

Abstract
The realization that experimentally observed functional motions of proteins can be predicted by coarse-grained normal mode analysis has renewed interest in applications to structural biology. Notable applications include the prediction of biologically relevant motions of proteins and supramolecular structures driven by their structure-encoded collective dynamics; the refinement of low-resolution structures, including those determined by cryo-electron microscopy; and the identification of conserved dynamic patterns and mechanically key regions within protein families. Additionally, hybrid methods that couple atomic simulations with deformations derived from coarse-grained normal mode analysis are able to sample collective motions beyond the range of conventional molecular dynamics simulations. Such applications have provided great insight into the underlying principles linking protein structures to their dynamics and their dynamics to their functions.
Introduction
Recent advances in sequencing and structural genomics indicate that the canonical sequence-to-structure-to-function paradigm is insufficient for understanding and controlling the mechanisms of biomolecular interactions and functions. Because molecular structures are dynamic rather than static, information regarding their dynamics is required to establish the link between structure and function. Normal mode analysis (NMA) has re-emerged in recent years as a powerful method for elucidating the structure-encoded dynamics of biomolecules. NMA has been applied to proteins since the early 1980s [1,2]. However, its usefulness in structural biology has been exploited only recently, after the observation that the collective motions predicted by NMA for folded structures are highly robust and bear functional significance [3,4,5••]. Although the actual motions of macromolecules in solution are very complex, involving transitions among innumerable conformations, the success of NMA hinges upon the fact that motions near native state conditions are much simpler and more robust. Structural changes are dominated by the inter-residue contact topology of the folded state, implying that the most probable deformations are those requiring the smallest energy ascent in the multidimensional energy landscape.
It is plausible that the motions NMA predicts are functional if one considers that each protein functions only if it is folded into its equilibrium/native structure and that each equilibrium structure encodes a unique equilibrium dynamics. Furthermore, NMA yields a unique analytical solution of the modes of motion accessible at equilibrium (near a global energy minimum). Thus, the equilibrium dynamics predicted by NMA, and the structure-encoded collective motions in general, ought to be functional, based on the premise that each protein has evolved to optimally achieve its biological function.
This review centers on the use of coarse-grained NMA methods to refine experimental data and predict biological functional features from macromolecular structures. The merits of several related methods are discussed, as well as recent successes in identifying the intrinsic motions of proteins and future prospects. Special attention is given to applications in which these models are used to predict motions, dynamics, and critical residues for function or folding.
EN models and coarse-grained NMA
Building upon the ability of NMA to predict the most probable cooperative motions of biomolecular structures, much of the increased utilization of NMA in recent years has resulted from the introduction of computationally simpler elastic network (EN) models. These EN models replace detailed atomic potentials with uniform harmonic potentials between interacting atom or residue pairs [6-8]. These and subsequent studies have demonstrated that the large-scale collective motions predicted by NMA are insensitive to both the model and the details of the force-fields used, provided that the topology of inter-residue contacts in the native structure is accurately modeled [6-11]. Given the computational efficiency of coarse-grained NMA, a convenient methodology has been to map the protein structure onto its EN model, perform a coarse-grained NMA using an EN model of suitable resolution to generate ‘alternative’ structures sampled during equilibrium fluctuations, and use the NMA-generated ‘alternative’ forms to characterize the natural dynamics or reconstruct structures at their atomic-level representation. This three-step procedure and associated applications are summarized in Figure 1. Below, we briefly describe the tasks indicated in the figure and discuss the various applications to structural biology.
Figure 1.
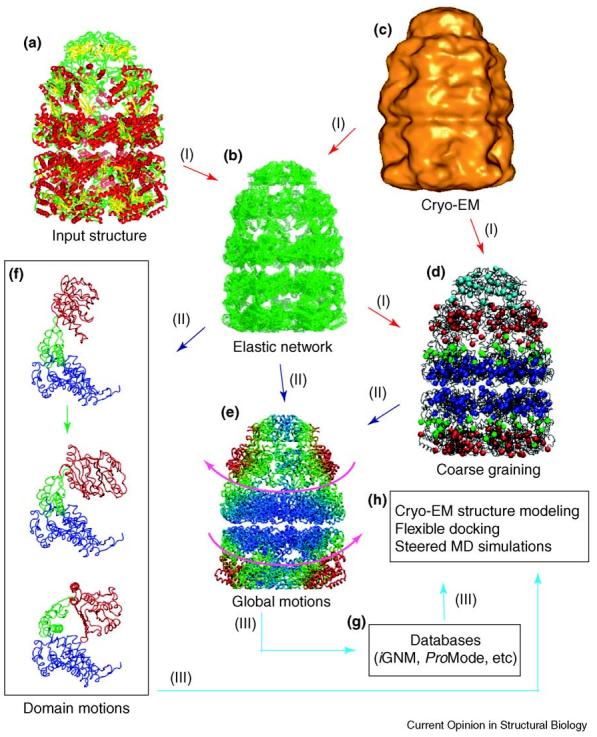
Overview of various methodologies and applications to the GroEL–GroES complex of EN models. The EN model (b) requires an initial input structure, typically an atomic-resolution structure such as in (a), colored according to secondary structure elements. As noted in the text, a lower-resolution structure, such as a cryo-EM map (c), can also be used as input for constructing an EN model. In order to process supramolecular assemblies, further coarse graining (d) is adopted. A low-resolution EN model in which only every 20th residue is used to define the nodes is shown. Once the EN model is constructed, various motions are calculable by NMA, ranging from the level of the entire molecule to domains and individual residues. (e) The global motions computed for the GroEL–GroES complex (PDB code 1gru) [68], revealing a counter-rotation of the GroES-bound (trans) ring with respect to the lower (cis) ring (as shown by the magenta arrows). The structure has been colored by increasing mobility from blue to red, showing that the mobility increases with increasing distance from the interface between the cis–trans rings and from the cylindrical axis of symmetry. (f) The motions of the individual subunits, each composed of three domains (apical, red; intermediate, green; equatorial, blue), obtained from analysis of the EN model. The top diagram shows the ATP-bound form of a subunit in the trans ring and the lowest diagram is its unliganded counterpart in the cis ring. Applying the deformations from the first (slowest) mode calculated by NMA to the trans ring monomer produces the middle structure, demonstrating the intrinsic (structure-encoded) ability of the subunit to reconfigure into the closed form assumed in the cis ring. This is consistent with successive interchange of the subunit conformations between the two forms upon binding of the cap to either ring and cap dissociation during the chaperonin cycle. From these calculations, (g) databases of global motions have been constructed, and (h) several important additional applications of these motions and deformations have been indicated.
Mapping the structure onto reduced models that maintain contact topology
The most common model adopted in coarse-grained NMA involves a single site per residue representation, in which the sites are identified by the Cα atoms and connected by uniform springs. The dynamics of such an interconnected bead-and-spring model can be described by the Gaussian network model (GNM) or an EN model using potentials of the form:
| (1) |
| (2) |
for the GNM and the EN model, respectively. Here, γ is the uniform spring constant, and Rij are the original and instantaneous distance vectors between residues i and j, and Rij are the corresponding magnitudes; the summation is performed over the pairs of residues/nodes filtered through the function , which selects the interacting pairs. is either the Heaviside function based on an interaction cut-off distance of Rc[ if and zero otherwise] [10,11] or an exponentially decaying function of distance [9].
Lower resolution models have been adopted in order to examine larger biomolecular assemblies, whereby groups of residues are clustered into unified sites [12,13] or rigid blocks (such as the rotations and translations of blocks [RTB] and block normal mode [BNM] methods) [14,15]. Related methods effectively quantize the shape of the structure without directly identifying specific residues or groups of residues [16,17]. A reduction in the number of nodes by one order of magnitude increases the computation speed by three orders of magnitude, as NMA computing time scales with N3. Notably, the global motions computed by such coarse-grained NMA maintain their fundamental characteristics and can be related to functional mechanisms [13].
Performing NMA with EN models: functional deformations and critical sites
NMA depends upon the eigenvalue decomposition of the Hessian matrix — a 3N × 3N matrix composed of the second derivatives of the potential (V ) with respect to residue fluctuations. Thus, for an EN model potential (Equation 2), one obtains 3N–6 normal mode vectors describing anisotropic deformations. In the case of the GNM, the Hessian is replaced by the N × N Kirchhoff matrix (Γ), which describes the inter-residue contact topology, such that N–1 isotropic modes are obtained. The B-factors computed by the GNM yield good agreement with X-ray crystallographic data [18] and NMR order parameters [19]. However, the mechanisms of deformations cannot be characterized unless a 3N-dimensional Hessian is used in NMA.
An exciting contribution of NMA to structural biology is its ability to provide insight into large-scale and long-time conformational motions of proteins, which tend to be inaccessible to standard molecular dynamics (MD) techniques. Recent applications to very large supramolecular assemblies include the ribosome [20,21] and viral capsids [22,23]. In general, a few of the low-frequency modes (uj) predicted by NMA exhibit a large degree of overlap:
| (3) |
with the vector describing the displacement between two known conformations (Δr) [11]. Overlap values exceeding 80% suggest that the structures (open and closed) have an intrinsic tendency to reconfigure along a small set of low-frequency modes, even if the fully evolved conformational change might involve passage over a conformational energy barrier. Recently, it has been shown that only minimal information about the target structure is required to drive one structure into the other through a linear combination of low-frequency normal modes [24].
The usefulness of NMA becomes particularly significant when combined with experimental data. Notable applications that provide insights into functional mechanisms include the study of muscle myosin ATPase regulation [25] and flexibility [26,27•], the modulation of protein flexibility during the RNA polymerase cycle [28] and the elucidation of the ribosomal machinery [20,21].
Although these coarse-grained Cα-based NMA methods lack any sequence specificity, there is increasing evidence of their ability to identify functional and structural roles of individual residues. Many studies have identified residues that impart inherent stability and are critical for folding [29-31], as well as residues that form binding ‘hot spots’ [32], catalytic residues [33•] and deformable residues [34].
Applications to structural biology: use in predicting structure and dynamics
Flexible docking
A major application of NMA is the identification of potential conformational changes (e.g. of enzymes upon ligand binding) [11,35]. In particular, it has been shown that over half of 3800 known protein motions (inferred from different conformations of the same protein) can be approximated by perturbing the original structures along the direction of their two lowest-frequency normal modes [36]. Such results suggest that protein structures may have evolved to accommodate or facilitate biologically functional conformational changes. Among the alternative mechanisms of motion accessible near the folded state, those along the smoothest ascent directions are the most readily explored. The biological functions will then be more readily achieved, provided that the associated motions coincide with those smoothest ascent directions (i.e. those along the lowest-energy modes). The fact that the observed changes coincide with those predicted by the slowest NMA modes should not be a coincidence, but a design principle favored by nature. Building on the notion that NMA can be used to identify potential motions induced by binding, a computationally tractable way to generate a set of docking targets has been proposed [35].
Cryo-EM structure modeling
Recently, there have been several applications of NMA to low-resolution cryo-electron microscopy (cryo-EM) structure modeling. Such experimental data are naturally low resolution, being reconstructed by averaging over multiple images of many molecules from several different angles. Additionally, the imaged molecules often undergo structural changes together with vibrations, making it very difficult to extract high-resolution structural information. Several groups [16,17,37•] have constructed EN models of pseudo-atomic representations for a given cryo-EM map and calculated the resulting distortions due to normal modes as an aid in the refinement of the raw cryo-EM data to produce higher resolution structural information. Alternatively, a procedure for the flexible docking of atomic or residue-level structures into cryo-EM maps has been suggested, using the NMA mode shapes calculated for either the pseudo-atomic EN models or homology-based structures [37•,38•,39,40••].
Domain identification
Because elastic networks quickly identify coupled motions, it is possible to partition a protein into various domains [9]. Recently, this idea of decomposing proteins into domains based on their structural topology has been automated [41], and applied to identifying domains that have been recombined or swapped during evolution [42].
Steering MD simulations and exploring non-equilibrium dynamics
As discussed above, the low-frequency modes from NMA are able to capture the collective dynamics of proteins. This fact has recently been applied to steer MD simulations along these dominant modes of motion using hybrid methods that combine MD and harmonic modes [43••,44,45]. Specifically, a hybrid MD/NMA simulation protocol has been implemented, whereby motions along the direction of the slowest few modes are coupled to a temperature bath and thus amplified to study the unfolding and large-scale domain motions of peptides and proteins [43••,44]. The inverse of this approach, namely that the normal modes of a protein can be extracted from an applied driving force in an MD simulation [46], has also recently been shown.
Drawing on similar insight, it has been suggested that one can minimize steric clashes and interpolate between two conformations of a protein using the modes from an EN model [47] to characterize this transition. Because the harmonic approximation of NMA remains valid only near the equilibrium structure, an alternative method for escaping the local minima surrounding the native state involves the iterative calculation of successive EN models deformed along one or several low-frequency modes [48]. This method allows ‘cracking’ or partial unfolding of the underlying EN structure, suggesting that such unfolding or ‘proteinquakes’ may be coupled to collective motions [49,50•].
High-throughput examination of families of proteins
Fold families, such as globins [51], and protein super-families [52••] in general have been compared using NMA-based methods to identify common and distinctive structural and dynamic features. For the test case of proteases, salient dynamic features derived from GNM calculations, combined with data-mining methods in an unsupervised learning technique, have been shown to identify the highly conserved catalytic triad [53]. More recently, the minima in the slowest modes (global hinge centers) have been shown to be co-localized near catalytic residues in a representative set of enzymes [33•]. These results indicate that a great deal of information about functional residues can be extracted from the comparative coarse-grained NMA of protein family members.
How are NMA predictions verified by experiments?
Inherent to many of these computational predictions is assignment of correlated or collective motions. Several experimental techniques, including hydrogen-deuterium (H/D) exchange, FRET probes and labeled NMR, have the capacity to verify such predictions by identifying pairs of residues that experience coupled motions. Key residues predicted to act as functional hinges or ligand-binding sites [33•], or critical to folding [29,30] are tested by site-directed mutagenesis (e.g. correlated mutations), H/D exchange data and other biochemical (e.g. cross-linking) experiments. The free energy changes associated with H/D exchange of individual amino acids measured near native state conditions for a series of proteins have been correlated, for example, with the entropic costs predicted by the GNM [54]; the experimentally observed structural changes of enzymes between their open and closed forms have been shown, in several applications, to correlate with the low-frequency motions predicted by coarse-grained NMA [11,19,33•,55]. As mentioned above, NMA results are particularly useful in providing insights into molecular mechanisms of biological function when interpreted in conjunction with experimental data [21,25,26,27•,28-32,33•].
Databases and servers of molecular motion
The logical extension of family analysis is the compilation and maintenance of web-accessible databases housing NMA-based calculations for all available protein structures. Several such databases have been constructed, including iGNM [56], ProMode [57], ElNémo [58], WEBnm [59] and MolMovDB [60], which allow the user to browse precalculated data and/or submit structures for NMA.
Conclusions and perspectives
The past five years have seen a renewed interest in NMA-inspired methods because they provide a biologically relevant and unique analytical solution of the equilibrium dynamics of biomolecules. The successes of NMA indicate that three-dimensional structures contain the requisite information to determine functional motions. The most collective, or global, modes of motion predicted by NMA are insensitive to the details of models and energy parameters, and instead depend on the topology of inter-residue contacts at equilibrium; this justifies the widespread use of the more efficient coarse-grained EN models described here. Such approaches are now being used, in conjunction with experimental studies, to unravel the supramolecular dynamics and long timescale motions of large structures that are otherwise inaccessible via conventional simulations.
These studies lead to emerging paradigms for a dual role for key structural elements in both chemical and mechanical activities of enzymes [33•,61], or in both folding and signaling properties of membrane proteins [30,62]. More recently, applications to membrane proteins have provided insights into their gating mechanisms [63,64]. The major future directions of this type of computational research and also the anticipated impact on structural biology lie in the elucidation of the functional dynamics of quaternary structures or supramolecular assemblies, as already suggested by the applications to the ribosome, viral capsids and motor proteins [20-23,25,26,27•,28]. Also, the development of hierarchical coarse-graining algorithms that reduce the complexity of the systems while maintaining their functional features will become increasingly important [65].
The utility of coarse-grained NMA partially stems from the use of EN models for analyzing structure-encoded dynamics. An important area of future research is deciphering the networks of communication in biomolecular systems and, in particular, understanding the allosteric mechanisms of signal transduction [66••]. EN-based models, combined with NMA and machine learning algorithms, appear to be promising tools for quantifying allosteric effects [65,67].
Acknowledgements
Partial support from the National Institutes of Health (P20-GM065805-02) and the National Science Foundation (ITR 0225636) is gratefully acknowledged by IB.
Footnotes
This review comes from a themed issue on Biophysical methods Edited by Wah Chiu and Keith Moffat
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
•of special interest
••of outstanding interest
- 1.Go N, Noguti T, Nishikawa T. Dynamics of a small globular protein in terms of low-frequency vibrational modes. Proc Natl Acad Sci USA. 1983;80:3696–3700. doi: 10.1073/pnas.80.12.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brooks B, Karplus M. Harmonic dynamics of proteins: normal modes and fluctuations in bovine pancreatic trypsin inhibitor. Proc Natl Acad Sci USA. 1983;80:6571–6575. doi: 10.1073/pnas.80.21.6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Case DA. Normal mode analysis of protein dynamics. Curr Opin Struct Biol. 1994;4:285–290. [Google Scholar]
- 4.Kitao A, Go N. Investigating protein dynamics in collective coordinate space. Curr Opin Struct Biol. 1999;9:164–169. doi: 10.1016/S0959-440X(99)80023-2. [DOI] [PubMed] [Google Scholar]
- 5••.Ma J. Usefulness and limitations of normal mode analysis in modeling dynamics of biomolecular complexes. Structure. 2005;13:373–380. doi: 10.1016/j.str.2005.02.002. This recent insightful review highlights many applications of coarse-grained NMA to large-scale conformational changes. The article discusses the relationship between NMA modes and the underlying energy landscape, along with some limitations of the methods. [DOI] [PubMed] [Google Scholar]
- 6.Tirion MM. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys Rev Lett. 1996;77:1905–1908. doi: 10.1103/PhysRevLett.77.1905. [DOI] [PubMed] [Google Scholar]
- 7.Haliloglu T, Bahar I, Erman B. Gaussian dynamics of folded proteins. Phys Rev Lett. 1997;79:3090–3093. [Google Scholar]
- 8.Bahar I, Atilgan AR, Erman B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold Des. 1997;2:173–181. doi: 10.1016/S1359-0278(97)00024-2. [DOI] [PubMed] [Google Scholar]
- 9.Hinsen K. Analysis of domain motions by approximate normal mode calculations. Proteins. 1998;33:417–429. doi: 10.1002/(sici)1097-0134(19981115)33:3<417::aid-prot10>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 10.Atilgan AR, Durell SR, Jernigan RL, Demirel MC, Keskin O, Bahar I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys J. 2001;80:505–515. doi: 10.1016/S0006-3495(01)76033-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tama F, Sanejouand YH. Conformational change of proteins arising from normal mode calculations. Protein Eng. 2001;14:1–6. doi: 10.1093/protein/14.1.1. [DOI] [PubMed] [Google Scholar]
- 12.Kurkcuoglu O, Jernigan RL, Doruker P. Mixed levels of coarse-graining of large proteins using elastic network model succeeds in extracting the slowest motions. Polymers. 2004;45:649–657. [Google Scholar]
- 13.Doruker P, Jernigan RL, Bahar I. Dynamics of large proteins through hierarchical levels of coarse-grained structures. J Comput Chem. 2002;23:119–127. doi: 10.1002/jcc.1160. [DOI] [PubMed] [Google Scholar]
- 14.Tama F, Gadea FX, Marques O, Sanejouand YH. Building-block approach for determining low-frequency normal modes of macromolecules. Proteins. 2000;41:1–7. doi: 10.1002/1097-0134(20001001)41:1<1::aid-prot10>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 15.Li G, Cui Q. A coarse-grained normal mode approach for macromolecules: an efficient implementation and application to Ca2+-ATPase. Biophys J. 2002;83:2457–2474. doi: 10.1016/S0006-3495(02)75257-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ming D, Kong YF, Lambert MA, Huang Z, Ma JP. How to describe protein motion without amino acid sequence and atomic coordinates. Proc Natl Acad Sci USA. 2002;99:8620–8625. doi: 10.1073/pnas.082148899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tama F, Wriggers W, Brooks CL., III Exploring global distortions of biological macromolecules and assemblies from low-resolution structural information and elastic network theory. J Mol Biol. 2002;321:297–305. doi: 10.1016/s0022-2836(02)00627-7. [DOI] [PubMed] [Google Scholar]
- 18.Kundu S, Melton JS, Sorensen DC, Phillips GN., Jr Dynamics of proteins in crystals: comparison of experiment with simple models. Biophys J. 2002;83:723–732. doi: 10.1016/S0006-3495(02)75203-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Temiz NA, Bahar I. E. coli adenylate kinase dynamics: comparison of elastic network model modes with mode-coupling 15N-NMR relaxation data. Proteins. 2004;57:468–480. doi: 10.1002/prot.20226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tama F, Valle M, Frank J, Brooks CL., III Dynamic reorganization of the functionally active ribosome explored by normal mode analysis and cryo-electron microscopy. Proc Natl Acad Sci USA. 2003;100:9319–9323. doi: 10.1073/pnas.1632476100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Rader AJ, Bahar I, Jernigan RL. Global ribosome motions revealed with elastic network model. J Struct Biol. 2004;147:302–314. doi: 10.1016/j.jsb.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 22.Tama F, Brooks CL., III Diversity and identity of mechanical properties of icosahedral viral capsids studied with elastic network normal mode analysis. J Mol Biol. 2005;345:299–314. doi: 10.1016/j.jmb.2004.10.054. [DOI] [PubMed] [Google Scholar]
- 23.Rader AJ, Vlad DH, Bahar I. Maturation dynamics of HK97 bacteriophage capsid. Structure. 2005;13:413–421. doi: 10.1016/j.str.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 24.Zheng W, Brooks BR. Normal-modes-based prediction of protein conformational changes guided by distance constraints. Biophys J. 2005;88:3109–3117. doi: 10.1529/biophysj.104.058453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tama F, Feig M, Liu J, Brooks CL, III, Taylor KA. The requirement for mechanical coupling between head and S2 domains in smooth muscle myosin ATPase regulation and its implications for dimeric motor function. J Mol Biol. 2005;345:837–854. doi: 10.1016/j.jmb.2004.10.084. [DOI] [PubMed] [Google Scholar]
- 26.Navizet I, Lavery R, Jernigan RL. Myosin flexibility: structural domains and collective vibrations. Proteins. 2004;54:384–393. doi: 10.1002/prot.10476. [DOI] [PubMed] [Google Scholar]
- 27•.Zheng W, Brooks BR. Identification of dynamical correlations within the myosin motor domain by the normal mode analysis of an elastic network model. J Mol Biol. 2005;346:745–759. doi: 10.1016/j.jmb.2004.12.020. A thorough analysis of the dynamics of the myosin motor domain using coarse-grained NMA. Two types of dynamic correlations, fluctuation based and density based, are examined using a subset of dominant modes. Key residues that control the dynamics are shown to be distributed over functional residues. [DOI] [PubMed] [Google Scholar]
- 28.Van Wynsberghe A, Li G, Cui Q. Normal-mode analysis suggests protein flexibility modulation throughout RNA polymerase's functional cycle. Biochemistry. 2004;43:13083–13096. doi: 10.1021/bi049738+. [DOI] [PubMed] [Google Scholar]
- 29.Rader AJ, Bahar I. Folding core predictions from network models of proteins. Polymers. 2004;45:659–668. [Google Scholar]
- 30.Rader AJ, Anderson G, Isin B, Khorana HG, Bahar I, Klein-Seetharaman J. Identification of core amino acids stabilizing rhodopsin. Proc Natl Acad Sci USA. 2004;101:7246–7251. doi: 10.1073/pnas.0401429101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Micheletti C, Lattanzi G, Maritan A. Elastic properties of proteins: insight on the folding process and evolutionary selection of native structures. J Mol Biol. 2002;321:909–921. doi: 10.1016/s0022-2836(02)00710-6. [DOI] [PubMed] [Google Scholar]
- 32.Haliloglu T, Keskin O, Ma B, Nussinov R. How similar are protein folding and protein binding nuclei? Examination of vibrational motions of energy hot spots and conserved residues. Biophys J. 2005;88:1552–1559. doi: 10.1529/biophysj.104.051342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33•.Yang LW, Bahar I. Coupling between catalytic site and collective dynamics: A requirement for mechanochemical activity of enzymes. Structure. 2005;13:893–904. doi: 10.1016/j.str.2005.03.015. GNM analysis applied to a set of 98 enzymes shows that the global hinge centers identified by NMA coincide with, or closely communicate with, catalytic sites, emphasizing the importance of conformational dynamics in assisting enzymatic activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kovacs JA, Chacon P, Abagyan R. Predictions of protein flexibility: first-order measures. Proteins. 2004;56:661–668. doi: 10.1002/prot.20151. [DOI] [PubMed] [Google Scholar]
- 35.Delarue M, Sanejouand Y-H. Simplified normal mode analysis of conformational transitions in DNA-dependent polymerases: the elastic network model. J Mol Biol. 2002;320:1011–1024. doi: 10.1016/s0022-2836(02)00562-4. [DOI] [PubMed] [Google Scholar]
- 36.Krebs WG, Alexandrov V, Wilson CA, Echols N, Yu H, Gerstein M. Normal mode analysis of macromolecular motions in a database framework: developing mode concentration as a useful classifying statistic. Proteins. 2002;48:682–695. doi: 10.1002/prot.10168. [DOI] [PubMed] [Google Scholar]
- 37•.Delarue M, Dumas P. On the use of low-frequency normal modes to enforce collective movements in refining macromolecular structural models. Proc Natl Acad Sci USA. 2004;101:6957–6962. doi: 10.1073/pnas.0400301101. Together with [38•,39,40••], this study investigates the use of low-frequency modes derived from NMA for refining structural models. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38•.Tama F, Miyashita O, Brooks CL., III Normal mode based flexible fitting of high-resolution structure into low-resolution experimental data from cryo-EM. J Struct Biol. 2004;147:315–326. doi: 10.1016/j.jsb.2004.03.002. This study uses a few of the slowest modes, determined from coarse-grained NMA of high-resolution structures, to generate new optimally deformed structures that are then fitted into cryo-EM maps. This method has been applied to cryo-EM structures of EF-G-bound ribosome, Escherichia coli RNA polymerase and the swollen state of cowpea chlorotic motile virus capsid. This type of EN/NMA-based flexible fitting seems to be a promising tool for predicting and refining cryo-EM structures, and determining the conformational changes of large structures using low-resolution structural data. [DOI] [PubMed] [Google Scholar]
- 39.Tama F, Miyashita O, Brooks CL., III Flexible multi-scale fitting of atomic structures into low-resolution electron density maps with elastic network normal mode analysis. J Mol Biol. 2004;337:985–999. doi: 10.1016/j.jmb.2004.01.048. [DOI] [PubMed] [Google Scholar]
- 40••.Hinsen K, Reuter N, Navaza J, Stokes DL, Lacapère JJ. Normal mode-based fitting of atomic structure into electron density maps: application to sarcoplasmic reticulum Ca-ATPase. Biophys J. 2005;88:818–827. doi: 10.1529/biophysj.104.050716. This insightful study of the use of coarse-grained NMA for docking high-resolution structures into cryo-EM densities examines the conformational transition between two forms of a large structure. The method, applied to two different atomic structures of sarcoplasmic reticulum Ca-ATPase, shows that only a few modes contribute to the transition between the two forms – a rotation and translation of the cytoplasmic domain with movements of the cytoplasmic loop. The paper also presents a comprehensive summary of how to select the modes that maximize the overlap with the real cryo-EM data, as well as the number of modes needed to optimize the fit. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kundu S, Sorensen DC, Phillips GN., Jr Automatic domain decomposition of proteins by a Gaussian network model. Proteins. 2004;57:725–733. doi: 10.1002/prot.20268. [DOI] [PubMed] [Google Scholar]
- 42.Kundu S, Jernigan RL. Molecular mechanism of domain swapping in proteins: an analysis of slower motions. Biophys J. 2004;86:3846–3854. doi: 10.1529/biophysj.103.034736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43••.Zhang Z, Shi Y, Liu H. Molecular dynamics simulations of peptides and proteins with amplified collective motions. Biophys J. 2003;84:3583–3593. doi: 10.1016/S0006-3495(03)75090-5. Global modes from NMA are imposed upon an atomic-level MD simulation in order to speed up the exploration of large-scale domain motion in T4 lysozyme and the refolding of an S-peptide analog. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He J, Zhang Z, Shi Y, Liu H. Efficiently explore the energy landscape of proteins in molecular dynamics simulations by amplifying collective motions. J Chem Phys. 2003;119:4005–4017. [Google Scholar]
- 45.Tatsumi R, Fukunishi Y, Nakamura H. A hybrid method of molecular dynamics and harmonic dynamics for docking of flexible ligand to flexible receptor. J Comput Chem. 2004;25:1995–2005. doi: 10.1002/jcc.20133. [DOI] [PubMed] [Google Scholar]
- 46.Kaledin M, Brown A, Kaledin AL, Bowman JM. Normal mode analysis using the driven molecular dynamics method. II. An application to biological macromolecules. J Chem Phys. 2004;121:5646–5653. doi: 10.1063/1.1777573. [DOI] [PubMed] [Google Scholar]
- 47.Kim MK, Jernigan RL, Chirikjian GS. Efficient generation of feasible pathways for protein conformational transitions. Biophys J. 2002;83:1620–1630. doi: 10.1016/S0006-3495(02)73931-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miyashita O, Onuchic JN, Wolynes PG. Nonlinear elasticity, proteinquakes, and the energy landscapes of functional transitions in proteins. Proc Natl Acad Sci USA. 2003;100:12570–12575. doi: 10.1073/pnas.2135471100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Itoh K, Sasai M. Dynamical transition and proteinquake in photoactive yellow protein. Proc Natl Acad Sci USA. 2004;101:14736–14741. doi: 10.1073/pnas.0402978101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50•.Miyashita O, Wolynes PG, Onuchic JN. Simple energy landscape model for the kinetics of functional transitions in proteins. J Phys Chem B. 2005;109:1959–1969. doi: 10.1021/jp046736q. The transition between two stable equilibrium structures is explored by modeling both end points as ENs. The theoretical basis of this iterative procedure, introduced in [48], is elaborated upon and a more realistic cracking (partial unfolding) model is employed to better estimate the energy barrier associated with adenylate kinase activity. [DOI] [PubMed] [Google Scholar]
- 51.Maguid S, Fernandez Alberti S, Ferrelli L, Echave J. Exploring the common dynamics of homologous proteins. Application to the globin family. Biophys J. 2005;89:3–13. doi: 10.1529/biophysj.104.053041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52••.Leo-Macias A, Lopez-Romero P, Lupyan D, Zerbino D, Ortiz AR. An analysis of core deformations in protein superfamilies. Biophys J. 2005;88:1291–1299. doi: 10.1529/biophysj.104.052449. A comparison of the structural dynamics within protein families indicates that the regions with the highest evolutionary fluctuations correspond to the most unconstrained regions. This suggests that structural topology and dynamics play a role in maintaining protein function during evolution. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen SC, Bahar I. Mining frequent patterns in protein structures: a study of protease families. Bioinformatics. 2004;20:i77–i85. doi: 10.1093/bioinformatics/bth912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bahar I, Wallqvist A, Covell DG, Jernigan RL. Correlation between native state hydrogen exchange and cooperative residue fluctuations from a simple model. Biochemistry. 1998;37:1067–1075. doi: 10.1021/bi9720641. [DOI] [PubMed] [Google Scholar]
- 55.Wang MM, Borchardt RT, Schowen RL, Kuczera K. Domain motions and the open-to-closed conformational transition of an enzyme: a normal mode analysis of S-adenosyl-L-homocysteine hydrolase. Biochemistry. 2005;44:7228–7239. doi: 10.1021/bi047524m. [DOI] [PubMed] [Google Scholar]
- 56.Yang LW, Liu X, Jursa CJ, Holliman M, Rader AJ, Karimi HA, Bahar I. iGNM: a database of protein functional motions based on Gaussian network model. Bioinformatics. 2005;21:2978–2987. doi: 10.1093/bioinformatics/bti469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wako H, Kato M, Endo S. ProMode: a database of normal mode analyses on protein molecules with a full-atom model. Bioinformatics. 2004;20:2035–2043. doi: 10.1093/bioinformatics/bth197. [DOI] [PubMed] [Google Scholar]
- 58.Suhre K, Sanejouand YH. ElNémo: a normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004;32:W610–W614. doi: 10.1093/nar/gkh368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hollup SM, Salensminde G, Reuter N. WEBnm@: a web application for normal mode analyses of proteins. BMC Bioinformatics. 2005;6:1–8. doi: 10.1186/1471-2105-6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Echols N, Milburn D, Gerstein M. MolMovDB: analysis and visualization of conformational change and structural flexibility. Nucleic Acids Res. 2003;31:478–482. doi: 10.1093/nar/gkg104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kondrashov DA, Phillips GN. Molecular mastication mechanics. Structure. 2005;13:836–837. doi: 10.1016/j.str.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 62.Klein-Seetharaman J. Dual role of interactions between membranous and soluble portions of helical membrane receptors for folding and signaling. Trends Pharma Sci. 2005;26:183–189. doi: 10.1016/j.tips.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 63.Taly A, DeLauro M, Grutter T, Nilges M, Le Novere N, Corringer PJ, Changeux JP. Normal mode analysis suggests a quaternary twist model for the nicotinic receptor gating mechanism. Biophys J. 2005;88:3954–3965. doi: 10.1529/biophysj.104.050229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Haider S, Grottesi A, Hall BA, Ashcroft FM, Sansom MSP. Conformational dynamics of the ligand-binding domain of inward rectifier K channels as revealed by molecular dynamics simulations: toward an understanding of Kir channel gating. Biophys J. 2005;88:3310–3320. doi: 10.1529/biophysj.104.052019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chennubhotla C, Rader AJ, Yang L-W, Bahar I. Elastic models for understanding biomolecular machinery: from enzymes to supramolecular assemblies. Phys Biol. 2005 doi: 10.1088/1478-3975/2/4/S12. in press. [DOI] [PubMed] [Google Scholar]
- 66••.Changeux J-P, Edelstein SJ. Allosteric mechanisms of signal transduction. Science. 2005;308:1424–1428. doi: 10.1126/science.1108595. An excellent review of models of allosteric mechanisms and their applications to understanding regulatory enzymes, nuclear receptors and various membrane receptors. The article emphasizes the intrinsic ability of unliganded structures to undergo conformational changes that accommodate ligand binding and the prior accessibility of functional conformers in the absence of ligand. [DOI] [PubMed] [Google Scholar]
- 67.Ming DM, Wall ME. Quantifying allosteric effects in proteins. Proteins. 2005;59:697–707. doi: 10.1002/prot.20440. [DOI] [PubMed] [Google Scholar]
- 68.Ranson NA, Farr GW, Roseman AM, Gowen B, Fenton WA, Horwich AL, Saibil HR. ATP-bound states of GroEL captured by cryo-electron microscopy. Cell. 2001;107:869–879. doi: 10.1016/s0092-8674(01)00617-1. [DOI] [PubMed] [Google Scholar]