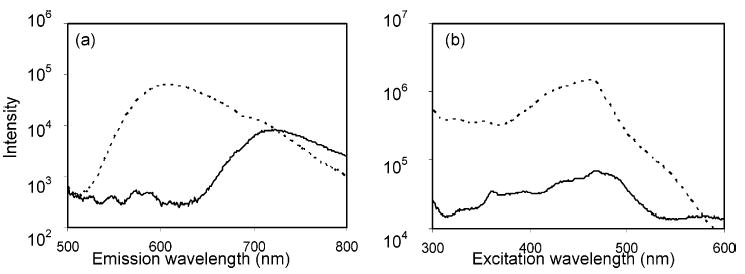
Emission and excitation photoluminescence spectra (log intensity vs wavelength) of complexes. (a) Emission spectra were measured with excitation at 488 nm. (b) Excitation spectra were measured at individual emission wavelength maxima while the wavelength of the excitation beam was scanned from 300 to 600 nm. Photoluminescence intensity on the Y-axis is arbitrary and plotted logarithmically: 1 (solid line) and 2 (dotted line).
Electrochemical Properties
Since 13+ is the oxidant in PFET, the electrochemical properties of 1 are important. Its redox potential basically defines an electrochemical potential window for detectable analytes and, thus, the detection selectivity.
Table 1 shows the measured electrochemical properties of various species. The redox potential of 13+/2+ is ~0.76 V. It can be seen that the ability of 1 to detect compounds is not clearly related to the anodic peak potential of the analytes investigated. 13+ oxidizes ferrocene, ferrocene carboxylic acid, Fe2+, and hydroquinone as well as the catechols dopamine, DOPAC, carbidopa, and norepinephrine, and the indoles serotonin and 5-hydroxyindole acetic acid.33 These oxidations may be expected based on a consideration of the peak potentials in cyclic voltammetry for the analytes. However, it is not always possible to use cyclic voltammetric behavior to infer reactivity of a particular analyte. Just as in standard electrochemical detection, it is probably necessary to test potential analytes experimentally. For example, we note that phenol, bromide, nitrite, and chlorite have rather high peak potentials, yet nitrite and chlorite give signals while phenol and bromide do not. We have not investigated this in detail, but we can say a few things about these reactions. The redox potential for phenol should be ~1.0 V (Ag/AgCl, 3 M NaCl) based on the data of Harriman.46 This is significantly more positive than the redox potential for the reagent. Bromide, on the other hand, has the more-modest redox potential of ~0.86 V (Ag/AgCl, 3 M NaCl),47 yet it does not give a signal. This is likely due to the mechanism of the oxidation. The redox potential for bromide/ bromine radical is ~2 V (Ag/AgCl, 3 M NaCl).47 It is the dimerization of two bromine radicals from bromine that lowers the free energy significantly. This is a second-order reaction; thus, its rate is diminished in homogeneous solution at low concentrations. The solutes nitrite and chorite react with 13+ despite showing very positive anodic peak potentials. No relevant literature could be found on chlorite, but it has been reported that nitrite is oxidized in a catalytic wave by Ce(IV)EDTA.48 The wave is near 0.75 V (Ag/AgCl, 3 M NaCl). This is consistent with its oxidation by 13+. Thus, the selectivity of PFET detection is governed not only by thermodynamics but also by kinetics. This is generally true for electrochemical detection as well. However, it cannot be assumed that the selectivities of the two approaches are the same.
Table 1.
Electrochemical Properties of Various Analytesa
| species | Epa | reaction with 13+ |
|---|---|---|
| Br− | 1.12 | N |
| phenol | 1.18 | N |
| Fe2+ | 0.803 | Y |
| ferrocene carboxylic acid | 0.566 | Y |
| p-hydroquinone | 0.638 | Y |
| ClO2− | >1.3 | Y |
| NO2− | >1.3 | Y |
Electrolyte solutions contain 0.1 M NaClO4 and 0.05% trifluoroacetic acid in 50:50 water/acetonitrile mixture to mimic the detection condition in the PFET system. All potentials are reported versus Ag/AgCl reference electrode.
Oxidation by PbO2
The oxidized complexes are used as a homogeneous oxidant in the PFET system. Previous studies29,30 utilized a packed-bed porous carbon electrode, but this is complicated. It requires a potentiostat and strategic placement of the auxiliary electrode to avoid reconversion of the oxidized product back to the reduced starting material. Any strongly oxidizing redox couple can achieve the same result more simply. PbO2 has been used by Rubenstein and Bard for oxidizing 2.49 Figure 3 shows the UV–vis and photoluminescence spectra of complexes after they were mixed with solid particles of PbO2 (dashed lines). Both spectroscopic measurements lead to the same conclusion: 1 is virtually completely oxidized by PbO2, while 2 was not oxidized at all under our conditions.
Figure 3.
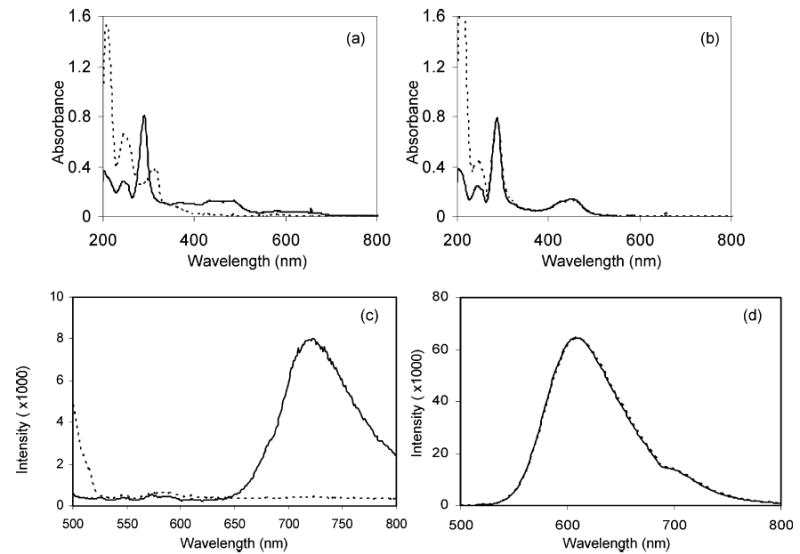
UV absorbance and photoluminescence emission spectra of complexes 1 and 2 before and after reaction with solid PbO2. Solid lines show spectra of the complexes’ original absorbance luminescence, while dashed lines show the absorbance luminescence when the complexes were mixed with PbO2 and subsequently filtered. Complexes are 10 μM in 50:50 (v/v) acetonitrile/water: (a) 1 absorbance, (b) 2 absorbance, (c) 1 photoluminescence, and (d) 2 photoluminescence.
Although clean 13+ can be prepared by simple oxidation with PbO2, it is possible that 13+ can be converted back to 12+ through the oxidation of residual impurities. The residual amount of 12+ is very important in chromatographic applications, since the level of residual 12+ defines the baseline of PFET detection. Figure 4 shows the reversion of 13+ in 0.2% TFA, 0.1 M NaClO4 in AN. This is the same composition as the reagent stream before mixing with the HPLC effluent. 13+ was prepared by reaction with PbO2 particles, and the absorbances at 480 nm were continuously measured to monitor the degree of reversion. Excess PbO2 particles were removed from 13+ by two methods: the solution was filtered by a syringe filter or only the supernatant was taken. The absorbance changes in the two solutions are quite different. The filtered solution showed steady reversion of 13+ over the 20 min, while in the supernatant solution, the reversion stopped after about 500 s. We found that the supernatant solution contained fine particles of PbO2; thus, they were continuously oxidizing 12+. To prevent PbO2 particles from entering the detection system in chromatography, commercial polyetheretherketone (PEEK) frits with a pore size of 0.45 μm (Upchurch, Oak Harbor, WA) were used. In the PFET system, the residence time of 13+ is in the order of 10 s; thus, it is expected that the reversion of 13+ is small in its application in the detection system.
Figure 4.
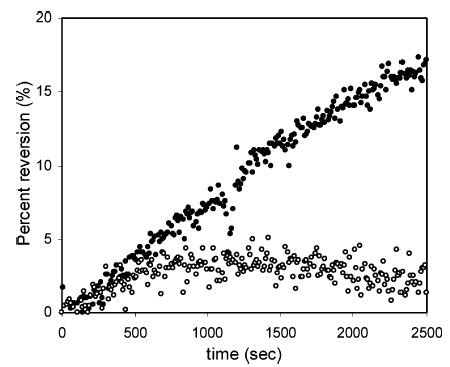
Reversion of 13+. 13+ was prepared from a reaction of 12+ with PbO2. Reversion was calculated by measuring absorbances of the solution at 480 nm. Solid circles are from the filtered mixture, and open circles are from the supernatant. Concentration of 1 is 10 μM in 0.2% TFA, 0.1 M NaClO4 acetonitrile solution.
When 13+ is in the PFET system, however, there is another source of the increased baseline: the introduction of mobile phase. The baseline increase due to the mobile phase was previously reported in a similar system with 23+ as the oxidant.29 Figure 5 shows the reversion of oxidized complexes, 1 and 2,29 when the PFET reagents are mixed with mobile phases at various pHs. It is clear that the reversion increases as the mobile phase becomes more basic. The increased reversion, and thus the baseline, can be due to hydroxide ion.39,50,51 Oxidizable impurities are another possibility. To prevent more than 10% reversion of 13+ requires a pH < 6. For 23+, a pH < 4 is needed. This result is also consistent with the difference in the redox potentials of complexes.34,52
Figure 5.
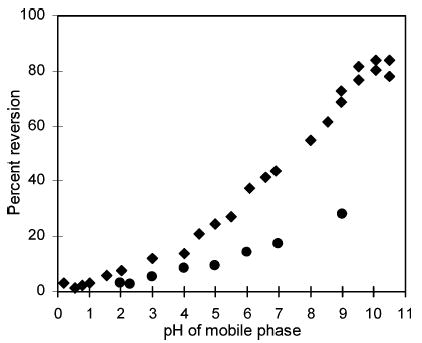
Reversion of 13+ (•) and 23+ (♦ from Woltman et al.29) after being mixed with buffered solutions at various pHs in a flow-injection system.
Control over Sensitivity and Selectivity
Most organic redox reactions are accompanied by proton-transfer steps, while the reduction of 13+ is not. Therefore, the difference in E° between solutes and 13+ may be pH sensitive. Thus, a thermodynamic control exists. There may also be a kinetic means of control by the same route. The second-order rate constants of 13+ with DA and DOPAC were measured in a fluorometer at various pHs. Figure 6 shows that the second-order rate constants at pHs below 4.0 decreased as pH decreased. At pH 4.5 and 5.0, the reaction rates are pH invariant for both species. The pKa for the redox-active functional group is certainly higher than the range of pH 4. This result can be explained by the “scheme of squares”.53–56 Catechols and similar hydroquinone analogues are known to undergo a proton-coupled 2-electron oxidation. It was found from previous studies that the oxidation of catechols follows an eHHe (e = electron transfer reaction, H = proton transfer reaction) mechanism at pH 4–6, while the mechanism changes to an eHeH mechanism at lower pHs.53,55 Rate-determining steps are electron-transfer steps. However, deprotonation at low pH is thermodynamically not favorable, thus making the overall reaction slow at low pH.
Figure 6.
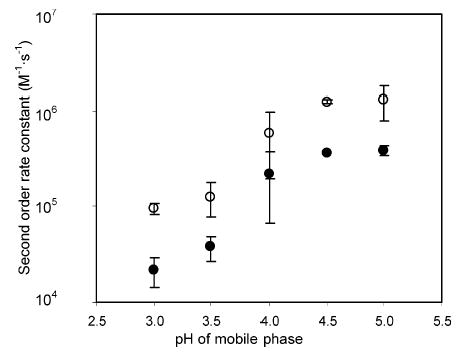
Change of second-order reaction rate constants as a function of mobile-phase pH. Rate constants of reactions between 13+ and dopamine (•) and DOPAC (○) were measured in a fluorometer. The error bars on the figure represent the standard errors of the mean (n = 3 for pH = 4.00 and n = 2 for all other pHs).
It is, therefore, generally desirable to operate the PFET detection system at a relatively high pH in proton-coupled electron-transfer reactions. For separation of certain compounds, however, acidic mobile phases may be beneficial. It was reported that metabolic acids such as DOPAC and amino acids with a pKa ≈ 2 exhibit more retention at pH values below 2.57 In such cases, in principle, increased temperature can increase the sensitivity.
The temperature dependence of the sensitivity was, thus, studied by analyzing chromatographic peaks of dopamine, carbidopa, and serotonin at various temperatures. The reactor, but not the column, was heated. Figure 7 shows the results. The peak area (panel a) increases for dopamine and carbidopa as temperature increases. The peak area for serotonin remains constant. We infer from this that the reaction with serotonin is not kinetically limited on the ~seconds time scale, whereas the reaction with dopamine and carbidopa is. Panel b in the same figure shows that, despite the increase in sensitivity, the detection limit does not change or decreases as temperature increases. However, the baseline, as well as its noise level, increased even more at elevated temperature. The overall effect was similar or worse detection limits at high temperatures.
Figure 7.
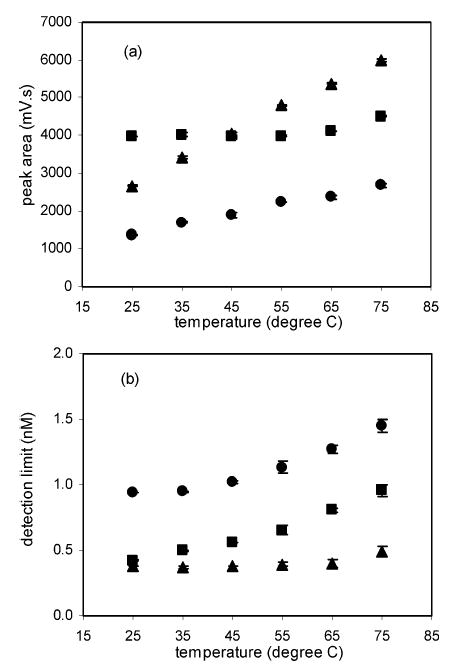
Reaction of 13+ with analytes at various temperatures: (a) peak area; (b) detection limits are compared for chromatographic peaks of dopamine (•), carbidopa (▴), and serotonin (▪). The error bars on the figure represent the standard errors of the mean (n = 4 for 75 °C and n = 2 for all other temperatures).
Figure 7a shows that the peak area of 5HT did not change much with temperature, while that of DA does change with temperature. The concentration of each species was much lower than that of 13+: 100 nM of solutes compared to 10 μM 13+(before mixing). DA was reported previously to undergo slow reaction with 13+ at similar reaction conditions.33 Therefore, one can expect that the reaction rate of DA, and thus the peak area of DA, will increase with temperature. On the other hand, the reaction of 13+ and 5HT might be fast enough to complete the oxidation of 5HT in the given reaction length (8 cm) at room temperature. As a result, the increase of reaction yield from increasing the temperature may be negligible despite an increase in reaction rate. The reason for the increase in baseline and background noise at high temperature, on the other hand, is not certain. The dependence of the noise on the background level is a clear indication that the presence of “background” 12+ is the determining factor in establishing detection limits.
Applications to Biological Separation
The PFET system was applied to detect biogenic monoamines: DA, 3MT, and 5HT. Figure 8a shows a chromatogram of a standard solution containing 100 nM of DOPAC, DA, 5-HIAA, 3MT, and 5HT. Detection limits (signal-to-noise ratio of 3) for DA, 3MT, and 5HT are 0.66, 1.1, and 0.58 nM at 500 nL injection volume (330, 550, and 290 amol), respectively. Detection limits for DOPAC and 5-HIAA are much lower, at 0.50 and 0.26 nM. The HPLC–PFET system was stable for ~6 h without changes in sensitivity.
Figure 8.
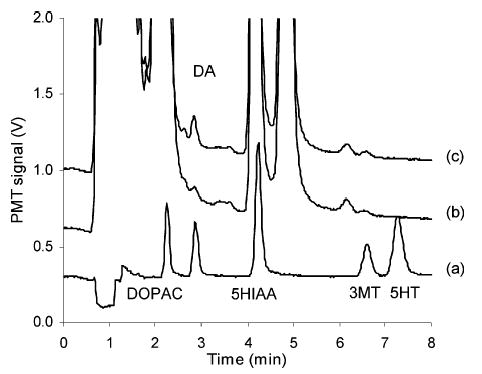
Chromatograms of monoamines: (a) 100 nM standard mixture, brain microdialysis sample from an anesthetized rat, (b) before and (c) after injection of nomifensine. Separation conditions were as follows: mobile phase, pH 4.7 aqueous buffer containing 100 mM sodium acetate aqueous buffer with 0.15 mM disodium EDTA and 0.15 mM sodium 1-octanesulfonate, mixed with methanol (90: 10, v/v); column, 100-μm i.d., 9.5-cm length capillary column packed with 3-μm AlltimaC18 reversed-phase material; flow rate, 1 μL/min; and injection volume, 500 nL.
Rat brain microdialysis samples were analyzed. The basal levels of DA and its metabolite, 3MT, were 16 and 10 nM, respectively. Upon the injection of nomifensine, a DA uptake blocker, extracellular concentrations of DA and 3MT increased significantly (56 and 15 nM, respectively).58
CONCLUSIONS
The reagent 13+ is effective despite its low quantum yield and the use of a red-sensitive photocathode. Having a pair of reagents (1 and 2) that can operate with the same excitation source adds to the flexibility and applicability of PFET. The PFET technique, as demonstrated herein and in the accompanying paper, is highly suited to the quantitative determination of neurotransmitters in microdialysate of rat brain. The major limitation of the technique is nonspecific reduction of the reagent to the photoluminescent form. This is equivalent to background current in electrochemical detection. Major advantages are compatibility with capillary separations, day-to-day reproducibility, selectivity, sensitivity, and the lack of a need to create derivatives of the analytes. Compared to ECL, the breadth of application is far greater.
Acknowledgments
This study was supported by NIH through Grant 2R01GM-044842-12. We acknowledge Dr. Eskil Sahlin for synthesis of 1. We thank Dr. Ed Bouvier at Waters Corp., Milford, MA, for the gift of the XTerra packing material.
References
- 1.Chen JG, Woltman SJ, Weber SG. Adv Chromatogr (N Y, NY, US) 1996;36:273–313. [PubMed] [Google Scholar]
- 2.Sahlin, E.; Beisler, A. T.; Sandberg, M.; Weber, S. G. In Electroanalytical Methods for Biological Materials; Brajter-Toth, A., Chambers, J. Q., Eds.; Marcel Dekker: New York, 2002; pp 367–398.
- 3.Dou L, Krull IS. Anal Chem. 1990;62:2599–2606. doi: 10.1021/ac00222a015. [DOI] [PubMed] [Google Scholar]
- 4.Albery WJ, Svanberg LR, Wood P. J Electroanal Chem Interfacial Electrochem. 1984;162:45–53. [Google Scholar]
- 5.Kissinger PT. J Pharm Biomed Anal. 1996;14:871–880. doi: 10.1016/s0731-7085(96)01781-5. [DOI] [PubMed] [Google Scholar]
- 6.Al-Arfaj NA. Talanta. 2004;62:255–263. doi: 10.1016/j.talanta.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 7.Monji H, Yamaguchi M, Aoki I, Ueno H. J Chromatogr, B. 1997;690:305–313. doi: 10.1016/s0378-4347(96)00412-4. [DOI] [PubMed] [Google Scholar]
- 8.Wei S, Zhao L, Cheng X, Lin JM. Anal Chim Acta. 2005;545:65–73. [Google Scholar]
- 9.Xi J, Shi Ba, Ai X, He Z. J Pharm Biomed Anal. 2004;36:237–241. doi: 10.1016/j.jpba.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 10.Holeman JA, Danielson ND. J Chromatogr, A. 1994;679:277–284. doi: 10.1016/0021-9673(94)80570-9. [DOI] [PubMed] [Google Scholar]
- 11.Noffsinger JB, Danielson ND. J Chromatogr. 1987;387:520–524. [Google Scholar]
- 12.Bolden ME, Danielson ND. J Chromatogr, A. 1998;828:421–430. doi: 10.1016/s0021-9673(98)00833-4. [DOI] [PubMed] [Google Scholar]
- 13.Kim Y-S, Park JH, Choi Y-s. Bull Korean Chem Soc. 2004;25:1177–1181. [Google Scholar]
- 14.Saito K, Murakami S, Yamazaki S, Muromatsu A, Hirano S, Takahashi T, Yokota K, Nojiri T. Anal Chim Acta. 1999;378:43–46. [Google Scholar]
- 15.Tsukagoshi K, Miyamoto K, Nakajima R, Ouchiyam N. J Chromatogr, A. 2001;919:331–337. doi: 10.1016/s0021-9673(01)00809-3. [DOI] [PubMed] [Google Scholar]
- 16.Yakabe T, Yoshida H, Nohta H, Yamaguchi M. Anal Sci. 2002;18:1375–1378. doi: 10.2116/analsci.18.1375. [DOI] [PubMed] [Google Scholar]
- 17.Takezawa K, Tsunoda M, Watanabe N, Imai K. Anal Chem. 2000;72:4009–4014. doi: 10.1021/ac0003697. [DOI] [PubMed] [Google Scholar]
- 18.Ishida J, Takada M, Hitoshi N, Iizuka R, Yamaguchi M. J Chromatogr, B. 2000;738:199–206. doi: 10.1016/s0378-4347(99)00503-4. [DOI] [PubMed] [Google Scholar]
- 19.Chen X, Sato M. Anal Sci. 1995;11:749–754. [Google Scholar]
- 20.Zorzi M, Pastore P, Magno F. Anal Chem. 2000;72:4934–4939. doi: 10.1021/ac991222m. [DOI] [PubMed] [Google Scholar]
- 21.Guo Z, Dong S. Electroanalysis. 2005;17:607–612. [Google Scholar]
- 22.Morita H, Konishi M. Anal Chem. 2002;74:1584–1589. doi: 10.1021/ac0109669. [DOI] [PubMed] [Google Scholar]
- 23.Uchikura K. Chem Pharm Bull. 2003;51:1092–1094. doi: 10.1248/cpb.51.1092. [DOI] [PubMed] [Google Scholar]
- 24.Miao W, Bard AJ. Anal Chem. 2004;76:5379–5386. doi: 10.1021/ac0495236. [DOI] [PubMed] [Google Scholar]
- 25.Miao W, Bard AJ. Anal Chem. 2004;76:7109–7113. doi: 10.1021/ac048782s. [DOI] [PubMed] [Google Scholar]
- 26.Miao W, Choi JP, Bard AJ. J Am Chem Soc. 2002;124:14478–14485. doi: 10.1021/ja027532v. [DOI] [PubMed] [Google Scholar]
- 27.Wightman RM, Forry SP, Maus R, Badocco D, Pastore P. J Phys Chem B. 2004;108:19119–19125. [Google Scholar]
- 28.Zhan W, Alvarez J, Sun L, Crooks RM. Anal Chem. 2003;75:1233–1238. doi: 10.1021/ac026294j. [DOI] [PubMed] [Google Scholar]
- 29.Woltman SJ, Even WR, Weber SG. Anal Chem. 1999;71:1504–1512. doi: 10.1021/ac981181e. [DOI] [PubMed] [Google Scholar]
- 30.Woltman SJ, Even WR, Sahlin E, Weber SG. Anal Chem. 2000;72:4928–4933. doi: 10.1021/ac000170u. [DOI] [PubMed] [Google Scholar]
- 31.Beisler AT, Schaefer KE, Weber SG. J Chromatogr, A. 2003;986:247–251. doi: 10.1016/s0021-9673(02)02018-6. [DOI] [PubMed] [Google Scholar]
- 32.Beisler AT, Sahlin E, Schaefer KE, Weber SG. Anal Chem. 2004;76:639–645. doi: 10.1021/ac034785d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jung MC, Weber SG. Anal Chem. 2005;77:974–982. doi: 10.1021/ac0486241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Juris A, Balzani V, Barigelletti F, Campagna S, Belser P, Von Zelewsky A. Coord Chem Rev. 1988;84:85–277. [Google Scholar]
- 35.Zhang C, Haruyama T, Kobatake E, Aizawa M. Anal Chim Acta. 2000;408:225–232. [Google Scholar]
- 36.Zakeeruddin SM, Fraser DM, Nazeeruddin MK, Graetzel M. J Electroanal Chem. 1992;337:253–283. [Google Scholar]
- 37.Weaver MJ. J Phys Chem. 1990;94:8608–8613. [Google Scholar]
- 38.Nakabayashi Y, Nakamura K, Kawachi M, Motoyama T, Yamauchi O. JBIC, J Biol Inorg Chem. 2003;8:45–52. doi: 10.1007/s00775-002-0385-8. [DOI] [PubMed] [Google Scholar]
- 39.Abruna HD. J Electroanal Chem Interfacial Electrochem. 1984;175:321–326. doi: 10.1016/0302-4598(87)85005-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sutin N, Creutz C. Adv Chem Ser. 1978;168:1–27. [Google Scholar]
- 41.Gaudiello JG, Bradley PG, Norton KA, Woodruff WH, Bard AJ. Inorg Chem. 1984;23:3–10. [Google Scholar]
- 42.Watts RJ, Crosby GA. J Am Chem Soc. 1971;93:3184–3188. [Google Scholar]
- 43.Kennedy RT, Jorgenson JW. Anal Chem. 1989;61:1128–1135. doi: 10.1021/ac00180a012. [DOI] [PubMed] [Google Scholar]
- 44.Sahlin E, Beisler AT, Woltman SJ, Weber SG. Anal Chem. 2002;74:4566–4569. doi: 10.1021/ac025622c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pellegrino, L. J.; Pellegrino, A. S.; Cushman, A. J. A Stereotaxic Atlas of the Rat Brain, 2nd ed.; Plenum Press: New York, 1979.
- 46.Harriman A. J Phys Chem. 1987;91:6102–6104. [Google Scholar]
- 47.Wardman P. J Phys Chem Ref Data. 1989;18:1637–1755. [Google Scholar]
- 48.Abbaspour A, Mehrgardi MA. Talanta. 2005;67:579–584. doi: 10.1016/j.talanta.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 49.Rubinstein I, Bard AJ. J Am Chem Soc. 1981;103:512–516. [Google Scholar]
- 50.Nord G, Wernberg O. J Chem Soc, Dalton Trans (1972–1999) 1975:845–849. [Google Scholar]
- 51.Shi M, Anson FC. Langmuir. 1996;12:2068–2075. [Google Scholar]
- 52.Kalyanasundaram, K. Photochemistry of polypyridine and porphyrin complexes; Academic Press Inc.: San Diego, CA, 1992.
- 53.Deakin MR, Wightman RM. J Electroanal Chem. 1986;206:167–177. [Google Scholar]
- 54.Laviron E. J Electroanal Chem. 1983;146:15–36. [Google Scholar]
- 55.Laviron E. J Electroanal Chem. 1984;164:213–227. [Google Scholar]
- 56.Lebeau EL, Binstead RA, Meyer TJ. J Am Chem Soc. 2001;123:10535–10544. doi: 10.1021/ja000517a. [DOI] [PubMed] [Google Scholar]
- 57.Freeman, K.; Lin, P.; Lin, L.; Blank, C. L. In High Performance Liquid Chromatography in Neuroscience Research; Holman, R. B., Cross, A. J., Joseph, M. H., Eds.; John Wiley & Sons: Chichester, U.K., 1993; pp 27–55.
- 58.Zahniser NR, Larson GA, Gerhardt GA. J Pharmacol Exp Ther. 1999;289:266–277. [PubMed] [Google Scholar]