

Abstract
The areca alkaloids comprise arecoline, arecaidine, guvacoline, and guvacine. Approximately 600 million users of areca nut products, for example, betel quid chewers, are exposed to these alkaloids, principally arecoline and arecaidine. Metabolism of arecoline (20 mg/kg p.o. and i.p.) and arecaidine (20 mg/kg p.o. and i.p.) was investigated in the mouse using a metabolomic approach employing ultra-performance liquid chromatography–time-of-flight mass spectrometric analysis of urines. Eleven metabolites of arecoline were identified, including arecaidine, arecoline N-oxide, arecaidine N-oxide, N-methylnipecotic acid, N-methylnipecotylglycine, arecaidinylglycine, arecaidinylglycerol, arecaidine mercapturic acid, arecoline mercapturic acid, and arecoline N-oxide mercapturic acid, together with nine unidentified metabolites. Arecaidine shared six of these metabolites with arecoline. Unchanged arecoline comprised 0.3–0.4%, arecaidine 7.1–13.1%, arecoline N-oxide 7.4–19.0%, and N-methylnipecotic acid 13.5–30.3% of the dose excreted in 0–12 h urine after arecoline administration. Unchanged arecaidine comprised 15.1–23.0%, and N-methylnipecotic acid 14.8%–37.7% of the dose excreted in 0–12 h urine after arecaidine administration. The major metabolite of both arecoline and arecaidine, N-methylnipecotic acid, is a novel metabolite arising from carbon–carbon double-bond reduction. Another unusual metabolite found was the monoacylglyceride of arecaidine. What role, if any, that is played by these uncommon metabolites in the toxicology of arecoline and arecaidine is not known. However, the enhanced understanding of the metabolic transformation of arecoline and arecaidine should contribute to further research into the clinical toxicology of the areca alkaloids.
Introduction
Areca nut is the seed of the oriental palm Areca catechu L. that contains the closely related alkaloids arecoline, arecaidine, guvacoline, and guvacine (1) (Figure 1). Combined with the leaf of the betel vine Piper betle and slaked lime, various preparations of areca nut are prepared in India, Taiwan, and Southeast Asia for the purpose of chewing. It has been estimated that areca nut chewing is the fourth most popular habit worldwide, after the use of tobacco, alcohol, and caffeine (2). Areca nut chewing manifests several pharmacological effects that are predominantly parasympathetic in nature, including euphoria, central nervous system stimulation, vertigo, salivation, miosis, tremor, and bradycardia (1), muscarinic effects that are thought to be largely attributable to the predominant alkaloid arecoline (3). Several studies have reported a dependency syndrome associated with areca nut chewing (1, 2), that is said to produce relaxation, improved concentration, mild euphoria, and enhanced postprandial satisfaction (1, 2). A withdrawal syndrome has also been described, comprising mood swings, anxiety, irritability, and insomnia (1, 2). The severity of dependence was reported to be similar to that associated with amphetamine use (1, 2).
Figure 1.
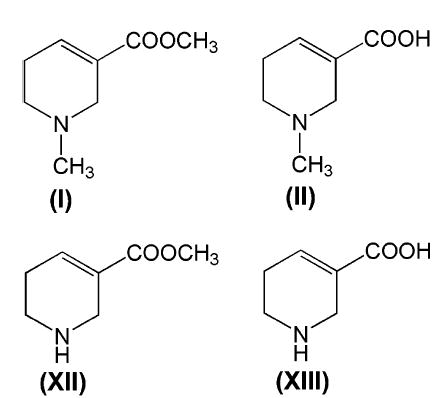
Structures of the areca alkaloids found in areca nut. (I), arecoline; (II), arecaidine; (XII), guvacoline; (XIII), guvacine.
Some areca nut preparations also contain tobacco. The habit of chewing areca nut preparations without tobacco has been associated with an increased risk of oral cancer in four epidemiologic studies, one each from India, Pakistan, Taiwan, and China (1). One dominant theory has been that various nitrosamines may be formed in the mouth from areca alkaloids and that these are causative of human oral cancer (4–6). These nitrosamines include the nitrosamines of guvacine and guvacoline (see Figure 1), together with 3-methylnitrosopropionitrile (1). What remains unclear is if arecoline is metabolized in the body to any of these other alkaloids or to other derivatives that may also form nitrosamines.
Despite its social and toxicological importance, relatively little is known about the metabolism of arecoline. Early reports suggested that rat liver homogenate was able rapidly and quantitatively to hydrolyze arecoline to arecaidine, provided the concentration of arecoline did not exceed 0.1% (6.5 mM) (3). Subsequently, the kinetics of this reaction was determined in mouse liver homogenate (7) and the Km estimated to be 9.6 mM. While a meager amount of arecaidine production occurred in mouse blood and brain, liver and kidney carried out this reaction virtually quantitatively. Inhibitor studies strongly suggested that the conversion of arecoline to arecaidine was mediated by carboxylesterase (EC 3.1.1.1) (7). Both arecoline and arecaidine have been reported to form mercapuric acids in the rat (8) and therefore presumably undergo glutathione conjugation. Moreover, the N-oxide of arecoline has been described in the rat (9). When the N-oxide was administered to rats, a pattern of metabolites was observed similar to that after arecoline administration, suggesting that the N-oxide is reduced back to arecoline in vivo (9).
Metabolomics is a technology that aims to identify and quantify the metabolome, the dynamic set of all small molecules present in an organism or a biological sample (10). Data-rich analytical chemical methods, such as nuclear magnetic resonance (NMR)1 and liquid chromatography–mass spectrometry (LC–MS), combined with chemometric analyses, such as principal components analysis (PCA) or partial least squares-discriminant analysis (PLS-DA), can be used to give insights into the changes in chemical composition of a particular metabolome that occur in response to a defined environmental stimulus. In the case of drug metabolism, metabolomics is ideally placed to yield novel information regarding the number and nature of drug metabolites that appear in a specified metabolome, for example, urine, after the administration of an exogenous drug molecule. In this report, we have harnessed the resolving power of ultra-performance liquid chromatography (UPLC), the accurate mass determination that is provided by time-of-flight mass spectrometry (TOFMS), and data deconvoluting chemometric software to identify the principal molecular ions that are separated when urines from arecoline and arecaidine treated mice were compared to untreated control mouse urines. This UPLC-TOFMS metabolomic protocol has led to a comprehensive evaluation of the metabolites of arecoline and arecaidine.
Experimental Procedures
Chemicals
Arecoline hydrobromide, arecaidine, l-pipecolic acid, formic acid, formaldehyde, N-acetylcysteine, peroxyacetic acid solution (39% w/v in acetic acid), titanium(III) chloride (TiCl3) solution (10% w/v in 20–30% w/v HCl), and caffeine were purchased from Sigma-Aldrich (St. Louis, MO). N-Methylnipecotic acid was purchased from Oakwood Products Inc. (West Columbia, SC). All solvents and inorganic reagents were of the highest obtainable grade.
Synthesis of (R,S)-1-Methyl-1,2,5,6-tetrahydropyridine-3-carboxylic Acid 1-Oxide Methyl Ester (Arecoline N-Oxide)
For purposes of quantitation of arecoline N-oxide in urine, authentic arecoline N-oxide was prepared by a modification of a published method (9). The viscous and pale yellow product (460 mg) was dried overnight in vacuo over P2O5, giving a pale yellow oil (284 mg, 20.8% yield). TiCl3 reduction is specific for N-oxides (11). Therefore, a sample of the synthesized product (~10 μg in 200 μL), treated with ice-cold TiCl3 in HCl (20 μL) and shaken at room temperature for 1 h, was analyzed by UPLC-TOFMS. The N-oxide peak (172.097 m/z) was completely reduced back to arecoline (156.103 m/z), demonstrating that the synthesized material was authentic arecoline N-oxide. This material was subjected to both 1H and 13C NMR analysis, which confirmed the identity of arecoline N-oxide. 1H NMR (399.7 MHz, D2O): δ 7.23 (1H, m), 4.30 (1H, ddd, J = 2.2, 3.4, 16.4 Hz), 4.22 (1H, ddd, J = 1.6, 3.9, 16.4 Hz), 3.80 (3H, s), 3.58–3.63 (2H, m), 3.43 (3H, s), 2.85 (1H, m), 2.66 (1H, m). 13C NMR (100.5 MHz, D2O): δ169.2, 140.6, 129.7, 66.5, 63.9, 60.2, 55.5, 25.5.
Synthesis of (S)-1-Methyl-2-piperidine Carboxylic Acid (N-Methyl- l-pipecolic Acid)
The reductive methylation of l-pipecolic acid was performed in refluxing formic acid and formaldehyde in the Corvallis laboratory, following a literature procedure (12). The product was purified as the HCl salt using cation exchange chromatography. 1H NMR (300 MHz, D2O): δ3.80 (1H, bd, J = 9.9 Hz), 3.51 (1H, d, J = 12.6 Hz), 3.06 (1H, dt, J = 2.5, 12.6 Hz), 2.87 (3H, s), 2.27 (1H, d, J = 14.4 Hz), 1.89 (2H, bdd, J = 14.4 Hz), 1.73 (2H, dt, J = 2.4, 12.0 Hz), 1.57 (1H, m). 13C NMR (75 MHz, D2O): δ 173.8, 68.4, 57.3, 44.7, 30.0, 24.7, 23.0.
Synthesis of N-Acetyl-S-(3-methoxycarbonyl-1-methylpiperid- 4-yl)-(2R)-cysteine (Arecoline Mercapturic Acid)
The mercapturic acid metabolite of arecoline was synthesized from arecoline hydrobromide and N-acetylcysteine according to published methods (8). A solution in water (~10 μg/mL) was analyzed by UPLC-TOFMS in positive-ion mode (see below) and gave a single peak with a [M + H]+ of 319.130 m/z corresponding to the correct empirical formula (C13H21N2O5S) for the target compound.
Animals and Treatments
Mice used were 6–7 week old FVB males. Mice were maintained under a standard 12 h light/12 h dark cycle with water and chow provided ad libitum. Handling was in accordance with an animal study protocol approved by the National Cancer Institute Animal Care and Use Committee. Arecoline hydrobromide (20 mg/kg) was administered by gavage to six mice, and urine (0–12 h) was collected from mice housed individually in glass metabolic chambers (Jencons, Leighton Buzzard, U.K.). Pre-dose control 0–12 h urines were similarly collected from each mouse, at least 12 h prior to arecoline administration. A further four mice were administered arecaidine (20 mg/kg p.o.), and both 0–12 h predose and postdose urines were collected. This complete experiment was repeated using intraperitoneal administration.
UPLC-TOFMS Analyses
Batches of pre-dose and post-dose urines, for each treatment (arecoline p.o., arecoline i.p., arecaidine p.o., arecaidine i.p.), were analyzed together by UPLC-TOFMS (13). Urine samples were diluted with 5 vol water and centrifuged at 14 000 rpm to remove particles and protein. A 200 μL aliquot of the supernatant was transferred to an auto-sampler vial for UPLC-TOFMS analysis. Urine samples (5 μL/injection) were separated on a 50 × 2.1 mm ACQUITY 1.7 μm C18 column (Waters Corp, Milford, MA) using an ACQUITY UPLC system (Waters) with a gradient mobile phase comprised of 0.1% formic acid (A) and acetonitrile containing 0.1% formic acid (B). A 0.6 mL min–1 flow rate was maintained in a 10 min run. The gradient comprised 100% A for 0.5 min, increasing to 100% B over the next 8.5 min. The eluent was directly introduced into the mass spectrometer by electrospray. Mass spectrometry was performed on aWaters Q-TOF Premier operating in positive-ion mode. The desolvation gas flow was set to 600 L h–1 at a temperature of 350 °C with the cone gas set to 50 L h–1 and the source temperature set to 120 °C. The capillary voltage and the cone voltage were set to 3000 and 60 V, respectively. Leucine-enkephalin was used as the lock mass (m/z 556.277) for accurate mass calibration and introduced using the LockSpray interface at 30 μL min–1 and a concentration of 0.2 ng μL–1 in 50% aqueous methanol. In MS scanning, data were acquired in centroid mode from 100 to 950 m/z. As for MS/MS fragmentation of target ions, collision energy ranging from 15 to 30 V was applied. Following data acquisition, UPLC-MS chromatograms and spectra were further analyzed by MassLynx application software (Waters).
Data Processing and Multivariate Data Analysis (MDA)
Following UPLC-TOFMS data acquisition, centroided and integrated mass chromatographic data were deconvoluted by Marker- Lynx software (Waters) to generate a multivariate data matrix. The intensity of each ion was calculated as the percentage of total ion counts in the whole chromatogram. The data matrix and sample list were further exported into SIMCA-P+ software (Umetrics, Kinnelon, NJ) for MDA. PCA and PLS-DA were conducted after data were transformed by mean-centering and Pareto optimization, a scaling technique that increases the importance of low concentration metabolites without significant amplification of noise. Different from unsupervised PCA, samples/observations were classified as the group of control and arecoline treated in PLS-DA. After PCA and PLS-DA processing, principal components were generated to highlight the major latent variables in the data matrix and were described in a scattering plot. Identification of potential arecoline metabolites was further performed by analyzing the loadings plot and contribution plot.
Identification of Metabolites
PLS-DA analyses reveal the ions that contribute most significantly to the group differences, for example, arecoline treated urines versus pretreatment urines. In general, the 20 most significant ions were examined in more detail. These were expected to be metabolites of the alkaloid administered or, alternatively, endogenous compounds whose levels were elevated by the administration of the alkaloid. Elemental compositions of each of the ions were generated by MarkerLynx, and those with best fits, and also C, H, N, O, and S compositions that could be related to the alkaloids, were noted. In all cases, the identity of the metabolite was apparent from its elemental composition. However, in one case, there existed the possibility that the particular ion was derived from an endogenous metabolite that had the same empirical formula as an arecoline metabolite. This was resolved through the use of authentic standards. In all cases where N-oxides were proposed as metabolites, experiments were carried out with TiCl3 reduction to confirm that the ion in question disappeared after this treatment.
Quantitation of Metabolites
Both identified metabolites and unidentified metabolites were treated similarly for the purposes of quantitation. Accurate mass single ion chromatograms were extracted for each ion, using a tolerance of 50 ppm, from the total ion chromatograms for each biological sample studied. The peaks of interest were quantitated by peak area integration using Mass- Lynx software. This generated relative areas for each peak of interest. These were used to calculated the relative percent of each metabolite in every biological sample, based upon the two following assumptions: (i) that there was little or no fragmentation of the pseudo-molecular ions under the instrumental conditions employed, and (ii) that all compounds of interest ionized similarly. Accordingly, therefore, it is appreciated that these estimates of relative excretion of metabolites are only approximate. In the case of certain metabolites, calibration curves were constructed using authentic standards dissolved in blank urine and concentrations of these metabolites in urine determined using the UPLC-TOFMS. In the case of N-methylnipecotic acid ([M + H]+= 144.102 m/z), an interfering endogenous ion with the same mass, but a different MS/ MS spectrum, prevented quantitation by UPLC-TOFMS. In this case, quantitation was performed using a triple quadrupole mass spectrometer operating in multiple reactions monitoring (MRM) mode.
Liquid Chromatography–TandemMass Spectrometry (LC–MS/ MS) Analysis for N-Methylnipecotic Acid
LC–MS/MS analysis was performed on an ABI API2000 mass spectrometer (Applied Biosystems, Foster City, CA) in ESI mode coupled to a PE 200 LC pump and autosampler (Perkin-Elmer Shelton, CT). Instrumentation was controlled by Analyst 1.4.1 software. Compounds were separated on a Luna C18 3μm 50 × 4.6 mm column (Phenomenex Torrance, CA) with the following LC gradient: 98% A (water containing 0.1% formic acid)/2% B (methanol), held for 1 min, then a linear gradient to 10% A/90% B to 8 min. The column was equilibrated with 98% A/2% B for 2 min between injections. The flow rate was 0.25 mL/min at ambient temperature. The mass spectrometer was operated in positive-ion mode utilizing multiple reactions monitoring (MRM) with the transitions m/z 141.1/125.2 for N-methylnipecotic acid, 195.1/137.3 for caffeine (used as internal standard). The turbo ion spray was maintained at 350 °C, with 5.0 kV applied to the spray needle. Nitrogen was used as the turbo spray, nebulizing, and collision gas. All raw data were processed using the Analyst software. The calibration curve for N-methylnipecotic acid was linear from 1 to 100 μM.
Results
Identification of the Urinary Metabolites of Arecoline Using Metabolomics
Multivariate data analysis on the ions produced by UPLC-TOFMS assay of control and arecoline treated (20 mg/kg p.o.) mouse urines is shown in Figure 2. The supervised PLS-DA scores (Figure 2A) reveal two clusters corresponding to the control and treated urines assayed. The loadings plot (Figure 2B) reveals the ions that contribute to this group separation. The 20 ions associated with arecoline treatment that contribute the greatest to group separation are listed in Table 1, together with arecaidine N-oxide, which did not emerge from the metabolomic analysis, but which was determined by extraction of the exact mass from the individual chromatograms. The presence of unchanged arecoline (I) in urine was determined from the occurrence of an ion of [M +H]+ = 156.104 m/z, which gave a match for C8H13NO2 with an error of 7.0 ppm. The presence of the de-esterified metabolite arecaidine (II) was determined from the presence of an ion of [M +H]+ = 142.087 m/z, which gave a match for C7H11NO2 with zero error. This ion is also clearly visible in the loadings plot (Figure 2B). The presence of arecoline N-oxide (III) was determined from the presence of an ion of [M +H]+ = 172.097 m/z, which gave a match for C8H14NO3 with an error of 5.2 ppm and corresponded to the N-oxide with a formula of C8H13- NO3. This ion appears in the loadings plot as one of the two major outliers and as the M +2 isotope ion (III-i in Figure 2B). Of interest was the observation of an additional ion at the same retention time (1.16–1.17 min, Table 1) with a mass of 343.187 m/z that gave a match for C20H26N2O4 with an error of 4.6 ppm. This ion corresponds to the protonated dimer of arecoline N-oxide. The formation under electrospray conditions of a dimer of the closely related pyridine N-oxide has recently been reported (14). Treatment of urines with TiCl3 resulted in the complete disappearance of the two ions, confirming that it was the N-oxide and not some other hydroxylated derivative. Additional proof was furnished from the synthesis of an authentic standard with the same retention time, accurate mass, and both MS and MS/MS spectra. The presence of arecaidine N-oxide (IV) could not be deduced from the 20 major ions associated with arecoline administration (Table 1). However, because this metabolite had been previously described to occur in the urine of rats fed arecoline (8), its presence in the urines of arecoline treated mice was determined. This revealed an ion of [M +H]+ = 158.083 m/z, which gave a match for C7H11- NO3 with an error of 5.1 ppm. Furthermore, treatment of urines with TiCl3 resulted in the disappearance of this ion, confirming that this ion was due to arecaidine N-oxide and not some other hydroxylated derivative. The presence of N-methylnipecotic acid (V; 1-methyl-3-piperidine carboxylic acid) was determined from the presence of an ion of [M +H]+ = 144.102 m/z, which gave a match for C7H13NO2 with an error of 2.1 ppm. This ion was also a prominent outlier in the loadings plot, and even the M +2 isotope ion (V-I in Figure 2B) is an outlier, suggesting that this compound is a main metabolite. The formula C7H13- NO2 could also be due to the isomer N-methylpipecolic acid (1-methyl-2-piperidine carboxylic acid), a potential metabolite of the endogenous compound pipecolic acid, that is itself derived from lysine catabolism (15, 16). Because the metabolomic analysis clearly showed that the ion 144.102 m/z was elevated in the urine of arecoline treated mice, relative to controls (Figure 2B), it was possible that arecoline administration was perturbing lysine metabolism and provoking an enhanced urinary excretion of N-methylpipecolic acid. By employing authentic standards for these two isomers, it was readily possible to distinguish between them on the basis of both UPLC retention times and MS/MS fragmentation patterns. As shown in Figure 3, blank mouse urine (panel A), urine from an arecoline treated mouse (panel B), and solutions of N-methylnipecotic acid (panel C) and N-methylpipecolic acid (panel D) all produce protonated ions at 144.1 m/z. Two features distinguish these four LC–MS/ MS analyses. First, N-methylpipecolic acid elutes later (0.38 min) than N-methylnipecotic acid or the urinary analytes (all 0.30 min). This excludes N-methylpipecolic acid as the source of the 144.102 ion in the arecoline treated urines. Second, the MS/MS fragmentation pattern for the control urine (panel A) is different from the other three spectra (panels B–D), in that it lacks the ion of mass 98.0962–98.0975 m/z that is present in all of the other spectra. The nature of the material in control urine was not further pursued. Nevertheless, these LC–MS/ MS experiments establish that the ion 144.102 m/z (Table 1) is due to the presence of N-methylnipecotic acid in the arecoline treated mouse urines. The presence of N-methylnipecotylglycine (VI), the glycine conjugate of (V), was determined from the presence of an ion of [M +H]+ = 201.123 m/z, which gave a match for C9H16N2O3 with an error of 1.0 ppm. In theory, this formula could have been due to N-methylpipecolylglycine, the glycine conjugate of N-methylpipecolic acid, but, given the finding (above) concerning N-methylnipecotic acid, this possibility was considered to be remote. The presence of arecaidinylglycine (VII), the glycine conjugate of arecaidine (II), was determined from the presence of an ion of [M +H]+ = 199.108 m/z, which gave a match for C9H14N2O3 with an error of 1.0 ppm. The presence of arecaidinylglycerol (VIII), the conjugate of arecaidine with glycerol, was deduced from the presence of an ion of [M +H]+ = 216.124 m/z, which gave a match for C10H17NO4 with an error of 4.2 ppm. The incorporation of xenobiotic acids into monacylglycerols has long been known (17) and will be discussed later. The presence of arecaidine mercapturic acid (IX) was determined from the presence of an ion of [M +H]+ = 305.115 m/z, which gave a match for C12H20N2O5S with an error of 1.0 ppm. This ion was also a prominent outlier in the loadings plot (Figure 2B). The presence of arecoline mercapturic acid (X) was determined from the presence of an ion of [M +H]+ = 319.133 m/z, which gave a match for C13H22N2O5S with an error of 2.5 ppm. For confirmation, a synthetic sample of this conjugate was compared to the urinary material by UPLC-TOFMS. Figure 4 shows the two mass spectra from the urinary metabolite (panel A) and the synthetic material (panel B). Both spectra contain the same protonated ion and fragmented ions, confirming the identity of this metabolite. The presence of arecoline N-oxide mercapturic acid (XI) was determined from the presence of an ion of [M +H]+ = 335.128 m/z, which gave a match for C13H22N2O6S with an error of 2.1 ppm. Reduction of urines with TiCl3 caused the complete disappearance of this ion, confirming that it corresponded to arecoline N-oxide mercapturic acid, and not the mercapturic acid of a hydroxylated arecoline derivative. Finally, ions of masses 158.027, 160.105, 166.088, 249.114, 263.125, 279.121, 343.296, 344.207, and 482.173 m/z were associated with arecoline treatment. Attempts were made to match all of these to empirical formulas, but no matches that made chemical sense and with errors less than 20 ppm could be determined. The average error in the 12 ions identified in Table 1 was <3 ppm. Accordingly, the identity of these nine ions was not further pursued and they were labeled as unknowns UK-1–UK-9, respectively.
Figure 2.
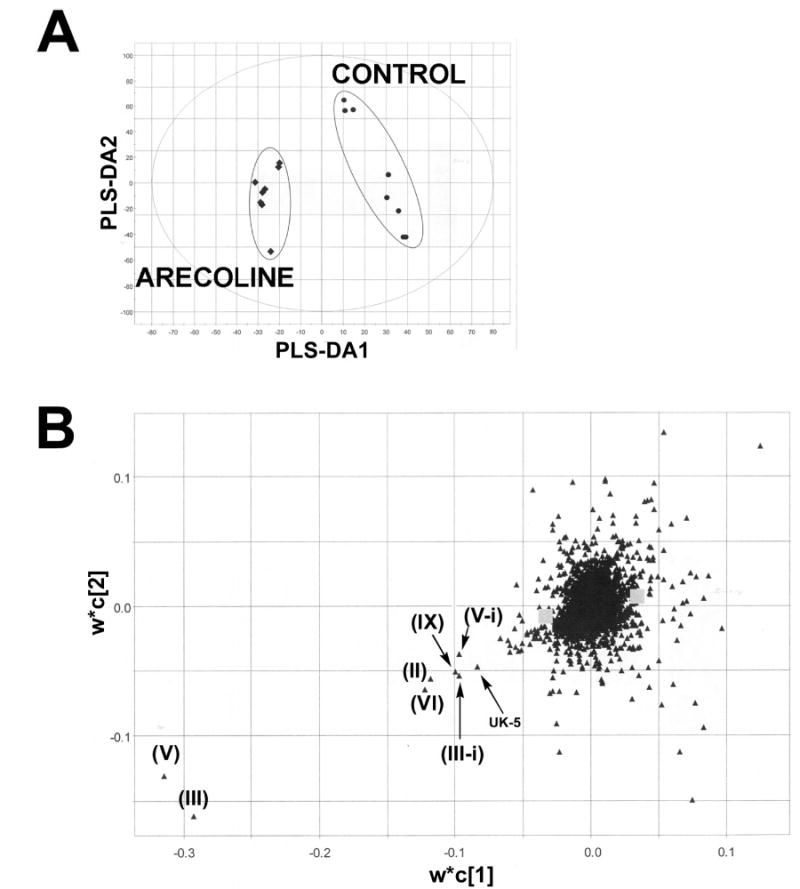
Multivariate data analysis of urinary arecoline metabolites. (A) Separation of control and arecoline treated (20 mg/kg p.o.) mouse urine samples in a PLS-DA scores plot (PLS-DA1/PLS-DA2). (B) Loadings plot of variables generated by PLS-DA. This loading plot represents the relationship between variables (ions) and observation groups (control and arecoline treated) with regard to the first and second components (PLS-DA1/ PLS-DA2) present in (A). W*C represents the combination of the weights of variables and the principal components.
Table 1.
Identification and Quantitation (Peak Areas) of the 0–12 h Urinary Metabolites of Arecoline and Arecaidine in the Mousea
| mass [M + H]+ | retention time (min) | deduced formula [M] | error (ppm) | identity (corresponding compound in Figure 2) | arecoline (p.o.) mean ± SD | arecoline (i.p.) mean ± SD | arecaidine (p.o.) mean ± SD | arecaidine (i.p.) mean ± SD |
|---|---|---|---|---|---|---|---|---|
| 142.087 | 0.28-0.30 | C7H11NO2 | 0 | arecaidine (II) | 36.1 ± 4.5 | 33.7 ± 17.8 | 64.8 ± 28.8 | 124.7 ± 35.3 |
| 144.102 | 0.30-0.31 | C7H13NO2 | 2.1 | N-methylnipecotic acid (V) | 384.0 ± 84.5 | 335.2 ± 142.9 | 446.3 ± 50.9 | 527.7 ± 54.7 |
| 156.104 | 0.63 | C8H13NO2 | 7.0 | arecoline (I) | 6.5 ± 3.8 | 20.3 ± 9.5 | 0 | 0 |
| 158.027 | 1.55 | n.d. | n.d. | UK-1 | 56.7 ±2.1 | 55.2 ± 10.7 | 61. 3 ±28.7 | 55.6 ± 35.4 |
| 158.083 | 0.33-0.35 | C7H11NO3 | 5.1 | arecaidine N-oxide (IV)* | 2.4 ± 1.4 | 12.7 ± 4.4 | 16.6 ±3.1 | 19.8 ±11.0 |
| 160.105 | 0.35 | n.d. | n.d. | UK-2 | 0 | 0 | 11.0 ±3.8 | 6.1 ± 3.4 |
| 166.088 | 1.58 | n.d. | n.d. | UK-3 | 0 | 76.9 ± 74.0 | 58.7 ± 43.2 | 76.1 ± 74.9 |
| 172.097 | 1.16 | C8H13NO3 | 5.2 | arecoline N-oxide (III)* | 428.6 ± 30.2 | 339.5 ± 53.6 | 0 | 0 |
| 199.108 | 0.31-0.32 | C9H14N2O3 | 1.0 | arecaidinylglycine (VII) | 9.7 ± 1.2 | 20.1 ±7.0 | 25.2 ± 6.8 | 49.6 ± 26.2 |
| 201.123 | 0.35 | C9H16N2O3 | 1.0 | N-methylnipecotylglycine (VI) | 32.2 ±4.1 | 69.9 ±27.1 | 70.4 ±12.1 | 187.9 ± 96.5 |
| 216.124 | 2.60 | C10H17NO4 | 4.2 | arecaidinylglycerol (VIII) | 15.2 ±7.1 | 30.3 ± 32.3 | 22.8 ± 0.4 | 15.8 ± 2.2 |
| 249.114 | 2.37 | n.d | n.d. | UK-4 | 2.9 ± 1.0 | 3.3 ± 1.3 | 2.8 ±3.1 | 2.9 ±1.8 |
| 263.125 | 2.35-2.44 | n.d. | n.d. | UK-5 | 14.0 ± 3.5 | 13.7 ± 6.6 | 22.4 ±11. 2 | 11.6 ±3.5 |
| 279.121 | 1.48 | n.d. | n.d. | UK-6 | 5.4 ± 5.2 | 7.4 ± 4.6 | 0 | 0 |
| 305.115 | 0.46-0.48 | C12H20N2O5S | 1.0 | arecaidine mercapturic acid (IX) | 14.8 ± 5.0 | 30.3 ± 25.6 | 45.1 ± 34.8 | 39.9 ± 9.2 |
| 319.133 | 1.22-1.23 | C13H22N2O5S | 2.5 | arecoline mercapturic acid (X) | 7.1 ±0.2 | 14.0 ± 6.1 | 0 | 0 |
| 335.128 | 1.99 | C13H22N2O6S | 2.1 | arecoline N-oxide mercapturic acid (XI)* | 7.9 ± 2.9 | 4.4 ± 2.1 | 0 | 0 |
| 343.187 | 1.17 | C20H26N2O4 | 4.6 | arecoline N-oxide (dimer) (III) | 15.0 ± 2.4 | 35.0 ± 20.6 | 0 | 0 |
| 343.296 | 5.77 | n.d. | n.d. | UK-7 | 0 | 0 | 9.4 ± 16.4 | 0 |
| 344.207 | 3.80 | n.d. | n.d. | UK-8 | 6.4 ± 2.8 | 14.5 ± 6.6 | 12.5 ± 8.6 | 14.4 ± 12.1 |
| 482.173 | 2.00 | n.d. | n.d. | UK-9 | 1.6 ±2.0 | 0 | 7.5 ± 7.9 | 37.6 ± 34.5 |
| total area | 1046 | 1116 | 876.8 | 1170 |
n.d., not determined; 0 area, peak not detected;
, peak disappears on reduction with TiCl3.
Figure 3.
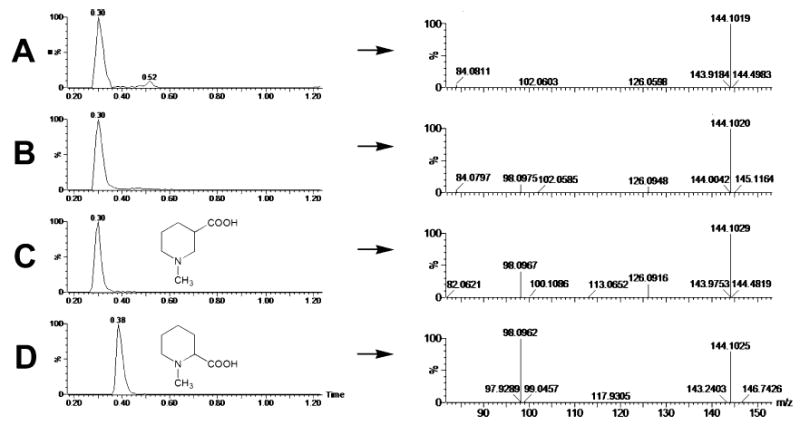
Identification of N-methylnipecotic acid as a metabolite of arecoline by LC–MS/MS. (A) Single-ion chromatogram (m/z = 144.1) of urine from an untreated mouse, showing a single major peak eluting at 0.30 min in 20 mM ammonium formate (pH 6.4). The positive-ion MS/MS spectrum shows a [M +H]+ion at 144.102 m/z and a fragment ion at 84.081 m/z. (B) Single-ion chromatogram (m/z = 144.1) of urine from a mouse treated with arecoline (20 mg/kg p.o.), showing a single peak eluting at 0.30 min in 20 mM ammonium formate (pH 6.4). The positive-ion MS/MS spectrum shows a protonated ion at 144.102 m/z and fragment ions at 98.098 and 126.095 m/z. (C) Single-ion chromatogram (m/z = 144.1) of a 10 μM aqueous solution of N-methylnipecotic acid, showing its chemical structure and a single peak eluting at 0.30 min in 20 mM ammonium formate (pH 6.4). The positive-ion MS/MS spectrum shows a protonated ion at 144.103 m/z and fragment ions at 98.097 and 126.092 m/z. (D) Single-ion chromatogram (m/z = 144.1) of a 10 μM aqueous solution of N-methylpipecolic acid, showing its chemical structure and a single peak eluting at 0.38 min in 20 mM ammonium formate (pH 6.4). The positive-ion MS/MS spectrum shows a protonated ion at 144.103 m/z and a large fragment ion at 98.096 m/z.
Figure 4.
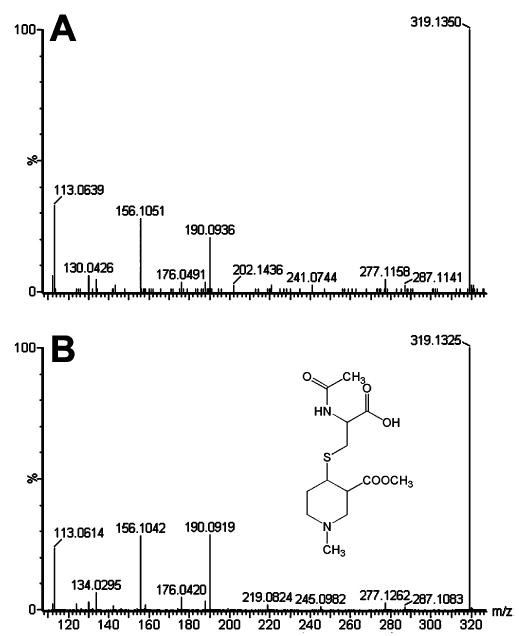
MS/MS analysis of arecoline mercapturic acid. (A) MS/ MS spectrum of the urinary peak eluting at 1.22–1.23 min with a mass of 319.1 m/z. (B) MS/MS spectrum of synthetic arecoline mercapturic acid that eluted at 1.22–1.23 min with a mass of 319.1 m/z.
The same multivariate analysis was performed on urines from mice administered arecoline intraperitoneally (20 mg/kg). All of the same ions associated with arecoline treatment were found as with the oral administration (Table 1). The only qualitative difference observed between these two routes of administration was that UK-3 was only found in the urines from i.p. administration. No new metabolites could be observed after i.p. administration.
Identification of the Urinary Metabolites of Arecaidine UsingMetabolomics
Multivariate data analysis was performed on data derived from UPLC-TOFMS analysis of control urines and urines from arecaidine treated, as described above. A number of qualitative differences were found between the arecaidine and arecoline data sets. Unknown compounds, UK-2 and UK-7, were present in the arecaidine data set but absent from the arecoline data set. Of more interest was the observation that the ions corresponding to arecoline (I, 156.104), arecoline N-oxide (III, 158.083; 343.187), arecoline mercapturic acid (X, 319.133), and arecoline N-oxide mercapturic acid (XI, 335.128) were all absent from the arecaidine data set (Table 1). This is added proof of the identity of these ions. Intraperitoneal and oral administration of arecaidine both gave qualitatively similar data sets.
The Arecoline and Arecaidine Metabolic Map
Figure 5 depicts the metabolic pathways that were found for both arecoline and arecaidine administration in the mouse, integrated into a single metabolic map. The majority of arecoline metabolites are formed after the compound is first hydrolyzed to arecaidine. N-Oxidation and mercapturic acid formation are the only two pathways of arecoline metabolism not involving prior hydrolysis. Arecaidine also undergoes N-oxidation and mercapturic acid formation but, in addition, participates in three other reactions, conjugation with both glycine and glycerol, and double-bond reduction to yield N-methylnipecotic acid, which is itself further conjugated with glycine. Therefore, 10 urinary metabolites of arecoline and 7 of arecaidine are hereby described in the mouse. Of these, only arecoline N-oxide (III) (9), arecaidine N-oxide (IV) (9), and arecaidine mercapturic acid (IX) (8, 9) have been previously described after acute and chronic administration of arecoline and arecaidine to rats. Arecaidine was also known to be produced from arecoline by rat liver homogenates and in the mouse after administration of 10 mg/kg s.c.
Figure 5.
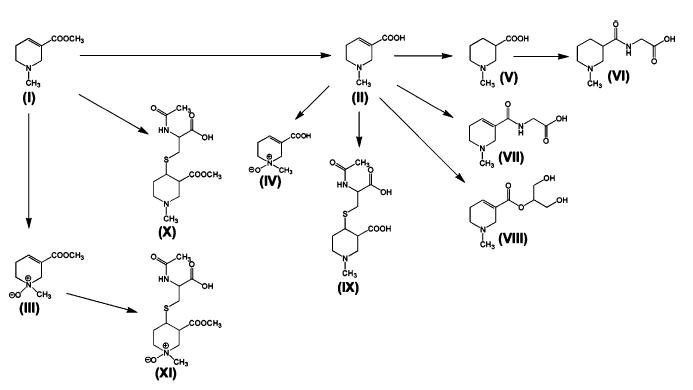
The metabolic map of arecoline and arecaidine in the mouse. Identity of urinary metabolites is as follows: arecoline (I), arecaidine (II), arecoline N-oxide (III), arecaidine N-oxide (IV), N-methylnipecotic acid (V), N-methylnipecotylglycine (VI), arecaidinylglycine (VII), arecaidinylglycerol (VIII), arecaidine mercapturic acid (IX), arecoline mercapturic acid (X), and arecoline N-oxide mercapturic acid (XI).
Quantitation of Urinary Metabolites of Arecoline and Arecaidine
Rough determinations were made of the relative urinary excretions of all of the identified metabolites of both arecoline and arecaidine to put some approximate weight on the various and competing pathways of metabolism. It is appreciated that such estimations are only approximate, due to differences in ionization of the various metabolites and variable ion suppression effects throughout the chromatograms. However, for the principal metabolites, quantitation was based upon calibration curves made with authentic standards.
Relative urinary concentrations of all of the identified and unknown metabolites of arecoline and arecaidine, after both oral and i.p. administration at 20 mg/kg, were quantitated from accurate mass single-ion chromatograms to yield the data shown in Table 1. It is apparent that the metabolites that had the most differentiated ions on the loadings plot (Figure 2B) are also those metabolites that were the most abundant when quantitated as described above. Of particular note, after arecoline administration, are arecoline N-oxide (III) and N-methylnipecotic acid (V), and these were also the two furthest outliers in the loadings plot (Figure 2B). After arecaidine administration, again N-methylnipecotic acid was the principal metabolite. These patterns of urinary metabolites after arecoline and arecaidine administration to mice, both by p.o. and by i.p. routes, are shown in Figure 6. Approximately 10–20% of the arecoline and arecaidine related compounds in urine remain unidentified. Approximately 30% of the balance of metabolism of arecoline is by doublebond reduction to N-methylnipecotic acid, and for arecaidine this value is closer to 50%. In the case of arecoline, there is also considerable N-oxidation comprising 30–40% of the metabolism. The patterns of metabolism for p.o. and i.p. administration for both compounds are broadly similar.
Figure 6.
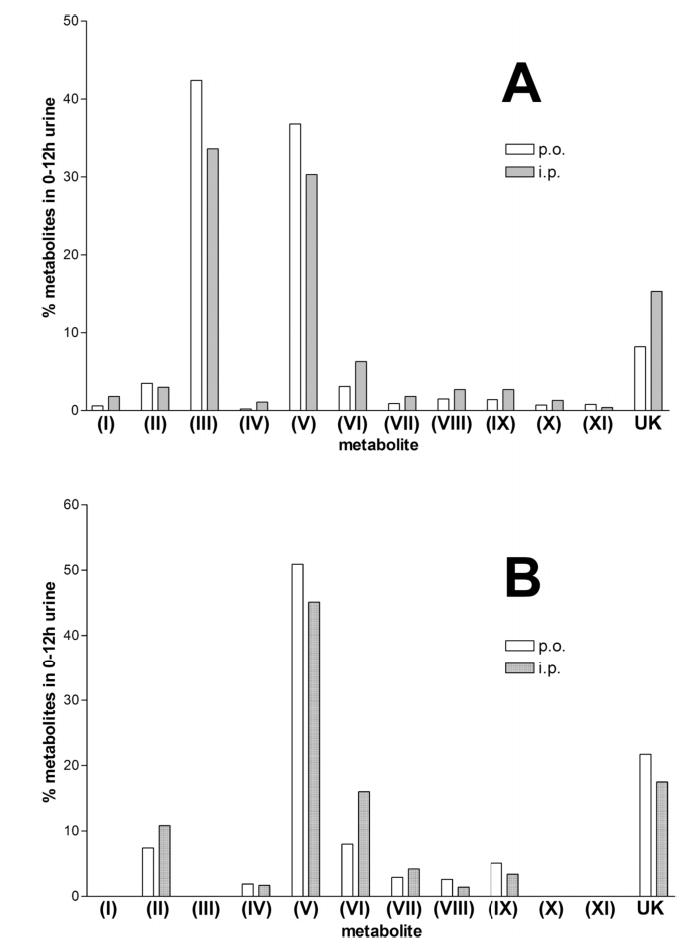
Relative approximate percentages of each urinary metabolite of arecoline and arecaidine in the mouse. (A) Urinary metabolite profile for arecoline (20 mg/kg p.o. and i.p.) in the mouse (for key to metabolites, see Figure 5). (B) Urinary metabolite profile for arecaidine (20 mg/kg p.o. and i.p.) in the mouse (for key to metabolites, see Figure 5).
These aforementioned metabolic data represent estimates of the relative proportions of each identified and unknown metabolite in mouse urine. It is likely that the method of estimation may contain assumptions that preclude accurate determination of metabolite levels. Such assumptions largely concern the ionization and fragmentation of individual compounds under experimental conditions. To obtain a more precise estimate of the urinary excretion of individual metabolites, calibration curves were constructed using the authentic standards arecoline, arecaidine, arecoline N-oxide, and N-methylnipecotic acid, the first three using UPLC-TOFMS and the last by MRM using a triple quadrupole LC–MS/MS. This maneuver was necessary because of the unidentified interfering substance in mouse urine that also had a mass of 144.1 m/z (Figure 3A). The calibration curves for each of these substances were linear and are shown in Figure 7. Analysis of mouse urines revealed that very little arecoline (0.3–0.4% dose) is excreted unchanged after arecoline administration. However, arecaidine (7.1–13.1% dose), arecoline N-oxide (7.4–19.0% dose), and N-methylnipecotic acid (13.5–30.3% dose) were all excreted as major metabolites of arecoline. After arecaidine administration, unchanged arecaidine (15.1–23.0% dose) and N-methylnipecotic acid (14.8–37.7% dose) were major excretion products. The difference between the estimates of relative proportions of metabolites (Table 1) and the calculated percent of dose excreted (Table 2) is minor in nature.
Figure 7.
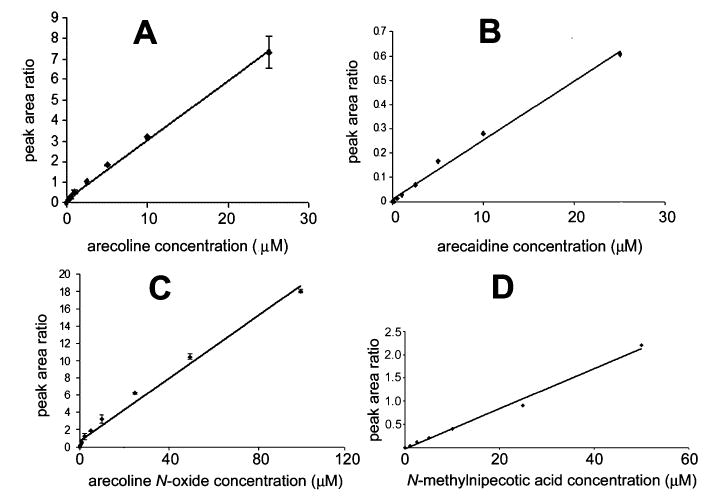
Calibration curves for authentic arecoline metabolites. Data are mean ± SD for duplicates. (A) Arecoline, linear from 0 to 25 μM, r2 = 0.997. (B) Arecaidine, linear from 0 to 25 μM, r2 = 0.992. (C) Arecoline N-oxide, linear from 0 to 100 μM, r2 = 0.998. (D) N-Methylnipecotic acid, linear from 0 to 50 μM, r2 = 0.992.
Table 2.
Calculated Percent of Dose of Metabolites Excreted (0–12 h) after Administration of Both Arecoline and Arecaidine to the Mousea
| % dose (mean ± SD) eliminated in 0-12 h urine as:
|
||||
|---|---|---|---|---|
| administration | arecoline | arecaidine | arecoline N oxide | N-Methylnipecotic acid |
| arecoline (20 mg/kg p.o.) | 0.3 ± 0.4 | 13.1 ±6.3 | 19.0 ± 9.0 | 30.3 ± 11.1 |
| arecoline (20 mg/kg i.p.) | 0.4 ± 0.4 | 7.1 ±6.2 | 7.4 ± 2.8 | 13.5±2.6 |
| arecaidine (20 mg/kg p.o.) | 0 | 23.0 ±11.2 | 0 | 37.7±0 |
| arecaidine (20 mg/kg i.p.) | 0 | 15.1 ±8.3 | 0 | 14.8±6.3 |
0, not detected.
Discussion
It has been estimated that globally there are as many as 600 million chewers of areca nut products, such as betel quid (18). IARC has evaluated betel quid without tobacco as causing oral cancer and has stated that (i) there is sufficient evidence in experimental animals for the carcinogenicity of areca nut, (ii) there is limited evidence in experimental animals for the carcinogenicity of arecoline, and (iii) there is insufficient evidence in experimental animals for the carcinogenicity of arecaidine (1). Despite the prevalence of its use and potential human carcinogenicity, relatively little is known about the metabolism and disposition of arecoline in humans or in animals. Hitherto, only four metabolites of arecoline have been described, arecaidine (3, 9), arecoline N-oxide (9), arecaidine N-oxide (9), and arecaidine mercapturic acid (8, 9). Studies have been limited to two in rats (8, 9) and one in mice (3), and nothing on this topic has been published in over 30 years. The metabolism of arecoline and arecaidine has not been reported in humans, despite clinical trials with arecoline in Alzheimer patients (19) and the widespread exposure to these compounds through betel quid and areca nut use. This compares very poorly to nicotine, for which 24 metabolites have been reported, and much is known about the biochemistry and pharmacogenetics of their production (20).
Here, we report a metabolomic evaluation of arecoline and arecaidine metabolism in the mouse. The chromatographic resolving power of UPLC, exact mass determination by TOFMS, and the handling and deconvolution by metabolomics software of large data sets, containing thousands of ions per sample, have been combined to isolate compounds that are associated with administration of these alkaloids to mice. Analysis of 21 ions led to the identification of unchanged arecoline and 11 urinary metabolites of arecoline and arecaidine (Figure 5). Of the nine unidentified compounds (UK-1–UK-9; Table 1), it is not known if these are metabolites of arecoline and/or arecaidine, or are endogenous substances whose excretion is enhanced by the administration of the alkaloids.
The principal pathways of arecoline metabolism appear to be hydrolysis to arecaidine, N-oxidation, together with double-bond reduction of the metabolite arecaidine. This last pathway also appears particularly dominant after administration of arecaidine itself. Regarding N-oxidation, three N-oxides were detected after arecoline administration, and one after arecaidine administration. The principal N-oxide found was arecoline N-oxide (III in Figure 5), which appeared to constitute 30– 40% of the excreted metabolites. This compound was synthesized and determined in each urine sample and was found to represent up to 19% of the dose (Table 2). If 50% of the dose were excreted in 0–12 h, these data would agree well. Arecaidine N-oxide and arecoline N-oxide mercapturic acid were relatively minor metabolites. The full extent of N-oxidation of arecoline and arecaidine is unknown. It has been reported (9) that the administration of arecoline N-oxide to rats gives a metabolic profile similar to that of arecoline itself, suggesting that the N-oxide is reduced back to arecoline in vivo. This phenomenon has been named “metabolic retroversion” (21) and was uncovered after trimethylamine N-oxide administration to subjects with “fish-odor syndrome” that were genetically deficient in trimethylamine N-oxidation due to a polymorphism in the FMO3 gene. These three N-oxide metabolites or arecoline might be produced by a flavin-containing mono-oxygenase (FMO), or alternatively by cytochromes P450.
The other major metabolite appears to be N-methylnipecotic acid. Metabolic reduction of carbon–carbon double bonds in xenobiotics is rare. In the areca alkaloid series (Figure 1), the double bond is in conjugation with the carbonyl function, and, in the case of arecoline, it reacts readily with N-acetylcysteine in hot ethanol to form a mercapturic acid in high yield (8). Metabolic cleavage of arecoline mercapturic acid between the arecoline and the sulfur atom would yield the methyl ester of N-methylnipecotic acid, a metabolite that was not observed in any of the treated mouse urines. Nor did we find evidence for the action of cysteine conjugate β-lyase, which would have yielded the thiol and/or its S-methyl derivative (22). It is unlikely, therefore, that N-methylnipecotic acid, and its glycine conjugate also reported here, were produced as further products of metabolism of the observed mercapturic acid conjugates. It is more likely that these novel metabolites arise from arecaidine, especially because their excretion is greater after arecaidine, rather than arecoline, administration (Tables 1 and 2). One additional clue comes from the observation that arecaidine is converted to both glycine and glycerol conjugates, and these pathways almost certainly require the formation of a CoA ester intermediate to become substrates for the glycine N-acyltransferase (23). Enzymes are known that can reduce double bonds in conjugated acyl-CoA esters, such as that present in arecaidinyl- CoA. One candidate is trans-2-enoyl thioester reductase (EC 1.3.1.38), an NADPH-dependent enzyme involved in mitochondrial fatty acid synthesis and mitochondrial maintenance (24, 25). It may be speculated that arecaidine may be a substrate for this enzyme, once it forms a CoA ester in mitochondria, of which there is evidence because arecaidinylglycine was detected as a urinary metabolite.
The finding of a glycerol conjugate of arecaidine in mouse urine was not unexpected. Arecaidine is a xenobiotic carboxylic acid, and a large number of such acids have been reported to form lipid conjugates (17). For example, the nonsteroidal antiinflammatory agent fenbufen (4-(4-biphenyl)-4-oxo-butanoic acid) forms a sn-2-monoacylglyceride in cultured adipocytes (17), analogous to the arecaidine metabolite (XIII in Figure 5). The monoacylglycerol of fenbufen can then form a triacylglycerol with palmitic acid (17). The fate and degradation of these xenobiotic-containing lipids is poorly understood.
Although arecoline and arecaidine are extensively metabolized to multiple products, none appears to be due to cytochrome P450. The oxidation reactions appear exclusively to be N-oxidations, certainly carried out by FMO enzymes. The other metabolites observed were either reductions or conjugations. No evidence was found for the formation of guvacoline (XII in Figure 1) from arecoline or guvacine (XIII) from arecaidine, by N-demethylation mediated by cytochrome P450. This is certainly unusual that a xenobiotic forms 11 metabolites, none of which is mediated by cytochrome P450.
Regarding the toxicology of arecoline and arecaidine, studies with nitrosamines derived from areca alkaloids have been inconclusive (1). However, it has been difficult to seek alternative mechanisms to explain the associations with various oral pathologies, including oral cancer, in users of areca nut containing products. There is a view that the areca alkaloids may be directly contributing to certain oral pathologies, but the proposed mechanisms are nebulous. For example, one report claims that arecoline is cytotoxic to human gingival fibroblasts in culture by virtue of glutathione depletion and inhibition of mitochondrial activity (26). Another group showed similar effects in both fibroblasts and keratinocytes (27). Whether or not any of these proposed mechanisms are related to the metabolism of arecoline, perhaps within the mitochondrion, remains a matter of speculation.
We have reported here the metabolic map of arecoline and arecaidine in the mouse. Novel pathways of metabolism for these compounds have been uncovered using the power of a metabolomic approach. This fuller appreciation of the disposition of the major areca alkaloids may contribute to a better understanding of the toxicology of the social customs involving areca nut products, habits that have an estimated 600 million consumers. Studies of the metabolism of areca nut alkaloids in other animal models for man, and in human volunteers also, are urgently required. In addition, it would be productive to investigate the biochemical toxicology of the newly described metabolites, rather than arecoline itself, especially if cell culture systems are employed that almost certainly have no competence to convert arecoline to its metabolites.
Acknowledgments
This work was supported by the National Cancer Institute Intramural Research Program of the NIH. J.R.I. is grateful to the U.S. Smokeless Tobacco Co. for a grant for collaborative research. S.G. was the recipient of a DBT Overseas Associateship (# BT/IN/BTOA/18/2004) from the Department of Biotechnology, Ministry of Science and Technology, Government of India.
Footnotes
Abbreviations: NMR, nuclear magnetic resonance; LC-MS, liquid chromatography-mass spectrometry; MS/MS, tandem mass spectrometry; MRM, multiple reactions monitoring; LC-MS/MS, liquid chromatography–tandem mass spectrometry; PCA, principal components analysis; PLS-DA, partial least squares-discriminant analysis; UPLC, ultra-performance liquid chromatography; TOFMS, time-of-flight mass spectrometry; MDA, multivariate data analysis; p.o., oral; i.p., intraperitoneal; s.c., subcutaneous.
References
- 1.IARC (2004) Betel-quid and Areca-nut Chewing and Some Areca-nut- derived Nitrosamines, Vol. 85, IARC, Lyon. [PMC free article] [PubMed]
- 2.Winstock A. Areca nut-abuse liability, dependence and public health. Addict Biol. 2002;7:133–138. doi: 10.1080/13556210120091509. [DOI] [PubMed] [Google Scholar]
- 3.Nieschulz O, Schmersahl P. On the pharmacology of active materials from betel. 2 Transformation of arecoline to arecaidine. Arzneimittelforschung. 1968;18:222–225. [PubMed] [Google Scholar]
- 4.Wenke G, Brunnemann KD, Hoffmann D, Bhide SV. A study of betel quid carcinogenesis. IV Analysis of the saliva of betel chewers: a preliminary report. J Cancer Res Clin Oncol. 1984;108:110–113. doi: 10.1007/BF00390981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wenke G, Rivenson A, Brunnemann KD, Hoffmann D, Bhide SV. A study of betel quid carcinogenesis. II. Formation of N-nitrosamines during betel quid chewing. IARC Sci Publ. 1984:859–866. [PubMed] [Google Scholar]
- 6.Wenke G, Rivenson A, Hoffmann D. A study of betel quid carcinogenesis. 3 3-(Methylnitrosamino)-propionitrile, a powerful carcinogen in F344 rats. Carcinogenesis. 1984;5:1137–1140. doi: 10.1093/carcin/5.9.1137. [DOI] [PubMed] [Google Scholar]
- 7.Patterson TA, Kosh JW. Elucidation of the rapid in vivo metabolism of arecoline. Gen Pharmacol. 1993;24:641–647. doi: 10.1016/0306-3623(93)90224-l. [DOI] [PubMed] [Google Scholar]
- 8.Boyland E, Nery R. Mercapturic acid formation during the metabolism of arecoline and arecaidine in the rat. Biochem J. 1969;113:123–130. doi: 10.1042/bj1130123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nery R. The metabolic interconversion of arecoline and arecoline 1-oxide in the rat. Biochem J. 1971;122:503–508. doi: 10.1042/bj1220503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weckwerth W, Morgenthal K. Metabolomics: from pattern recognition to biological interpretation. Drug Discovery Today. 2005;10:1551–1558. doi: 10.1016/S1359-6446(05)03609-3. [DOI] [PubMed] [Google Scholar]
- 11.Kulanthaivel P, Barbuch RJ, Davidson RS, Yi P, Rener GA, Mattiuz EL, Hadden CE, Goodwin LA, Ehlhardt WJ. Selective reduction of N-oxides to amines: application to drug metabolism. Drug Metab Dispos. 2004;32:966–972. [PubMed] [Google Scholar]
- 12.Patrick KS, Singletary JL. Relative configuration of thioridazine enantiomers. Chirality. 1991;3:208–211. [Google Scholar]
- 13.Wilson ID, Nicholson JK, Castro-Perez J, Granger JH, Johnson KA, Smith BW, Plumb RS. High resolution “ultra performance” liquid chromatography coupled to oa-TOF mass spectrometry as a tool for differential metabolic pathway profiling in functional genomic studies. J Proteome Res. 2005;4:591–598. doi: 10.1021/pr049769r. [DOI] [PubMed] [Google Scholar]
- 14.March RE, Stadey CJ, Lewars EG. Pyridine N-oxide and pyridine-d5 N-oxide: an electrospray/tandem mass spectrometric study carried out at high mass resolution. Rapid Commun Mass Spectrom. 2005;19:984–1004. doi: 10.1002/rcm.1877. [DOI] [PubMed] [Google Scholar]
- 15.Fujii T, Mukaihara M, Agematu H, Tsunekawa H. Biotransformation of l-lysine to l-pipecolic acid catalyzed by l-lysine 6-aminotransferase and pyrroline-5-carboxylate reductase. Biosci Biotechnol Biochem. 2002;66:622–627. doi: 10.1271/bbb.66.622. [DOI] [PubMed] [Google Scholar]
- 16.Fujita T, Hada T, Higashino K. Origin of d- and l-pipecolic acid in human physiological fluids: a study of the catabolic mechanism to pipecolic acid using the lysine loading test. Clin Chim Acta. 1999;287:145–156. doi: 10.1016/s0009-8981(99)00129-1. [DOI] [PubMed] [Google Scholar]
- 17.Dodds PF. Incorporation of xenobiotic carboxylic acids into lipids. Life Sci. 1991;49:629–649. doi: 10.1016/0024-3205(91)90110-w. [DOI] [PubMed] [Google Scholar]
- 18.Gupta PC, Warnakulasuriya S. Global epidemiology of areca nut usage. Addict Biol. 2002;7:77–83. doi: 10.1080/13556210020091437. [DOI] [PubMed] [Google Scholar]
- 19.Asthana S, Greig NH, Holloway HW, Raffaele KC, Berardi A, Schapiro MB, Rapoport SI, Soncrant TT. Clinical pharmacokinetics of arecoline in subjects with Alzheimer’s disease. Clin Pharmacol Ther. 1996;60:276–282. doi: 10.1016/S0009-9236(96)90054-5. [DOI] [PubMed] [Google Scholar]
- 20.Hukkanen J, Jacob P, III, Benowitz NL. Metabolism and disposition kinetics of nicotine. Pharmacol Rev. 2005;57:79–115. doi: 10.1124/pr.57.1.3. [DOI] [PubMed] [Google Scholar]
- 21.Al-Waiz M, Ayesh R, Mitchell SC, Idle JR, Smith RL. Disclosure of the metabolic retroversion of trimethylamine N-oxide in humans: a pharmacogenetic approach. Clin Pharmacol Ther. 1987;42:608–612. doi: 10.1038/clpt.1987.207. [DOI] [PubMed] [Google Scholar]
- 22.Stevens J, Jakoby WB. Cysteine conjugate beta-lyase. Mol Pharmacol. 1983;23:761–765. [PubMed] [Google Scholar]
- 23.van der Westhuizen FH, Pretorius PJ, Erasmus E. The utilization of alanine, glutamic acid, and serine as amino acid substrates for glycine N-acyltransferase. J Biochem Mol Toxicol. 2000;14:102–109. doi: 10.1002/(sici)1099-0461(2000)14:2<102::aid-jbt6>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 24.Miinalainen IJ, Chen ZJ, Torkko JM, Pirila PL, Sormunen RT, Bergmann U, Qin YM, Hiltunen JK. Characterization of 2-enoyl thioester reductase from mammals. An ortholog of YBR026p/MRF1′p of the yeast mitochondrial fatty acid synthesis type II. J Biol Chem. 2003;278:20154–20161. doi: 10.1074/jbc.M302851200. [DOI] [PubMed] [Google Scholar]
- 25.Airenne TT, Torkko JM, Van den plas S, Sormunen RT, Kastaniotis AJ, Wierenga RK, Hiltunen JK. Structure–function analysis of enoyl thioester reductase involved in mitochondrial maintenance. J Mol Biol. 2003;327:47–59. doi: 10.1016/s0022-2836(03)00038-x. [DOI] [PubMed] [Google Scholar]
- 26.Chang YC, Hu CC, Lii CK, Tai KW, Yang SH, Chou MY. Cytotoxicity and arecoline mechanisms in human gingival fibroblasts in vitro. Clin Oral Invest. 2001;5:51–56. doi: 10.1007/s007840000085. [DOI] [PubMed] [Google Scholar]
- 27.Chang MC, Ho YS, Lee PH, Chan CP, Lee JJ, Hahn LJ, Wang YJ, Jeng JH. Areca nut extract and arecoline induced the cell cycle arrest but not apoptosis of cultured oral KB epithelial cells: association of glutathione, reactive oxygen species and mitochondrial membrane potential. Carcinogenesis. 2001;22:1527–1535. doi: 10.1093/carcin/22.9.1527. [DOI] [PubMed] [Google Scholar]