

Abstract
Perinatal infection with hepatitis C virus (HCV) is characterized by a wide range of alanine aminotransferase (ALT) levels. The mechanisms responsible for this variability are unknown. We examined whether the evolution of the HCV quasispecies was associated with different ALT profiles in perinatally infected children. Sequences within HCV envelope 1 and 2 genes, inclusive of the hypervariable region 1, the viral load, and the nascent humoral immunity were analyzed in serial serum samples from 12 perinatally infected children prospectively followed for a median of 53 months. These patients were selected to represent two different ALT patterns during the first year of life: 6 had high levels (maximum values ranging from 4.2 to 30 times the normal upper limit), and 6 had normal or slightly elevated levels (<2 times the normal upper limit). Two patterns of viral evolution were identified according to the ALT profiles. Biochemical evidence of hepatic injury was invariably associated with a mono- or oligoclonal viral population, whereas mild or no liver damage correlated with the early emergence of a heterogeneous viral quasispecies. Consistent with selective immune pressure, amino acid changes occurred almost exclusively within the hypervariable region 1 and were temporally associated with antibody seroconversion; at this time, the difference in genetic diversity between the two groups was highly significant (P = 0.002). The two patterns of viral evolution persisted over time and did not correlate with viral load or genotype. Our study demonstrates that, in perinatally infected children, the evolution of HCV quasispecies correlates with hepatic injury.
Keywords: perinatal hepatitis C virus infection, genetic variability, alanine aminotransferase levels
Maternal–infant transmission is the dominant route for acquisition of hepatitis C virus (HCV) infection in children in developed countries, with an estimated transmission rate of ≈5% (1–3). Age and mode of infection are assumed to influence the progression of HCV disease in adults, but limited data are available on the natural history of perinatally acquired HCV infection (4, 5). Recent studies suggest that 80% of these children develop chronic infection (6–8), a rate similar to that documented in adults (9) but higher than that reported in children with posttransfusion HCV infection (10). Persistent HCV infection in infants and children is associated with minimal or mild liver damage and very rarely with advanced liver disease (5, 11–14). However, perinatal HCV infection is associated with a wide range of aminotransferase levels during the first year of life (5, 7). Whereas some infants reach serum concentrations of alanine aminotransferase (ALT) compatible with acute hepatitis, others show normal or slightly elevated levels (5, 7).
The mechanisms responsible for the different ALT profiles seen in perinatal HCV infection are unknown but are likely the result of a complex interplay between viral factors and host immunity. One of the most effective strategies enacted by HCV for escaping immune surveillance is genetic variability, specifically the simultaneous presence within the same individual of different, but closely related, viral variants exhibiting a distribution that follows the model referred to as a quasispecies (15). In adults, the early evolution of the viral quasispecies was shown to predict the clinical outcome of acute hepatitis C (16, 17). Likewise, the long-term viral evolution was shown to correlate with the severity of liver disease (18). In perinatally infected children, very limited information is available on the early evolution of the viral quasispecies and on its possible correlation with different ALT profiles (19–22). Access to a well defined cohort of children with perinatal infection prospectively followed by the European Pediatric HCV Network provided a unique opportunity to examine the evolution of the HCV quasispecies in relation to the development of the nascent humoral immune response, the viral kinetics, and the ALT patterns.
Results
Characteristics of the Children.
We studied 12 children (3 males and 9 females) with perinatally acquired HCV infection. These patients were selected because they represented two different ALT profiles during the first year of life, had serial serum samples available for virologic investigations, and had a prolonged follow-up. The first group included 6 children with high ALT levels (with a maximum value greater than four times the upper limit of the normal values; range, 4.2–30 times); the second group included 6 children with no or minimal increase in ALT values (with a maximum value lower than two times the upper limit of the normal values; range, normal to 1.8 times). Table 1 shows the features of the 12 children studied, grouped according to their ALT patterns during the first year of life. Six patients had high ALT values, with a geometric mean of 163.0 IU/liter (Fig. 1). The mean of their maximum values was 262.7 IU/liter (range, 161–1,213) (Table 1). The remaining 6 children had normal or slightly elevated ALT values (mean, 38.9 IU/liter), with a mean of their maximum values of 65.5 IU/liter (range, 56–75). When the patients of the first group (with high ALT values) were compared with those of the second group (with normal or low ALT values), there were no significant differences in the mean time to ALT peak (9.0 and 8.2 months, respectively), to serum HCV RNA peak (7.0 and 6.3 months, respectively), and to anti-HCV seroconversion (8.7 and 9.5 months, respectively). Evidence of seroconversion was considered as either the first appearance of, or a consistent increase in the intensity of, the reactivity against at least two HCV antigens after the loss of maternal antibodies, as determined by RIBA-3. There were no correlations between the biochemical profile and mode of delivery, breast feeding, or HCV genotype (Table 1). In both groups, HCV started to replicate rapidly, reaching high titers of viremia within the first month of life. The peak and mean levels of serum HCV RNA during the first year of life were similar in the two groups, and no significant changes were observed during follow-up (Table 1 and Fig. 1). In the first group of patients (Fig. 1A), ALT levels were significantly higher not only during the first (P = 0.002), but also during the second (P = 0.004) year of life when compared with those of the second group with normal or near-normal ALT values, whereas this difference was no longer significant after the third year of life. In the second group of patients (Fig. 1B), ALT values remained persistently normal or low after the first year of life. The levels of viremia did not correlate with the ALT values (Fig. 1). During the study period, none of the children had clinical symptoms or signs related to HCV infection, and all had regular growth.
Table 1.
Demographic, clinical, and serologic evaluation of hepatitis C in prospectively followed children with perinatal HCV infection grouped according to their ALT pattern during the first year of life
| Patient no. | Sex | Viral genotype | Maternal HIV status | Type of delivery | Breast feeding | Age at presentation, month | Peak ALT, IU/liter (months of age) | Peak of viremia, IU/ml (months of age) | Anti-HCV seroconversion RIBA-III,* months of age |
|---|---|---|---|---|---|---|---|---|---|
| High ALT levels | |||||||||
| 1 | F | 4d | Pos | Vaginal | No | 1 | 208 (7) | 325,000 (1) | 5 |
| 2 | M | 5a | Neg | Vaginal | Yes | 2 | 166 (12) | 1,226,000 (2) | 7 |
| 3 | M | 1a | Pos | Cesarean | No | 2 | 299 (9) | 735,000 (12) | 9 |
| 4 | F | 1b | Pos | Vaginal | No | 4 | 161 (4) | 112,000 (4) | 7 |
| 5 | F | 1a | Pos | Cesarean | No | 9 | 163 (12) | 151,000 (11) | 12 |
| 6 | F | 1a | Neg | Cesarean | No | 9 | 1,213 (10) | 880,000 (12)† | 12† |
| Low ALT levels | |||||||||
| 7 | F | 3a | Neg | Vaginal | No | 9 days | 56 (6) | 720,000 (1) | 6 |
| 8 | F | 2c | Neg | Vaginal | Yes | 6 | 58 (12) | 1,570,000 (12) | 9 |
| 9 | F | 3a | Pos | Cesarean | No | 1 | 67 (7) | 1,838,000 (2) | 7 |
| 10 | F | 3a | Pos | Vaginal | No | 3 | 75 (6) | 36,800 (9) | 13 |
| 11 | F | 1a | Pos | Cesarean | No | 10 days | 65 (7) | 8,700 (10) | 7 |
| 12 | M | 1a | Neg | Vaginal | Yes | 4 | 72 (11) | 880,000 (4) | 15 |
*RIBA-III denotes antibodies detectable on a third-generation recombinant immunoblot assay. When the interval between blood samples was > 3 months, the value represents the midpoint between the last negative sample and the first positive sample.
†Serum samples were not available for HCV RNA levels and RIBA testing before the 12th month of age.
Fig. 1.

Mean ALT values and serum HCV RNA during long-term follow-up in perinatally infected children with high (A) and low (B) ALT levels. Bars indicate the geometric mean ± SE of ALT values and the mean ± SE of serum HCV RNA (measured on a log10 scale), calculated from the means of each patient. Mean values for each patient were calculated from all available values per year. The number of patients analyzed is shown inside the bars. The difference in the mean ALT levels between the two groups of patients was statistically significant during the first (P = 0.002) and second (P = 0.004) years of life.
Evolution of the HCV Quasispecies in Children with Different ALT Patterns.
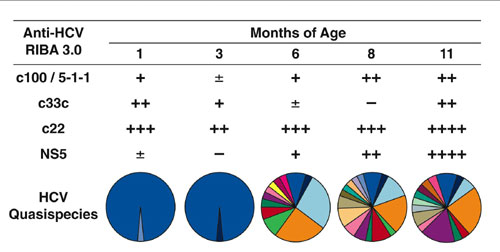
Before anti-HCV seroconversion, neither the genetic diversity nor the number of viral variants within the hypervariable region 1 (HVR1) differed significantly between children with high and low ALT levels (Fig. 2A and B). The viral diversity was generally low in all but two patients (Patients 1 and 12), 1 in each group, who showed high levels of genetic diversity (Hamming distance, 17.65 and 15.52, respectively), most likely reflecting the diversity of the HCV quasispecies acquired from their mothers. By contrast, at the time of antibody seroconversion, two distinct patterns of viral evolution were identified according to the ALT profiles (Figs. 2 and 3). In children with high ALT levels, the viral population became highly homogeneous, with the presence of a monoclonal virus in all patients, whereas, in the others, a complex HCV quasispecies emerged, with a marked increase in both HVR1 genetic diversity and in the number of variants. At this time, the difference in genetic diversity and in the number of variants between the two groups of patients was highly significant (P = 0.002 for both parameters) (Fig. 2 A and B).
Fig. 2.

Genetic diversity (distance among variants) and number of quasispecies variants within and outside HVR1 in perinatally infected children with high and low ALT levels. (A and C) Genetic diversity within the viral quasispecies, as measured by mean Hamming distance both within and outside HVR1. (B and D) Number of quasispecies variants. The values indicate the number of variants per 27 amino acids both within and outside HVR1. The data represent the mean ± SE of the results obtained from all of the patients within each group at different time points. In patients with high ALT levels, “Pre-seroconversion” denotes the results obtained after a mean ± SE of 4.6 ± 1.7 months; “Post-seroconversion” denotes the results obtained after a mean ± SE of 12.8 ± 2.0 months; “Late-follow-up” denotes the results obtained after a mean ± SE of 54.2 ± 5.5 months. In patients with low ALT levels, “Pre-seroconversion” denotes the results obtained after a mean ± SE of 3.6 ± 1.1 months; “Post-seroconversion” denotes the results obtained after a mean ± SE of 13.8 ± 2.3 months; “Late-follow-up” denotes the results obtained after a mean ± SE of 53.5 ± 1.2 months. The analysis before seroconversion was performed on 10 patients because, for 2 (1 in each group), insufficient serum was available; after antibody seroconversion, the analysis was extended to all 12 patients; at the late time point, the analysis was performed on the 8 patients (4 in each group) for whom a long-term follow-up was available.
Fig. 3.

Clinical course and evolution of the HCV quasispecies during the first 2 years of perinatally acquired HCV infection in four representative children, two with high ALT levels (Patients 1 and 2) and two with normal or low ALT levels (Patients 7 and 8). Patient numbers are the same as in Table 1. (A, C, E, and G) Clinical course of hepatitis C. The light blue areas denote ALT levels. The red horizontal bars denote positive assays for serum HCV RNA by PCR. The red lines denote the titer of serum HCV RNA on a logarithmic scale. The yellow horizontal bars denote the infant’s nascent antibody response to HCV, as detected by third generation RIBA. (B, D, F, and H) Number of viral variants and genetic diversity (genetic distance among variants) of the HCV quasispecies within the 27 amino acids of the HVR1. The vertical bars indicate the number and the proportion of viral variants within each sample. The dominant viral variant found in each patient at the first time point is indicated in blue; other variants are indicated by additional colors. Within the vertical bars, each variant is identified by a different color. The same color indicates identity between viral variants detected at different time points within each patient and not between different patients. The viral population diversity (black line) was calculated by mean Hamming distance from the predicted amino acid sequences obtained from each sample.
In children with high ALT values, the presence of a mono- or oligoclonal viral population preceded the biochemical evidence of hepatitis. In these children, the HVR1 consensus sequence of the virus recovered at the time of the ALT peak was identical (Patients 3 and 5) or nearly identical (one amino acid difference, Patient 4) to that obtained at baseline; whereas, in two patients (Patients 1 and 2), a highly divergent monoclonal virus emerged, which differed by 9 and 15 amino acids, respectively, from the baseline (Fig. 3). The rapid emergence of divergent variants in these two children suggests that these mutants were already present in the virus acquired from their mothers rather than representing new variants that had evolved in vivo.
In contrast to the pattern of viral evolution seen in children with biochemical evidence of hepatitis, those with normal or low ALT levels showed a progressive increase in both genetic diversity and the number of quasispecies variants (Fig. 2 A and B). Although several children initially harbored a homogeneous viral population (Fig. 3), a dramatic genetic evolution occurred concomitant with seroconversion (see Fig. 5, which is published as supporting information on the PNAS web site). The degree of genetic variability did not correlate with the levels of viremia. The two different patterns of viral evolution are clearly illustrated by two children first analyzed within the first month of life. In Patient 1, with an ALT peak of 208 IU/liter, the viral population was highly heterogeneous 1 month after birth but became monoclonal 2 months later (Fig. 3 A and B). By contrast, in Patient 7, with a maximum ALT value of 56 IU/liter, the viral population was highly homogeneous during the first 3 months of life; but, 3 months later, concomitant with the nascent humoral immunity, a complex quasispecies emerged with the simultaneous presence of 12 different viral variants (Fig. 3 E and F).
During follow-up, the different patterns of viral evolution continued to be associated with the ALT profiles, although, at a mean age of 54 months, this trend was no longer significant, most likely because of the low number of patients evaluated (Fig. 2). Three representative cases of long-term viral evolution are shown in Fig. 4. In Patient 6, who had the highest ALT peak (1,213 IU/liter at 10 months), the viral population remained highly homogenous, with persistence of the same dominant strain until the last analysis, performed at 57 months of age (Fig. 4 C and D).
Fig. 4.

Long-term clinical course and evolution of the HCV quasispecies during long-term follow-up of perinatally acquired HCV infection in three representative children, two with high ALT levels (Patients 1 and 6), one of whom had an unusually severe hepatitis, with an ALT peak of 1,213 IU/liter (Patient 6), and one with low ALT levels (Patient 8). Patient numbers are the same as in Table 1. (A, C, and E) Long-term clinical course of hepatitis C. (B, D, and F) Long-term evolution of the HCV quasispecies. The symbols are the same as in the legend to Fig. 3.
When we analyzed the genetic diversity and the number of viral variants in the envelope glycoprotein (E)1/E2 region outside HVR1, both were consistently lower than within HVR1 and did not change over time in the two groups (Fig. 2 C and D).
Synonymous and Nonsynonymous Substitutions Within and Outside HVR1.
To investigate whether the different patterns of viral evolution were due to positive selection, we measured the number of synonymous (silent) and nonsynonymous nucleotide substitutions both within and outside the HVR1. This analysis revealed a difference in virus evolution, according to the ALT pattern. Comparison of genetic distances between the earliest and the latest sample from each patient showed that, in children with normal or low ALT levels, the mean rate of nonsynonymous substitutions per site per year within the HVR1 was significantly higher (mean ± SE, 34.8 ± 5.5 × 10−3) than that of synonymous substitutions (mean ± SE, 3.4 ± 1.5 × 10−3) (P = 0.002). By contrast, in patients with high ALT levels, no significant differences were observed between the mean rate of nonsynonymous (mean ± SE, 13.6 ± 4.8 × 10−3) and synonymous substitutions (mean ± SE, 7.3 ± 3.9 × 10−3) (P = 0.131). When this analysis was conducted outside the HVR1, no differences were observed in either group of patients (data not shown).
Phylogenetic Analysis.
Phylogenetic analysis of the HVR1 amino acid sequences obtained from each patient at different time points showed different patterns, according to the ALT profile. In children with biochemical evidence of hepatitis, there was usually a monophyletic population, with intermingling of sequences derived from different time points (see Fig. 6 A and B, which is published as supporting information on the PNAS web site), whereas, in children with low ALT levels, sequential shifts in the viral population were observed at comparable time points (Fig. 6C). Phylogenetic analysis of the rest of the E1/E2 region, after the exclusion of the HVR1, showed no difference between the two groups (data not shown).
Discussion
This study provides important insights into the host–virus interactions in the setting of perinatal HCV infection. We found that, in infants, HCV starts to replicate rapidly, as documented in adults (23) and chimpanzees (24), reaching high titers of viremia before antibody seroconversion and the onset of hepatitis. Interestingly, the initial presence of maternal antibodies, and then of infant antibodies, did not prevent the appearance and persistence of high viral titers. Furthermore, the levels of viremia were disjoined from the ALT values, providing further evidence that HCV is not, by itself, cytopathic. But the most important finding of our study was a significant association between viral evolution and hepatic injury. Strikingly, biochemical evidence of liver damage was invariably associated with the presence of a mono- or oligoclonal viral population, an unexpected finding thus far documented only in patients with fulminant hepatitis C (16, 25). By contrast, in children with normal or slightly elevated ALT values, we documented the generation of a complex quasispecies, with a progressive increase in both the number of viral variants and their genetic diversity. The different patterns of viral evolution coincided with the appearance of anti-HCV antibodies, whereas the patterns did not correlate with the viral load or genotype. Thus, the viral evolution and the early liver damage observed in our children may reflect changes in the host environment, such as the appearance of the adaptive immune responses to HCV.
Several lines of evidence support the view that the immune response plays a central role in the pathogenesis of HCV-associated liver disease (26). In adults, the onset of acute hepatitis C coincides with the appearance of the CD8+ T cell responses and not with viral replication (23). Recent studies show that neonates may mount an effective T cell response, albeit with great variability (27). Even though, in our children, cellular samples were not available for analysis, we can infer that the development of biochemical evidence of hepatitis was the result of the nascent cell-mediated immunity, with the different magnitude of ALT alterations reflecting a different strength in the specific cytotoxic T cell response. By contrast, normal or low ALT levels were likely the result of a relative failure of specific cytotoxic T cells, with a consequent dominance of the antibody response, which exerted a strong selective pressure leading to a dramatic HCV diversification. Consistent with this hypothesis, amino acid substitutions in children with normal or low ALT levels occurred almost exclusively within the HVR1 of the E2 gene, a critical target of selective immune pressure (28) and were temporally associated with antibody seroconversion. These findings are in agreement with the model proposed by Klenerman et al. (29) based on the murine lymphocytic choriomeningitis virus system in which, after depletion of cytotoxic CD8+ T lymphocytes, antibodies become the dominant defense mechanism, leading to rapid virus diversification and the emergence of escape mutants (30).
The long-term outcome of perinatal HCV infection remains to be elucidated (5, 7, 14). Although biochemical evidence of hepatitis during the first year of life has been shown recently to be associated with a greater likelihood of viral clearance (7), none of the children in our study cleared the virus. Continued follow-up of perinatally infected children will permit us to determine whether viral clearance may occur later in childhood or adolescence. This study has documented an association between early evolution of the HCV quasispecies and hepatic injury that persisted over time. Whether the two distinct patterns of viral evolution and ALT might differentially influence the long-term outcome of perinatal HCV infection remains to be established.
In conclusion, our study demonstrates that, in perinatally infected children, the pattern of viral evolution, but not the level of HCV replication, correlates with the ALT profile. Biochemical evidence of liver damage is associated with limited viral diversity and dominance of a single viral strain, whereas mild or no liver damage correlates with the emergence of a heterogeneous viral population, temporally associated with the appearance of anti-HCV antibodies. These results provide insights into the virus–host interactions, which may have important implications for understanding pathogenesis and for devising therapeutic strategies against HCV in children. The identification of perinatally infected children who are most likely to benefit from therapy still represents a major challenge in the management of this special population.
Methods
Patients and Design of the Study.
A total of 12 children (3 males and 9 females) with perinatally acquired HCV infection enrolled in the European Pediatric HCV Network (6) followed at pediatric centers in Turin, Rome, Naples, and Cagliari were selected because they represented two different ALT profiles during the first year of life. The criteria for the diagnosis of HCV infection have been reported in ref. 6. Physical examination and laboratory testing were usually done every 3–6 months during the first 2 years of life and then every 6–12 months. Nine children were first observed within the first 4 months of life (mean ± SE, 2 ± 0.5 months), one at 6 months and two at 9 months (Table 1). All children were followed up to at least 2 years of age (median, 53 months; range, 24–180 months; mean ± SE, 65 ± 14.1 months). All patients were viremic at the last observation. No child in this study was coinfected with HIV, hepatitis B virus, or hepatitis D virus or had a history of blood transfusions or surgery or other types of liver diseases.
The number of quasispecies variants, the genetic distance between the variants (genetic diversity), and the evolution of the HCV quasispecies were assessed by examining viral sequences spanning the envelope genes (E1 and E2) both within and outside the HVR1 [nt 968–1,494 from the sequence of the HCV-1 prototype (31) and nt 1,306–1,833 from the sequence of HCV3a, strain NZL1 (32)]. For each patient, we examined one or two samples before antibody seroconversion, one at the time of seroconversion and then every 6–12 months. In 9 children, the first available PCR-positive sample for analysis of the HCV quasispecies was obtained at 3.3 ± 0.7 months of age; in Patient 11, the first sample was at 9 months of age; in Patient 5, at 10 months; and, in Patient 6, at 19 months of age. This patient started to be tested for anti-HCV antibodies and viremia at 9 months of age and developed the highest ALT peak (1,213 IU/liter at 10 months) among all patients; however, a serum sample for HCV quasispecies analysis was available only from 19 months of age.
All sera were tested for the presence and levels of serum HCV RNA and for anti-HCV throughout the follow-up period. The HCV genotype was determined in all children.
Tests for Anti-HCV.
Anti-HCV antibodies were tested by third-generation ELISA (ELISA-3; Ortho Diagnostics) and immunoblot assay (RIBA-3; Chiron).
Test for HCV RNA and Genotype.
Total RNA was extracted from 100 μl of serum by using TRIzol reagent (GIBCO/BRL) and reverse transcribed in a volume of 20 μl. The resulting cDNA was amplified by using one of two sets of nested primers spanning the E1 and E2, according to the HCV genotype: the first set (33) was derived from the sequence of the HCV-1 prototype (31); the second set was deduced from the sequence of HCV3a, strain NZL1 (32) (see Supporting Materials and Methods, which is published as supporting information on the PNAS web site). The presence and levels of serum HCV RNA were measured by commercial assays (Cobas Amplicor HCV 2.0 and Cobas Amplicor HCV Monitor 2.0; Roche Molecular Systems, Branchburg, NJ). The HCV genotype was determined by sequence analysis of part of the E1 gene (34).
Molecular Cloning and Sequencing.
The PCR products amplified from the E1 and E2 region of the HCV genome were purified by using a QIAquick gel extraction kit (Qiagen, Hilden, Germany), cloned into pCR2.1-TOPO vector (Invitrogen Life Technologies, Carlsbad, CA), and transformed into Escherichia coli strain J109. Plasmid DNA was extracted with the QIAprep spin miniprep kit (Qiagen) and sequenced by using Applied Biosystems 373 automated DNA sequencer with a modified Sanger method. A total of 2,672 molecular clones, each 528 nucleotides long, were sequenced, with a mean ± SE of 34.3 ± 0.41 sequences from each sample; 24 sequences were excluded from the analysis because they were defective.
Sequence Analysis.
The genetic diversity of HCV was assessed by the analysis of 176 amino acids and calculating the Hamming distance, which is defined as the number of amino acid differences between two sequences (35). The mean Hamming distance, which is the average of the values taken for all sequence pairs derived from a single sample, was separately calculated on HVR1 (27 amino acids) and on the entire E1 and E2 sequence outside the HVR1 (149 amino acids). The average number of synonymous (silent) nucleotide substitutions per synonymous site (Ks) and the number of nonsynonymous (amino acid replacement) nucleotide substitutions per nonsynonymous (Ka) site relative to the ancestral consensus sequence were calculated (36) by using the program mega (Molecular Evolutionary Genetic Analysis) (37). Within a single patient, sequences from each time point were compared with the consensus (reference) sequence of the first time point. The phylogenetic trees were constructed with the neighbor program in the phylip package (38).
Statistical Analysis.
Data were expressed as the mean ± SE. The ALT levels were calculated as geometric means because of the wide variability in values. The levels of serum HCV RNA were calculated on a log10 scale. Differences in ALT values, levels of viremia, genetic diversity, and number of viral strains were evaluated by Mann–Whitney U test. Differences between the average rates of synonymous (Ks) and nonsynonymous (Ka) substitutions were evaluated by paired t test. All statistical tests were two-sided and considered statistically significant when P values were <0.05.
Supplementary Material
Acknowledgments
We thank Dr. Robert H. Purcell for his support for automated sequence analysis and Karin Prims and Harry Newman for editorial assistance. This study was supported by grants from Regione Autonoma della Sardegna and from COFIN 2003, Italy.
Abbreviations
- ALT
alanine aminotransferase
- E
envelope glycoprotein
- HCV
hepatitis C virus
- HVR1
hypervariable region 1.
Footnotes
References
- 1.Ohto H., Terazawa S., Sasaki N., Sasaki N., Hino K., Ishiwata C., Kako M., Ujiie N., Endo C., Matsui A., et al. N. Engl. J. Med. 1994;330:744–750. doi: 10.1056/NEJM199403173301103. [DOI] [PubMed] [Google Scholar]
- 2.Zanetti A. R., Tanzi E., Paccagnini S., Principi N., Pizzocolo G., Cacciamo M. L., D’Amico E., Cambie G., Vecchi L. Lancet. 1995;345:289–291. doi: 10.1016/s0140-6736(95)90277-5. [DOI] [PubMed] [Google Scholar]
- 3.Resti M., Azzari C., Mannelli F., Moribondo M., Novembre E., de Martino M., Vierucci A. Br. Med. J. 1998;317:437–441. doi: 10.1136/bmj.317.7156.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwimmer J. B., Balistreri W. F. Semin. Liver Dis. 2000;20:37–46. doi: 10.1055/s-2000-9257. [DOI] [PubMed] [Google Scholar]
- 5.Jonas M. M. Hepatology. 2002;36:S173–S178. doi: 10.1053/jhep.2002.36799. [DOI] [PubMed] [Google Scholar]
- 6.Tovo P. A., Pembrey L. J., Newell M. L. J. Infect. Dis. 2000;181:419–424. doi: 10.1086/315264. [DOI] [PubMed] [Google Scholar]
- 7.Resti M., Jara P., Hierro L., Azzari C., Giacchino R., Zuin G., Zancan L., Pedditzi S., Bortolotti F. J. Med. Virol. 2003;70:373–377. doi: 10.1002/jmv.10405. [DOI] [PubMed] [Google Scholar]
- 8.Mast E. E., Hwang L. Y., Seto D. S., Nolte F. S., Nainan O. V., Wurtzel H., Alter M. J. J. Infect. Dis. 2005;192:1880–1889. doi: 10.1086/497701. [DOI] [PubMed] [Google Scholar]
- 9.Alter H. J., Seeff L. B. Semin. Liver Dis. 2000;20:17–35. doi: 10.1055/s-2000-9505. [DOI] [PubMed] [Google Scholar]
- 10.Vogt M., Lang T., Frosner G., Klingler C., Sendl A. F., Zeller A., Wiebecke B., Langer B., Meisner H., Hess J. N. Engl. J. Med. 1999;341:866–870. doi: 10.1056/NEJM199909163411202. [DOI] [PubMed] [Google Scholar]
- 11.Palomba E., Mancini P., Fiammingo P., Maderni P., Saracco G., Tovo P. A. Clin. Infect. Dis. 1996;23:47–50. doi: 10.1093/clinids/23.1.47. [DOI] [PubMed] [Google Scholar]
- 12.Bortolotti F., Resti M., Giacchino R., Azzari C., Gussetti N., Crivellaro C., Barbera C., Mannelli F., Cancan L., Bertolini A. J. Pediatr. 1997;130:990–993. doi: 10.1016/s0022-3476(97)70289-0. [DOI] [PubMed] [Google Scholar]
- 13.Guido M., Rugge M., Jara P., Hierro L., Giacchino R., Larrauri J., Cancan L., Leandro G., Marino C. E., Balli F., et al. Gastroenterology. 1998;115:1525–1529. doi: 10.1016/s0016-5085(98)70032-0. [DOI] [PubMed] [Google Scholar]
- 14.European Paediatric Hepatitis C Virus Network. Clin. Infect. Dis. 2005;41:45–51. doi: 10.1086/430601. [DOI] [PubMed] [Google Scholar]
- 15.Martell M., Esteban J. I., Quer J., Genesca J., Weiner A., Esteban R., Guardia J., Gomez J. J. Virol. 1992;66:3225–3229. doi: 10.1128/jvi.66.5.3225-3229.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farci P., Shimoda A., Coiana A., Diaz G., Peddis G., Melpolder J. C., Strazzera A., Chien D. Y., Munoz S. J., Balestrieri A., et al. Science. 2000;288:339–344. doi: 10.1126/science.288.5464.339. [DOI] [PubMed] [Google Scholar]
- 17.Laskus T., Wilkinson J., Gallegos-Orozco J. F., Radkowski M., Adair D. M., Nowicki M., Operskalski E., Buskell Z., Seeff L. B., Vargas H., et al. Gastroenterology. 2004;127:764–776. doi: 10.1053/j.gastro.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 18.Qin H., Shire N. J., Keenan E. D., Rouster S. D., Eyster M. E., Goedert J. J., Koziel M. J., Sherman K. E., Multicenter Hemophilia Cohort Study Group Blood. 2005;105:533–541. doi: 10.1182/blood-2004-04-1452. [DOI] [PubMed] [Google Scholar]
- 19.Murakami J., Okamoto M., Miyata H., Nagata I., Shiraki K., Hino S. Pediatr. Res. 2000;48:450–456. doi: 10.1203/00006450-200010000-00006. [DOI] [PubMed] [Google Scholar]
- 20.Manzin A., Solforosi L., Debiaggi M., Zara F., Tanzi E., Romano L., Zanetti A. R., Clementi M. J. Virol. 2000;74:4327–4334. doi: 10.1128/jvi.74.9.4327-4334.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caudai C., Battiata M., Riccardi M. P., Toti M., Bonazza P., Padula M. G., Pianese M., Valensin P. E. J. Clin. Microbiol. 2003;41:3955–3959. doi: 10.1128/JCM.41.8.3955-3959.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pollack H., Hou Z., Hughes A. L., Borkowsky W. J. Acquir. Immune Defic. Syndr. 2004;36:890–899. doi: 10.1097/00126334-200408010-00002. [DOI] [PubMed] [Google Scholar]
- 23.Thimme R., Oldach D., Chang K. M., Steiger C., Ray S. C., Chisari F. V. J. Exp. Med. 2001;194:1395–1406. doi: 10.1084/jem.194.10.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thimme R., Bukh J., Spangenberg H. C., Wieland S., Pemberton J., Steiger C., Govindarajan S., Purcell R. H., Chisari F. V. Proc. Natl. Acad. Sci. USA. 2002;99:15661–15668. doi: 10.1073/pnas.202608299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farci P., Alter H. J., Shimoda A., Govindarajan S., Cheung L. C., Melpolder J. C., Sacher R. A., Shih J. W., Purcell R. H. N. Engl. J. Med. 1996;335:631–634. doi: 10.1056/NEJM199608293350904. [DOI] [PubMed] [Google Scholar]
- 26.Bowen D. G., Walker C. M. Nature. 2005;436:946–952. doi: 10.1038/nature04079. [DOI] [PubMed] [Google Scholar]
- 27.Adkins B., Leclerc C., Marshall-Clarke S. Nat. Rev. Immunol. 2004;4:553–564. doi: 10.1038/nri1394. [DOI] [PubMed] [Google Scholar]
- 28.Farci P., Shimoda A., Wong D., Cabezon T., De Gioannis D., Strazzera A., Shimizu Y., Shapiro M., Alter H. J., Purcell R. H. Proc. Natl. Acad. Sci. USA. 1996;93:15394–15399. doi: 10.1073/pnas.93.26.15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klenerman P., Lechner F., Kantzanou M., Ciurea A., Hengartner H., Zinkernagel R. Science. 2000;289:2003. doi: 10.1126/science.289.5487.2003a. [DOI] [PubMed] [Google Scholar]
- 30.Ciurea A., Hunziker L., Zinkernagel R. M., Hengartner H. Immunogenetics. 2001;53:185–189. doi: 10.1007/s002510100314. [DOI] [PubMed] [Google Scholar]
- 31.Choo Q. L., Richman K. H., Han J. H., Berger K., Lee C., Dong C., Gallegos C., Coit D., Medina-Selby R., Barr P. J., et al. Proc. Natl. Acad. Sci. USA. 1991;88:2451–2455. doi: 10.1073/pnas.88.6.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sakamoto M., Akahane Y., Tsuda F., Tanaka T., Woodfield D. G., Okamoto H. J. Gen. Virol. 1994;75:1761–1768. doi: 10.1099/0022-1317-75-7-1761. [DOI] [PubMed] [Google Scholar]
- 33.Farci P., Alter H. J., Govindarajan S., Wong D. C., Engle R., Lesniewski R. R., Mushahwar I. K., Desai S. M., Miller R. H., Ogata N., et al. Science. 1992;258:135–140. doi: 10.1126/science.1279801. [DOI] [PubMed] [Google Scholar]
- 34.Bukh J., Purcell R. H., Miller R. H. Proc. Natl. Acad. Sci. USA. 1993;90:8234–8238. doi: 10.1073/pnas.90.17.8234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ganeshan S., Dickover R. E., Korber B. T., Bryson Y. J., Wolinsky S. M. J. Virol. 1997;71:663–677. doi: 10.1128/jvi.71.1.663-677.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nei M., Gojobori T. Mol. Biol. Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- 37.Kumar S., Tamura K., Nei M. mega: Molecular Evolutionary Genetics Analysis. University Park, PA: Pennsylvania State Univ; 1993. [Google Scholar]
- 38.Felsenstein J. phylip: Phylogeny Inference Package. Seattle: Univ. of Washington; 2000. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}