

Abstract
Cu-catalyzed cross-dehydrogenative coupling (CDC) methodologies were developed based on the oxidative activation of sp3 C–H bonds adjacent to a nitrogen atom. Various sp, sp2, and sp3 C–H bonds of pronucleophiles were used in the Cu-catalyzed CDC reactions. Based on these results, the mechanisms of the CDC reactions also are discussed.
Keywords: C–H activation, catalysis, Baylis–Hillman reaction, Mannich reaction, Friedel–Crafts reaction
C–C bond formation reactions are among the most important processes in organic chemistry. Transition metal-catalyzed cross-coupling reactions of various reactive functional groups are newer and more powerful methods for the formation of C–C bonds [Scheme 1, Path A (1)]. However, these well developed cross-coupling reactions must use prefunctionalized starting materials. Therefore, in the formation of a single chemical bond, an extra step(s) is required to prepare the functionalized starting material from a raw material. Accordingly, transition metal-catalyzed C–H bond activation and subsequent C–C bond formations have attracted much interest in recent years [Scheme 1, Path B (2–6)]. Compounds containing heteroatoms, such as nitrogen, are widely present in nature. The syntheses of these compounds have attracted much attention in industrial and academic research because of their biological and pharmaceutical properties. Recently, some excellent examples based on the direct sp3 C–H bond activation adjacent to a nitrogen atom for C–C bond formations were reported by Murahashi et al. (7), Murai and coworkers (8–11), Ishii and coworkers (12), Sames and coworkers (13–15), Davies et al. (16, 17), Yoshimitsu et al. (18), Yoshida and coworkers (19–22), and Yi and coworkers (23). Although these are elegant methodologies, most of them required another functionalized substrate. Furthermore, expensive metal catalysts, such as Ru, Rh, and Ir, were generally used. The direct utilization of only C–H bonds represents the next generation of C–C bond formations and is highly desirable because such a coupling will avoid the preparation of functional groups to make synthetic schemes shorter and more efficient (24). On the basis of these advances, we recently developed a Cu-catalyzed cross-dehydrogenative coupling (CDC) methodology to construct functional molecules by directly using two different C–H bonds [Scheme 1, Path C (25–29)]. In this work, we will present a detailed description of this method.
Scheme 1.

Various cross-coupling methods for the formation of C–C bonds.
Results and Discussions
CDC Mannich-Type Reaction.
Vicinal diamines are important structural motifs in biologically active natural products and are used as core units of chiral auxiliaries and chiral ligands in asymmetric catalysis (30). An efficient synthetic approach toward such compounds is through the nitro-Mannich (aza-Henry) reaction. The nucleophilic addition of nitroalkanes to imines generates β-nitroamine derivatives (31), which are readily converted into 1,2-diamines or α-amino carbonyl compounds by the reduction of the nitro group (32, 33) or through the Nef reaction (34).
Our initial efforts on this coupling were focused on the reaction of 1,2,3,4-tetrahydroisoquinoline 1a with nitromethane, with nitromethane serving as both the reagent and solvent. Various copper catalysts, such as CuCl, CuBr, CuI, Cu(OTf), CuCl2, CuBr2, Cu(OTf)2, and Cu(OAc)2·H2O, were examined at room temperature (RT), and the desired product was obtained in all cases. CuBr was found to be the most effective catalyst in this CDC reaction. However, nitroalkanes, being used as both a reactant and solvent, diminished the efficiency of this reaction (27). Therefore, further optimizations were made. To our delight, when a stoichiometric amount of nitromethane was used, the desired products were obtained in good yields (Table 1, entries 1 and 7). The use of 2 equivalents (equiv) of nitroalkane increased amine conversion and improved the product yields (Table 1). The use of nitroethane instead of nitromethane also gave the desired compounds with good yields (the ratios of the two diastereoisomers are 2:1; Table 1, entries 3, 5, and 8). With N,N-dimethylaniline derivatives, an excess amount of nitromethane was necessary to obtain the desired product with reasonable yields (Table 1, entries 9 and 10). The relatively low yield of product 3g was attributed to the formation of a demethylated compound and other unidentified by-products.
Table 1.
CDC reaction of tertiary amines with nitroalkanes
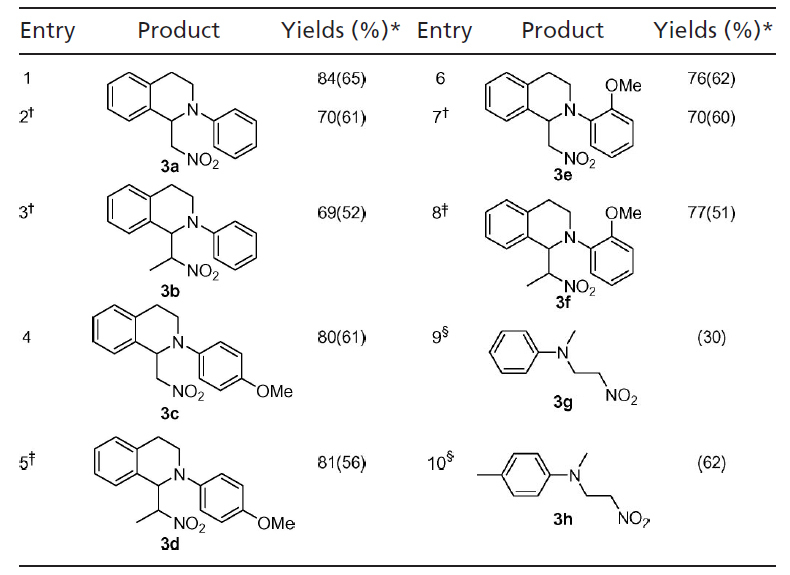
Amine (0.2 mmol), nitroalkane (0.4 mmol), and TBHP (0.24 mmol, 5.5 M in decane); otherwise are mentioned.
*NMR yields are based on amines and determined by 1H NMR using an internal standard; isolated yields are given in parentheses.
†1 equiv (0.2 mmol) of nitromethane was used.
‡The ratio of two isomers is 2.1.
§1 ml of nitromethane was used.
Other cyclic amines such as 1-phenylpyrrolidine also generated the desired product in good yields (Scheme 2). Although a large excess of nitromethane was used (as solvent), the bis-CDC product 6 was formed in only 4% yield along with the mono-CDC product. The ratio of trans-6 and cis-6 is 1:1 as determined by 1H NMR (35).
Scheme 2.

Reaction of 1-phenylpyrrolidine with nitromethane.
Dialkyl malonates are important synthons and are widely used in organic syntheses. The ester groups in malonates can be readily transformed into other functional groups or used in further reactions. Therefore, it is attractive to develop new methods to introduce a malonate unit into a molecule efficiently. We evaluated the use of dialkyl malonates as pronucleophiles in the CDC reaction. The desired products, β-diester amine derivatives, were obtained by the reaction of tetrahydroisoquinolines and various dialkyl malonates with 5 mol % CuBr at RT (Table 2). Moreover, the desired product 8a was obtained in 72% isolated yield even when only 0.5 mol % CuBr was used (Table 2, entry 2).
Table 2.
CDC reaction of tertiary amines with malonates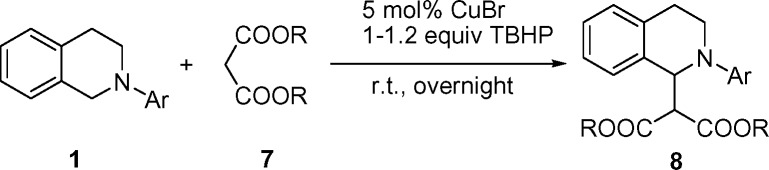
Tetrahydroisoquinoline (0.1 mmol), malonate (0.1 mmol), and TBHP (0.02 ml, 5–6 M in decane). [Reproduced with permission from ref. 25 (Copyright 2005, Wiley).]
*Isolated yields.
†CuBr (0.005 mmol, 0.5 mol%), tetrahydroisoquinoline (1.0 mmol), malonate (1.0 mmol), and TBHP (0.2 ml, 5–6 M in decane); the reaction time is 43 h.
‡Malonate (0.2 mmol).
Next, we investigated the synthesis of β-dicyano tetrahydroisoquinolines by using malononitrile as the pronucleophile under the standard reaction conditions. The desired product, β-dicyano tetrahydroisoquinoline 10, was obtained in 29% isolated yield (Table 3, entry 1). Surprisingly, α-cyano product 11 (7) was also obtained as one of the unexpected by-products. The yield of 10 was increased to 46% along with 6% of 11 when 6 equiv of malononitrile were used. Interestingly, when excess tert-butyl hydroperoxide (TBHP) (2 equiv) was used, 11 was obtained as the main product, and 10 was not observed by 1H NMR (Table 3, entry 3). To form 11, −CN anion must be generated during the reaction. It is likely that the reaction of some malononitrile and TBHP catalyzed by CuBr generated 2-oxo-malononitrile (36) and −CN (37).
Table 3.
CDC reaction of tetrahydroisoquinoline with malononitrile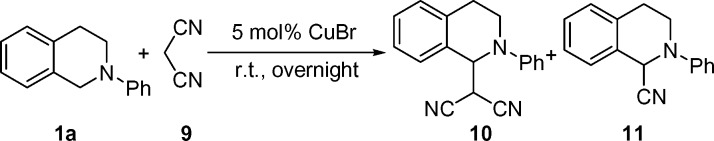
Tetrahydroisoquinoline (0.1 mmol) was used.
*Isolated yields were based on tetrahydroisoquinoline.
When free (NH) 1,2,3,4-tetrahydroisoquinoline 12 was used instead of N-phenyl tetrahydroisoquinolines, the desired reaction did not occur. However, the desired Mannich product was obtained when 12 was reacted with the Meldrum’s acid (13) (Scheme 3). Nonpolar solvent such as hexane was found to be helpful in mixing the reactants and improving the product yield. The highly enolizable ability of 13 appeared responsible for its higher reactivity under these conditions. Single crystal x-ray diffraction (Fig. 1a, which is published as supporting information on the PNAS web site) and its packing (Fig. 1b) display the betaine structure and its π-stacking.
Scheme 3.

CDC reaction of tetrahydroisoquinoline with Meldrum’s acid.
CDC Morita–Baylis–Hillman (MBH) Reaction.
Highly functionalized products formed in one step are important for organic synthesis and material science. MBH reaction is an organocatalytic coupling of sp2 C–H bond of electron-efficient alkenes with sp2 hybridized carbon electrophiles, including aldehydes, aldimines, and various ketones (38). Allylic carbonates (39), 2-bromomethyl acrylate (40), and allylic alcohols (41) have also been used in MBH reactions as alternative electrophiles. More recently, intramolecular α-alkylation of enones using sp3 hybridized electrophiles, alkyl halides, was established by using stoichiometric trialkyl phosphines and a base (42). Directly using sp3 C–H bonds in electrophiles would be a more desirable MBH reaction. To challenge this question, our attention was focused on integrating our CDC method with the MBH reaction by combining organo- and transition metal-catalyzed CDC–MBH-type reactions.
Triphenylphosphine and 1,4-diazabicyclo[2.2.2]octane (DABCO) were chosen as the organocatalysts. Triphenylphosphine was found to be ineffective in the CDC–MBH reaction (Table 4, entries 1 and 2). After the reactions, 31P NMR of the reaction mixture showed that the triphenylphosphine was oxidized during the reaction and lost its catalytic ability. DABCO was found to be an effective catalyst in this reaction. A reasonable yield was obtained with 5 mol % CuBr and 10 mol % DABCO at 50°C (Table 4, entry 6).
Table 4.
CDC reaction of tetrahydroisoquinoline with MVK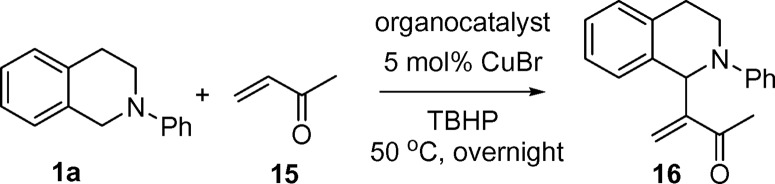
| Entry | Organocatalyst | Temperature, °C | Yield, %* |
|---|---|---|---|
| 1 | PPh3, 30 mol% | RT | 10 |
| 2 | PPh3, 30 mol% | 50 | 20 |
| 3 | DABCO, 5 mol% | 50 | 30 |
| 4 | DABCO, 30 mol% | 50 | 28 |
| 5 | DABCO, 10 mol% | RT | 24 |
| 6† | DABCO, 10 mol% | 50 | 53 |
Tetrahydroisoquinoline (0.1 mmol). Methylvinylketone (MVK) (0.2 mmol). TBHP (0.1 mmol, 5.5 M in decane), and CuBr (5 mol%).
*Reported yields were based on tetrahydroisoquinoline and determined by 1H NMR using an internal standard.
†4-Å molecular sieve (30 mg) was added.
Subsequently, various MBH products were obtained by means of the CDC–MBH reaction (Table 5). When acrylonitrile was used, the desired products were obtained in good yields (Table 5, entries 2 and 4).
Table 5.
Aza–Baylis–Hillman type CDC reaction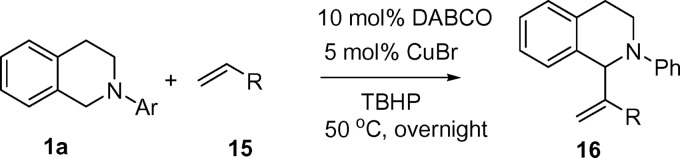
| Entry | Ar | R | Product | Yield, %* |
|---|---|---|---|---|
| 1 | Ph | Acyl | 16a | 53 (30) |
| 2 | Ph | CN | 16b | 61 (58) |
| 3 | 4-MeOC6H4 | Acyl | 16c | 31 (29) |
| 4 | 4-MeOC6H4 | CN | 16d | 74 (69) |
Tetrahydroisoquinoline (0.2 mmol), alkene (0.4 mmol), TBHP (0.2 mmol, 5.5 M in decane), CuBr (5 mol%), DABCO (10 mol%), and 4-éÅ molecular sieve (60 mg).
*NMR yields are based on tetrahydroisoquinoline and determined by 1H NMR using an internal standard; isolated yields are given in parentheses.
CDC Friedel–Crafts Reaction.
Friedel–Crafts reaction is an important and powerful method to introduce aromatic moieties into the structures of organic compounds and thus has attracted much attention in organic synthesis (43–45). Brönsted acid or Lewis acid-catalyzed imine or iminium Friedel–Crafts reactions (aza-Friedel–Crafts reactions) are important methods to construct a variety of nitrogen-containing compounds (46–50). We have been interested in the synthesis of indolyl tetrahydroisoquinolines for biological studies. There are two types of methods to synthesize such compounds: (i) the reactions of indoles with cotarnine (51), and (ii) the reactions of N-imidolylcycloimmonium salts with indoles followed by catalytic hydrogenation (52). However, there are limitations to these methods: for the first example, the natural tautomeric pseudobase, cotarnine, was used [the practical synthesis of itself also needs several steps (53)]; for the second example, a relatively long synthetic scheme is required. To address these challenges as well as to extend the scope of the CDC methodology, our attention was focused on the synthesis of such alkaloids by directly using free (NH)-indoles and tetrahydroisoquinolines. The desired product 18 was successfully obtained when tetrahydroisoquinoline was reacted with indole under our CuBr/TBHP system (Scheme 4). The yield was improved when the temperature was raised from RT to 50°C.
Scheme 4.

CDC reaction of tetrahydroisoquinoline with indole.
Various indoles were reacted with tetrahydroisoquinolines under the optimized reaction conditions to give the desired products in good to excellent yields (Table 6). The reactions occurred selectively at the C3 position of indoles, if both C2 and C3 positions of indoles are unoccupied. When the C3 position of indoles is substituted, the C2 substituted products were obtained (Table 6, entries 9 and 13). Indoles with electron-withdrawing or electron-donating groups also worked well under the optimized reaction conditions. This methodology provides the simplest way to construct indolyl tetrahydroisoquinolines as well as opens a different way to construct and study more complex alkaloids.
Table 6.
CDC reaction of indoles with tetrahydroisoquinolines
Tetrahydroisoquinolines (0.1 mmol), indoles (0.12 mmol), CuBr (0.005 mmol, 5 mol%), and TBHP (0.13 mmol, 5–6 M in decane).
*NMR yields are based on tetrahydroisoquinolines and determined by 1H NMR using an internal standard; isolated yields are given in parentheses.
2-Naphthol is another electron-rich aromatic compound. We envisioned that a new type of Betti base (54, 55) would be formed through the CDC reaction of tetrahydroisoquinoline with 2-naphthol. Recently, chiral tertiary amino-naphthol ligands were shown to be highly enantioselective catalysts in phenyl transfer reactions (56). Indeed, the desired product 20 was obtained when tetrahydroisoquinoline was reacted with 2-naphthol under our CuBr/TBHP system (Table 7, entry 3) in 58% NMR yield. CuI and CuCl gave similar results (Table 7, entries 1 and 2). Interestingly, the yield of 20 was slightly higher when CuBr2 was used as the catalyst (Table 7, entry 4). The highest yield was obtained when 2 equiv of tetrahydroisoquinoline was used (Table 7, entry 6). Other Cu(II)-catalysts gave low yields of 20, and 2,2′-binaphthol (BINOL) was formed as the by-product (57). Representative results are shown in Table 8, which is published as supporting information on the PNAS web site, when CuBr2 was used as the catalyst.
Table 7.
CDC reaction of tetrahydroisoquinoline with 2-naphthol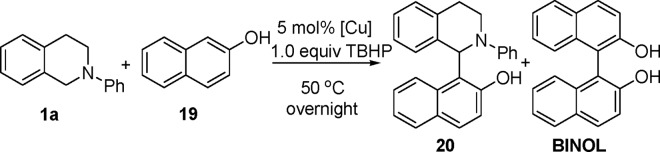
| Entry | [Cu] | Yield of 20* | Yield of BINOL* |
|---|---|---|---|
| 1 | Cul | 57 | 13 |
| 2 | CuCl | 51 | 10 |
| 3 | CuBr | 58 | 23 |
| 4 | CuBr2 | 63 | 10 |
| 5† | CuBr | 63 | 10 |
| 6† | CuBr2 | 72 | 11 |
| 7 | CuSO4 | 53 | 11 |
| 8 | Cu(OTf)2 | 55 | 15 |
Tetrahydroisoquinoline (0.1 mmol), 2-naphthol (0.1 mmol), TBHP (0.1 mmol, 5.5 M in decane), and [Cu] (5 mol%): otherwise are mentioned.
*Reported yields were NMR yields using an internal standard.
†Tetrahydroisoquinoline (0.2 mmol).
Alkynylation.
Propargylic amines are of great pharmaceutical interest and are synthetically useful intermediates for various nitrogen compounds as well as carbohydrate derivatives (58). Two main methods have been used to construct propargylic amines (Scheme 5): Path A represents stoichiometric nucleophilic reactions (59, 60), and Path B represents transition metal-catalyzed reactions of alkynes with imines generated from aldehydes and amines. There are many excellent examples of these two methods. For examples, we (61) and others (62–65) have described the direct addition of a terminal alkyne to aldehyde and imines to afford propargyl alcohols and propargyl amines. With Cu(I)–pybox as a chiral catalyst, a highly enantioselective alkyne-imine addition in either water or toluene also was reported by us (66, 67). We also developed the coupling of alkynes with N-acylimines and N-acyliminium ions by using a CuBr catalyst, and the gold- or silver-catalyzed coupling reactions of alkyne, aldehyde, and amine in water (68–75). Although these are effective methods, these methods need a leaving group or an imine formed from aldehyde and amine. With the desire to develop more efficient methods, we investigated the direct construction of propargylic amines by the catalytic coupling of sp3 C–H bond adjacent to nitrogen with a terminal alkyne (Path C).
Scheme 5.

Various methods for forming propargylamines.
As a model study, we used N,N-dimethylaniline and phenylacetylene as standard starting materials to identify the optimal reaction conditions. The desired product was obtained in good yield with the combination of a copper catalyst and TBHP. CuBr, CuBr2, CuCl, and CuCl2 were found to be the most effective catalysts among the copper catalysts examined. No reaction was observed in the absence of either a copper catalyst or TBHP. The best yield was obtained when the N,N-dimethylaniline:alkynes:TBHP ratio was 2:1:1.
With the optimized reaction conditions in hand, various alkynes were reacted with amines. The reaction of N,N-dimethylaniline (2 equiv) with phenylacetylene in the presence of a CuBr catalyst (5 mol %) and TBHP (1.0 equiv) at 100°C for 3 h gave N-methyl-N-(3-phenylprop-2-ynyl)aniline in 74% isolated yield (entry 1 of Table 9, which is published as supporting information on the PNAS web site). For aromatic alkynes, the reaction often provided good yields of the desired products. For aliphatic alkynes, the corresponding products were formed in lower yields (Table 9, entries 6 and 7).
When benzyldimethylamine was reacted with phenylacetylene under the standard conditions, alkynylation of the methyl group was the major product (Scheme 6). The minor product could not be isolated.
Scheme 6.

Reaction of N,N-dimethylbenzylamine with alkynes.
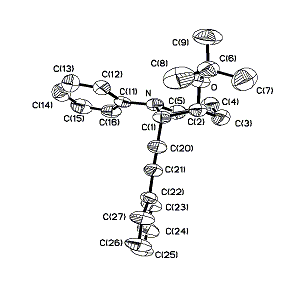
Cyclic amines such as tetrahydroisoquinoline can be selectively converted into the corresponding α-alkynylation compounds 25a and 25b in 73% and 74% isolated yields, respectively (Scheme 7). Interestingly, 1-phenyl-piperidine reacted with phenylacetylene in the presence of a catalytic amount of CuBr and 1 equiv of TBHP to give the desired direct alkynylation product in 12% yield together with a tert-butyloxide alkynylation compound 28 (12%) (Scheme 8). The structure of 28 was determined by x-ray crystallography measurement (Fig. 2, which is published as supporting information on the PNAS web site).
Scheme 7.

Reaction of cyclic benzylamine with alkynes.
Scheme 8.

Reaction of simple cyclic amine.
Mechanism of CDC Reactions.
Based on the above results and discussion, two main mechanistic aspects are informative: (i) an iminium-type intermediate is most likely involved in the reaction, and (ii) the reaction of the iminium intermediate with a pronucleophile is the rate-limiting step. In the CDC reaction of a sp3 C–H bond (adjacent to a nitrogen atom) with the sp3 C–H bond (in a pronucleophile), such as the Mannich-type reaction, the reaction occurs smoothly at RT. However, in the CDC reaction of the C–H bonds in same amines with the sp2 C–H bond in a pronucleophile, such as the MBH and Friedel–Crafts reactions, the CDC reactions must be carried out at 50°C. Furthermore, in the CDC reaction of the C–H bonds in the amines with the sp C–H bond in a pronucleophile, such as alkynylation, the CDC reactions occurred only at 100°C [when a ligand was used the reaction could be performed at 50°C, albeit the reaction time was 2 days (28)]. These findings correlate well with their abilities to generate the reactive nucleophile in those pronucleophiles. Therefore, the rate of the formation of the iminium-type intermediate appeared to be the faster step compared with the reaction of the iminium intermediate with a pronucleophile.
Based on the literature reports of oxidation of an amine to an iminium, there are two possible CDC mechanisms: either a radical mechanism (Scheme 9) or an ionic mechanism [Scheme 10 (76)]. In a radical mechanism, the iminium type intermediate 32 could be formed through a single-electron transfer (SET) from a α-carbon-centered radical 31, which was generated from the H-abstraction by tert-butoxyl radical (formed by copper-catalyzed decomposition of TBHP). Alternatively, an initial SET gave an aminyl cation radical 30, which then lost a H radical to generate 32 (77, 78). Subsequently, coupling of 32 with the pronucleophile resulted in the desired CDC product 33 and regenerated the copper catalyst (79).
Scheme 9.

Radical mechanism for the CDC reaction of amine with nucleophile.
Scheme 10.

Ionic mechanism for the CDC reaction of amine with nucleophile.
In an ionic mechanism (Scheme 10), copper catalyst is oxidized to oxy-copper 34 (80) by TBHP (81). The high Lewis-acidic copper species 34 coordinates to the nitrogen atom and forms a intermediate 35. Then, the iminium-type intermediate 32 is formed from 35 by elimination of water through a five-membered transition state. Finally, the CDC product 33 is formed by the nucleophilic attack of 32.
Although a radical mechanism is often invoked for metal-catalyzed oxidations by TBHP, such as the Kharasch-type oxidation (82), an attempt to gain more insight into the mechanism of CDC reactions was carried out (Scheme 11). A radical scavenger, 2,6-di-tert-butyl-4-methyl phenol (BHT), was used in the reaction between tetrahydroisoquinoline 1a and nitromethane 2a. The desired product 3a was still obtained in 70% yield (measured by 1H NMR). This result suggests that a free radical process is not a requirement for the present CDC reaction. Alternatively, tert-butylperoxide compounds (83, 84) are involved as intermediates. Further mechanistic studies for the CDC reaction are needed.
Scheme 11.

CDC reaction of amine with nitromethane in the presence of 2,6-di-tert-butyl-4-methyl phenol (BHT).
Conclusion
As an effort to develop highly efficient and selective reactions for chemical synthesis, a concept of cross-coupling reaction, CDC reaction, was developed. Various nitrogen-containing compounds were obtained efficiently through CDC reactions under mild reaction conditions. CDC reactions represent the most direct and efficient methods for C–C bond formations and provide the pillar for the next-generation chemical synthesis with an eye on green chemistry.
Experimental Procedures
General Information.
1H NMR spectra were recorded on 300-, 400-, and 500-MHz spectrometers (Varian), and the chemical shifts were reported in parts per million (ppm) (δ) relative to internal standard tetramethylsilane (TMS) (0 ppm) for CDCl3 or the center peak of residual DMSO (2.49 ppm). The peak patterns are indicated as follows: s, singlet; d, doublet; t, triplet; dt, doublet of triplet; dq, doublet of quartet; ddd, doublet of doublet of doublet; dddd, doublet of doublet of doublet of doublet; m, multiplet; q, quartet. The coupling constants, J, are reported in Hz. 13C NMR spectra were obtained at 75, 100, and 125 MHz and referenced to the internal solvent signals (central peak is 77.0 ppm in CDCl3 or 40.4 ppm in DMSO-d6). NMR spectra were obtained in CDCl3 unless stated otherwise. X-ray diffraction data were measured on a D8 diffractometer (Bruker, Billerica, MA). MS data were obtained by a MS25RFA mass spectrometer (Kratos Analytical Instruments). High-resolution mass spectra (HRMS) analysis was performed at by McGill University. IR spectra were recorded by an MB100 instrument (ABB Bomem, Quebec). Melting points were recorded by a melting point apparatus (Gallenkamp, Loughborough, U.K.). Thin-layer chromatography (TLC) was performed by using Silica Gel 60 F254 TLC plates (Sorbent Technologies, Atlanta) and visualized with UV light. Flash-column chromatography was performed over Sorbent silica gel 30–60 μm. All reagents were weighed and handled in air at RT. All reagents were purchased from Aldrich, Strem Chemicals (Newburyport, MA), and Acros (Geel, Belgium) and used without further purification.
General Procedure for Preparing 2-Aryl-1,2,3,4-Tetrahydroisoquinolines.
Copper(I) iodide (200 mg, 1.0 mmol) and potassium phosphate (4.25 g, 20.0 mmol) were put into a schlenk-tube. The tube was evacuated and back filled with nitrogen. 2-Propanol (10.0 ml), ethylene glycol (1.11 ml, 20.0 mmol), 1,2,3,4-tetrahydroisoquinoline (2.0 ml, 15 mmol), and iodobenzene (1.12 ml, 10.0 mmol) were added successively by microsyringe at RT. The reaction mixture was heated to 85–90°C and kept for 24 h and then allowed to cool to RT. Diethyl ether (20 ml) and water (20 ml) then were added to the reaction mixture. The organic layer was extracted by diethyl ether (2 × 20 ml). The combined organic phases were washed with brine and dried over magnesium sulfate. The solvent was removed by rotary evaporation, and the crude product was purified by column chromatography on silica gel (hexane/ethyl acetate = 20:1), and the fraction with Rf = 0.7 was collected to give the desired product 1a.
Supporting Information.
For general reaction procedures, diffraction details, and characterization of products, see Supporting Text, which is published as supporting information on the PNAS web site.
Supplementary Material
Acknowledgments
This work was supported by the Canada Research Chair (Tier I) foundation (to C.-J.L. and D.S.B.), the Canada Foundation for Innovation, the Natural Sciences and Engineering Research Council (Canada), Merck Frosst, and McGill University.
Abbreviations
- CDC
cross-dehydrogenative coupling
- DABCO
diazabicyclo[2.2.2]octane
- MBH
Morita–Baylis–Hillman
- RT
room temperature
- TBHP
tert-butylhydroperoxide.
Footnotes
Conflict of interest statement: Part of the research results will be filed for patent protection.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.De Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. 2nd Ed. Germany: Wiley–VCH, Weinheim; 2004. [Google Scholar]
- 2.Crabtree R. H. J. Organomet. Chem. 2004;689:4083–4091. [Google Scholar]
- 3.Ritleng V., Sirlin C., Pfeffer M. Chem. Rev. 2002;102:1731–1770. doi: 10.1021/cr0104330. [DOI] [PubMed] [Google Scholar]
- 4.Jia C., Kitamura T., Fujiwara Y. Acc. Chem. Res. 2001;34:633–639. doi: 10.1021/ar000209h. [DOI] [PubMed] [Google Scholar]
- 5.Dyker G. Angew. Chem. Int. Ed. 1999;38:1698–1712. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1698::AID-ANIE1698>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 6.Shilov A. E., Shul’pin G. B. Chem. Rev. 1997;97:2879–2932. doi: 10.1021/cr9411886. [DOI] [PubMed] [Google Scholar]
- 7.Murahashi S.-I., Komiya N., Terai H., Nakae T. J. Am. Chem. Soc. 2003;125:15312–15313. doi: 10.1021/ja0390303. [DOI] [PubMed] [Google Scholar]
- 8.Doye S. Angew. Chem. Int. Ed. 2001;40:3351–3353. doi: 10.1002/1521-3773(20010917)40:18<3351::aid-anie3351>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 9.Chatani N., Asaumi T., Yorimitsu S., Ikeda T., Kakiuchi F., Murai S. J. Am. Chem. Soc. 2001;123:10935–10941. doi: 10.1021/ja011540e. [DOI] [PubMed] [Google Scholar]
- 10.Chatani N., Asaumi T., Ikeda T., Yorimitsu S., Ishii Y., Kakiuchi F., Murai S. J. Am. Chem. Soc. 2000;122:12882–12883. doi: 10.1021/ja011540e. [DOI] [PubMed] [Google Scholar]
- 11.Jun C.-H., Hwang D.-C., Na S.-J. Chem. Commun. 1998:1405–1406. [Google Scholar]
- 12.Sakaguchi S., Kubo T., Ishii Y. Angew. Chem. Int. Ed. 2001;40:2534–2536. doi: 10.1002/1521-3773(20010702)40:13<2534::aid-anie2534>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 13.Sezen B., Sames D. J. Am. Chem. Soc. 2005;127:5284–5285. doi: 10.1021/ja0699586. [DOI] [PubMed] [Google Scholar]
- 14.Sezen B., Sames D. J. Am. Chem. Soc. 2004;126:13244–13246. doi: 10.1021/ja069957d. [DOI] [PubMed] [Google Scholar]
- 15.DeBoef B., Pastine S. J., Sames D. J. Am. Chem. Soc. 2004;126:6556–6557. doi: 10.1021/ja049111e. [DOI] [PubMed] [Google Scholar]
- 16.Davies H. M. L., Jin Q. Org. Lett. 2004;6:1769–1772. doi: 10.1021/ol0495467. [DOI] [PubMed] [Google Scholar]
- 17.Davies H. M. L., Venkataramani C., Hansen T., Hopper D. W. J. Am. Chem. Soc. 2003;125:6462–6468. doi: 10.1021/ja0290072. [DOI] [PubMed] [Google Scholar]
- 18.Yoshimitsu T., Arano Y., Nagaoka H. J. Am. Chem. Soc. 2005;127:11610–11611. doi: 10.1021/ja053855q. [DOI] [PubMed] [Google Scholar]
- 19.Maruyama T., Suga S., Yoshida J.-I. J. Am. Chem. Soc. 2005;127:7324–7325. doi: 10.1021/ja0511218. [DOI] [PubMed] [Google Scholar]
- 20.Suga S., Nishida T., Yamada D., Nagaki A., Yoshida J.-I. J. Am. Chem. Soc. 2004;126:14338–14339. doi: 10.1021/ja0455704. [DOI] [PubMed] [Google Scholar]
- 21.Suga S., Suzuki S., Yoshida J.-I. J. Am. Chem. Soc. 2002;124:30–31. doi: 10.1021/ja0171759. [DOI] [PubMed] [Google Scholar]
- 22.Yoshida J.-I., Suga S., Suzuki S., Kinomura N., Yamamoto A., Fujiwara K. J. Am. Chem. Soc. 1999;121:9546–9549. [Google Scholar]
- 23.Yi C. S., Yun S. Y., Guzei I. A. Organometallics. 2004;23:5392–5395. [Google Scholar]
- 24.Li Z., Li C.-J. J. Am. Chem. Soc. 2006;128:56–57. doi: 10.1021/ja056541b. [DOI] [PubMed] [Google Scholar]
- 25.Li Z., Li C.-J. Eur. J. Org. Chem. 2005:3173–3176. [Google Scholar]
- 26.Li Z., Li C.-J. J. Am. Chem. Soc. 2005;127:6968–6969. doi: 10.1021/ja0516054. [DOI] [PubMed] [Google Scholar]
- 27.Li Z., Li C.-J. J. Am. Chem. Soc. 2005;127:3672–3673. doi: 10.1021/ja050058j. [DOI] [PubMed] [Google Scholar]
- 28.Li Z., Li C.-J. Org. Lett. 2004;6:4997–4999. doi: 10.1021/ol047814v. [DOI] [PubMed] [Google Scholar]
- 29.Li Z., Li C.-J. J. Am. Chem. Soc. 2004;126:11810–11811. doi: 10.1021/ja0460763. [DOI] [PubMed] [Google Scholar]
- 30.Lucet D., Le Gall T., Mioskowski C. Angew. Chem. Int. Ed. 1998;37:2580–2627. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2580::AID-ANIE2580>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 31.Baer H. H., Urbas L. In: in The Chemistry of the Nitro and Nitroso Group, Fruer H., editor. New York: Interscience; 1970. pp. 75–200. Part 2. [Google Scholar]
- 32.Bernardi L., Bonini B. F., Capito E., Dessole G., Comes-Franchini M., Fochi M., Ricci A. J. Org. Chem. 2004;69:8168–8171. doi: 10.1021/jo0488762. [DOI] [PubMed] [Google Scholar]
- 33.Adams H., Anderson J. C., Peace S., Pennell A. M. K. J. Org. Chem. 1998;63:9932–9934. [Google Scholar]
- 34.Ballini R., Petrini M. Tetrahedron. 2004;60:1017–1047. [Google Scholar]
- 35.Breuer E., Melumad D. J. Org. Chem. 1973;38:1601–1602. [Google Scholar]
- 36.Choudary B. M., Reddy P. N. J. Mol. Cat. A Chem. 1996;112:385–388. [Google Scholar]
- 37.Kharasch M. S., Sosnovsky G. Tetrahedron. 1958;3:105–112. [Google Scholar]
- 38.Basavaiah D., Rao A. J., Satyanarayana T. Chem. Rev. 2003;103:811–892. doi: 10.1021/cr010043d. [DOI] [PubMed] [Google Scholar]
- 39.Jellerichs B. G., Kong J.-R., Krische M. J. J. Am. Chem. Soc. 2003;125:7758–7759. doi: 10.1021/ja0301469. [DOI] [PubMed] [Google Scholar]
- 40.Basavaiah D., Sharada D. S., Kumaragurubaran N., Reddy R. M. J. Org. Chem. 2002;67:7135–7137. doi: 10.1021/jo0257952. [DOI] [PubMed] [Google Scholar]
- 41.Krafft M. E., Haxell T. F. N. J. Am. Chem. Soc. 2005;127:10168–10169. doi: 10.1021/ja052146+. [DOI] [PubMed] [Google Scholar]
- 42.Krafft M. E., Seibert K. A., Haxell T. F. N., Hirosawa C. Chem. Commun. 2005:5772–5774. doi: 10.1039/b512665g. [DOI] [PubMed] [Google Scholar]
- 43.Olah G. A. Friedel-Crafts and Related Reactions. New York: Wiley; 1963. [Google Scholar]
- 44.Olah G. A. Friedel-Crafts Chemistry. New York: Wiley; 1973. [Google Scholar]
- 45.Bandini M., Melloni A., Umani-Ronchi A. Angew. Chem. Int. Ed. 2004;43:550–556. doi: 10.1002/anie.200301679. [DOI] [PubMed] [Google Scholar]
- 46.Luo Y., Li C.-J. Chem. Commun. 2004:1930–1931. doi: 10.1039/b404509b. [DOI] [PubMed] [Google Scholar]
- 47.Jorgensen K. A. Synthesis. 2003:1117–1125. [Google Scholar]
- 48.Ishii A., Soloshonok V. A., Mikami K. J. Org. Chem. 2000;65:1597–1599. doi: 10.1021/jo991691o. [DOI] [PubMed] [Google Scholar]
- 49.Johannsen M. Chem. Commun. 1999:2233–2234. [Google Scholar]
- 50.Olah G. A., Khrisnamurti R., Prakash G. K. S. In: in Comprehensive Organic Synthesis, 1st Ed. Trost B. M., Fleming I., editors. Vol. 3. New York: Pergamon; 1991. pp. 293–340. [Google Scholar]
- 51.Krasnov K. A., Kartsev V. G. Russ. J. Org. Chem. 2002;38:430–436. [Google Scholar]
- 52.Ryan R. P., Hamby R. A., Wu Y.-H. J. Org. Chem. 1975;40:724–728. [Google Scholar]
- 53.Shirasaka T., Takuma Y., Shimpuku T., Imaki N. J. Org. Chem. 1990;55:3767–6771. [Google Scholar]
- 54.Wang X., Dong Y., Sun J., Xu X., Li R., Hu Y. J. Org. Chem. 2005;70:1897. doi: 10.1021/jo0480444. [DOI] [PubMed] [Google Scholar]
- 55.Cardellicchio C., Ciccarella G., Naso F., Perna F., Tortorella P. Tetrahedron. 1999;55:14685–14692. [Google Scholar]
- 56.Ji J.-X., Wu J., Au-Yeung T. T.-L., Yip C.-W., Haynes R. K., Chan A. S. C. J. Org. Chem. 2005;70:1093–1095. doi: 10.1021/jo048395i. [DOI] [PubMed] [Google Scholar]
- 57.Chen Y., Yekta S., Yudin A. K. Chem. Rev. 2003;103:3155–3212. doi: 10.1021/cr020025b. [DOI] [PubMed] [Google Scholar]
- 58.Nakamura H., Kamakura T., Ishikura M., Biellmann J.-F. J. Am. Chem. Soc. 2004;126:5958–5959. doi: 10.1021/ja039175+. [DOI] [PubMed] [Google Scholar]
- 59.Murai T., Mutoh Y., Ohta Y., Murakami M. J. Am. Chem. Soc. 2004;126:5968–5969. doi: 10.1021/ja048627v. [DOI] [PubMed] [Google Scholar]
- 60.Ahn J. H., Joung M. J., Yoon N. M., Oniciu D. C., Katritzky A. R. J. Org. Chem. 1999;64:488–492. [Google Scholar]
- 61.Wei C., Li C.-J. Green Chem. 2002;4:39–41. [Google Scholar]
- 62.Fassler R., Frantz D. E., Oetiker J., Carreira E. M. Angew. Chem. Int. Ed. 2002;41:3054–3056. doi: 10.1002/1521-3773(20020816)41:16<3054::AID-ANIE3054>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 63.Fischer C., Carreira E. M. Org. Lett. 2001;3:4319–4321. doi: 10.1021/ol017022q. [DOI] [PubMed] [Google Scholar]
- 64.Frantz D. E., Fassler R., Carreira E. M. J. Am. Chem. Soc. 1999;121:11245–11246. [Google Scholar]
- 65.Miura M., Enna M., Okuro K., Nomura M. J. Org. Chem. 1995;60:4999–5004. [Google Scholar]
- 66.Wei C., Mague J. T., Li C.-J. Proc. Natl. Acad. Sci. USA. 2004;101:5749–5754. doi: 10.1073/pnas.0307150101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Koradin C., Polborn K., Knochel P. Angew. Chem. Int. Ed. 2002;41:2535–2538. doi: 10.1002/1521-3773(20020715)41:14<2535::AID-ANIE2535>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 68.Zhang J., Wei C., Li C.-J. Tetrahedron Lett. 2002;43:5731–5733. [Google Scholar]
- 69.Wei C., Li C.-J. J. Am. Chem. Soc. 2003;125:9584–9585. doi: 10.1021/ja0359299. [DOI] [PubMed] [Google Scholar]
- 70.Yao X., Li C.-J. Org. Lett. 2006;8:1953–1955. doi: 10.1021/ol060645p. [DOI] [PubMed] [Google Scholar]
- 71.Wei C., Li C.-J. Lett. Org. Chem. 2005;2:410–414. [Google Scholar]
- 72.Wei C., Li Z., Li C.-J. Org. Lett. 2003;5:4473–4475. doi: 10.1021/ol035781y. [DOI] [PubMed] [Google Scholar]
- 73.Yao X., Li C.-J. Org. Lett. 2005;7:4395–4398. doi: 10.1021/ol051575+. [DOI] [PubMed] [Google Scholar]
- 74.Li Z, Wei C, Chen L., Varma R. S., Li C.-J. Tetrahedron Lett. 2004;45:2443–2446. [Google Scholar]
- 75.Luo Y., Li Z., Li C.-J. Org. Lett. 2005;7:2675–2678. doi: 10.1021/ol050826b. [DOI] [PubMed] [Google Scholar]
- 76.Nicolaou K. C., Mathison C. J. N., Montagnon T. J. Am. Chem. Soc. 2004;126:5192–5201. doi: 10.1021/ja0400382. [DOI] [PubMed] [Google Scholar]
- 77.Wang F., Sayre L. M. J. Am. Chem. Soc. 1992;114:248–255. [Google Scholar]
- 78.Leonard N. J., Leubner G. W. J. Am. Chem. Soc. 1949;71:3408–3411. [Google Scholar]
- 79.Black D. A., Arndtsen B. A. Org. Lett. 2004;6:1107–1110. doi: 10.1021/ol036462+. [DOI] [PubMed] [Google Scholar]
- 80.Murahashi S.-I. Angew. Chem. Int. Ed. 1995;34:2443–2465. [Google Scholar]
- 81.Barton D. H. R., Beviere S. D., Chavasiri W., Csuhai E., Doller D. Tetrahedron. 1992;48:2895–2910. [Google Scholar]
- 82.Kharasch M. S., Fono A. J. Org. Chem. 1959;24:72–78. [Google Scholar]
- 83.Murahashi S.-I., Naota T., Kuwabara T., Saito T., Kumobayashi H., Akutagawa S. J. Am. Chem. Soc. 1990;112:7820–7822. [Google Scholar]
- 84.Murahashi S.-I., Naota T., Yonemura K. J. Am. Chem. Soc. 1988;110:8256–8258. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}