

Abstract
Extracellular matrix abnormalities have been found in both human and animal models of polycystic kidney disease (PKD). We have produced a new mouse PKD model through insertion of a PGKneo cassette in an intron of the gene encoding laminin α5, a major tubular and glomerular basement membrane component important for glomerulogenesis and ureteric bud branching. Lama5neo represents a hypomorphic allele due to aberrant splicing. Lama5neo/neo mice exhibit PKD, proteinuria, and death from renal failure by 4 weeks of age. This contrasts with mice totally lacking laminin α5, which die in utero with multiple developmental defects. At 2 days of age, Lama5neo/neo mice exhibited mild proteinuria and microscopic cystic transformation. By 2 weeks, cysts were grossly apparent in cortex and medulla, involving both nephron and collecting duct segments. Tubular basement membranes appeared to form normally, and early cyst basement membranes showed normal ultrastructure but developed marked thickening as cysts enlarged. Overall, laminin α5 protein levels were severely reduced due to mRNA frameshift caused by exon skipping. This was accompanied by aberrant accumulation of laminin-332 (α3β3γ2; formerly called laminin-5) in some cysts, as also observed in human PKD. This constitutes the first evidence that a primary defect in an extracellular matrix component can cause PKD.
Nonstandard abbreviations: BM, basement membrane; E, embryonic day; GBM, glomerular BM; P, postnatal day; PKD, polycystic kidney disease; PC1, polycystin-1
Introduction
Laminins are heterotrimeric glycoproteins present in all basement membranes (BMs), the specialized extracellular matrices associated with all epithelia, endothelia, muscle cells, fat cells, and peripheral nerves. At least 15 distinct laminin trimers, containing various combinations of five α, four β, and three γ subunits, have been found in mammals (1). We previously identified the laminin α5 chain as a novel subunit with widespread expression (2). Both laminin α5 and α1 chains are especially prevalent in kidney (3, 4). Laminin-511 (consisting of the α5β1γ1 chains and formerly known as laminin-10; see (5) for new nomenclature) is found in BMs underlying tubules and parietal epithelium of Bowman’s capsule and in the mesangial matrix. Laminin-521 (α5β2γ1; previously laminin-11) has a more restricted distribution in the kidney, being limited primarily to the glomerular basement membrane (GBM) and vascular smooth muscle BM. Laminin-111 (α1β1γ1, previously laminin-1) is found in the mesangium and in BMs of proximal tubule, Bowman’s capsule, and loop of Henle, as well as in the immature GBM (6).
Mice homozygous for a null mutation in the gene encoding laminin α5 (Lama5) die at embryonic day (E) 14–19 with multiple developmental abnormalities. The kidney phenotypes include avascular glomeruli, impaired branching morphogenesis, and occasional renal agenesis (7). In addition, Lama5−/− mice exhibit defects in neural tube closure, digit septation, lung morphogenesis, neural crest migration, and placentation (8–11).
In polycystic kidney disease (PKD), abnormalities in cyst BMs, including thickening and lamination, have been widely reported (12–15). Altered BM composition has also been described. For example, immunofluorescence and biochemical data have shown decreased levels and synthesis and altered post-translational modification of a heparan sulfate proteoglycan now thought to be perlecan (16). In contrast, type IV collagen and laminin have been reported to be focally increased (17). Ectopic deposition of laminin-332 (α3β3γ2, formerly laminin-5) in cyst BMs has also been reported (18). The significance of these findings is difficult to discern; the BM alterations reported have been observed primarily at late stages of disease, after cysts have become well developed and enlarged, and thus may represent secondary changes.
The focus of PKD research has recently turned to the structure and function of cilia, yet the predicted structure of polycystin-1 (PC1), the product of one of the two genes responsible for human autosomal dominant PKD, suggests that it could serve as a receptor for extracellular matrix components (19). Recent in vitro data using polycystin-1 fusion proteins has shown that PC1 fragments are capable of binding laminin (20). However, in vivo data implicating altered cell/matrix interactions in the pathogenesis of PKD has been lacking. Here we report the generation of a hypomorphic allele of Lama5 resulting in greatly reduced Lama5 gene expression and the consequent development of PKD, proteinuria, and death at 3 to 4 weeks of age from renal failure. Our results show for the first time that a primary defect in a non-cilia-associated matrix protein can cause PKD.
Results
During the course of making a conditionally mutant allele of Lama5, we generated mice carrying a loxP site and a PGKneo cassette flanked by frt sites in intron 21 of Lama5, and a second loxP site in an upstream intron (the Lama5neo allele; Figure 1). Mating to a mouse expressing FLPe recombinase results in removal of the neo cassette, leaving the loxP site adjacent to a single frt site in intron 21, and producing a functional but conditionally mutant allele (10).
Figure 1. An insertion in Lama5 causes PKD.
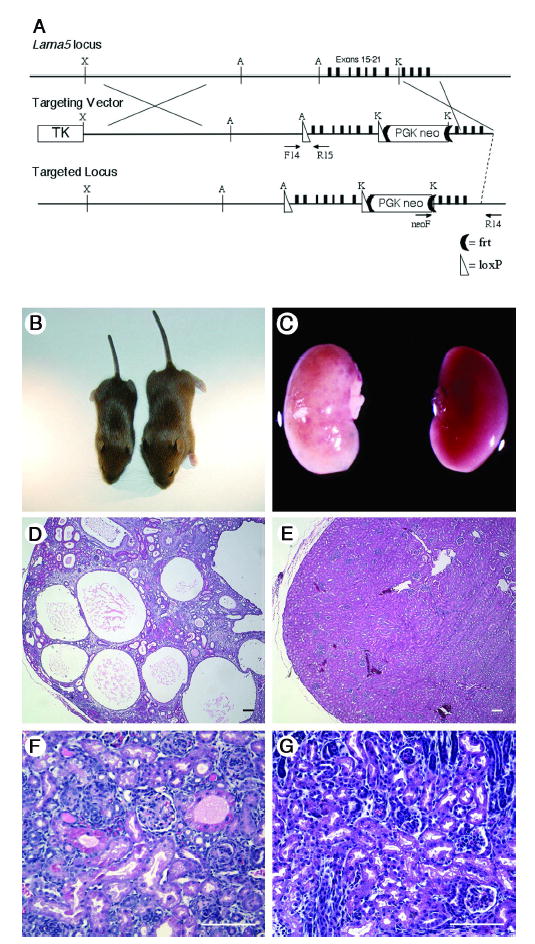
(A) Targeting of Lama5. A loxP-frt-PGKneo-frt cassette was inserted into intron 21 at a KpnI site (K), and a second loxP site was inserted into intron 14 at an AflII site (A). The HSV thymidine kinase gene was inserted at the XbaI site (X) for negative selection. Primers used to identify targeted clones (neoF and R14) and to genotype mice (F14 and R15) are indicated. (B) Three week old Lama5neo/neo (left) and littermate control mice. (C) Kidneys from these same mice. Despite the smaller size of the mutant mouse, its kidneys are as large as controls and exhibit grossly apparent cysts. (D and E) H&E stained paraffin sections show multiple cysts in the Lama5neo/neo kidney but normal architecture in the littermate control at P23. (F and G) Early cystic transformations were already apparent by P2 in the mutant (F) compared to control (G). Scale bars: 100 μm.
To determine whether the Lama5neo allele might exhibit partial activity that would provide novel insights into the biology of laminin α5, we intercrossed heterozygous Lama5+/neomice to produce homozygous Lama5neo/neo offspring. At postnatal day (P) 7, the time when tail clips were collected for genotyping, there were about half the number of expected Lama5neo/neo pups. Moreover, whereas heterozygotes were indistinguishable from wild-type, Lama5neo/neo mice were smaller (Figure 1) and exhibited PKD, progressive renal failure, and death by P28. The kidneys of Lama5neo/neo mice were similar in size to or larger than controls (Figure 1). This is consistent with the enlarged kidneys observed in human PKD, and contrasts with the smaller kidneys seen in cystic dysplasia or acquired cystic disease, for example.
Histopathology and renal function defects
Examination of H&E-stained paraffin sections at various ages revealed the presence of multiple large cysts by P14 and evidence of tubular dilation as early as P1-P2 (Figure 1). Interstitial fibrosis was also prominent at later ages. In some cysts epithelial cells lost their normal unilayered orientation, as cells were piled on top of one another with apparent loss of apposition to the BM.
Most cysts originated from proximal tubules, as shown by staining with the Lotus tetragonolobus lectin (Figure 2A). In addition, antibodies to Tamm-Horsfall protein stained some cyst epithelia, indicating an ascending limb of loop of Henle origin, whereas cytokeratin 8 and aquaporin-2 antibodies stained others, denoting collecting duct involvement (Figure 2B,C). Consistent with the development of a de-differentiated phenotype, some large cysts failed to label with any of the markers, although a distal tubule origin for these cannot be ruled out.
Figure 2. Immunofluorescence analysis of cyst phenotype and laminin α5 expression.
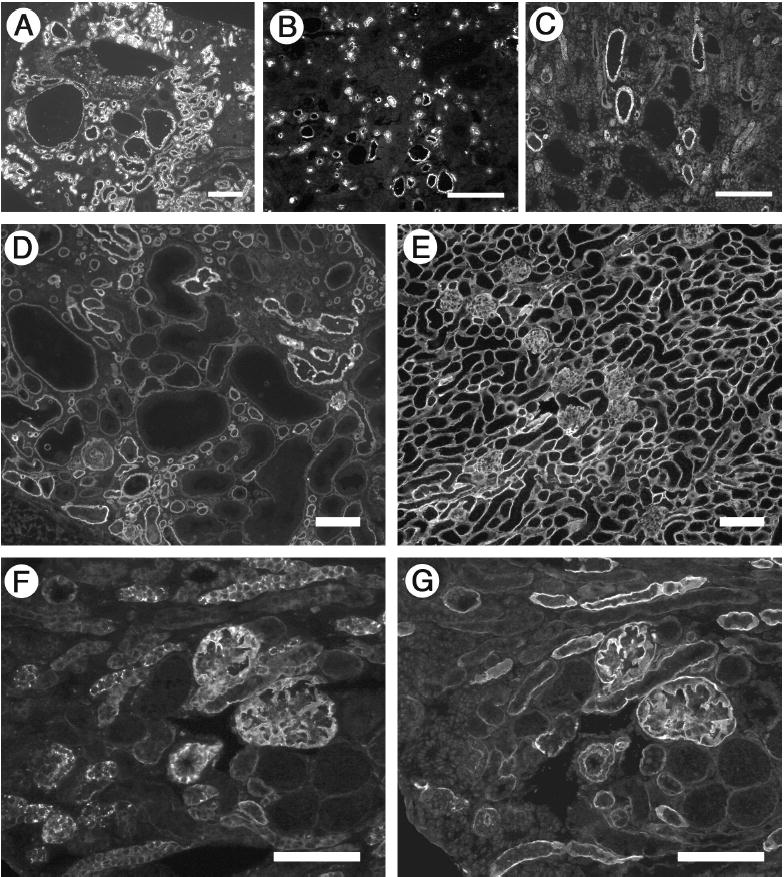
(A–C) Cysts derive from multiple tubular segments. Frozen sections of P23 cystic kidneys were stained with FITC-conjugated Lotus tetragonolobus lectin to label proximal tubules (A), antibody to Tamm-Horsfall protein to label thick ascending limb of the loop of Henle (B), and antibody to cytokeratin 8 to label collecting ducts (C). Consistent with a de-differentiated phenotype, some large cysts failed to label with any of the markers. (D and E) Laminin α5 levels were reduced in Lama5neo/neo kidney (D) compared to control (E) at P21. Laminin α5 deposition was uniform in the control but heterogeneous in the mutant; most cysts showed decreased to nearly undetectable levels of α5. (F and G) Truncated laminin α5 accumulates inside cells. Serial sections from a Lama5neo/neo kidney were stained with antibodies whose epitopes are either NH2-terminal (F) or COOH-terminal (G) of the truncation in domain LEa. Scale bars in A–C: 200 μm; in D-G: 100 μm.
By P21, the affected mice showed evidence of renal failure; the mean+/−s.d. serum urea nitrogen was 106+/−71, compared with 19+/−6.5 in controls. Urine protein to creatine (Upr/UCr) ratios were elevated at 95+/−0.5 mg protein/mg creatinine vs. 12+/−1.9 in controls. Increased protein excretion was an early event, as P2-P4 mutant mice had UPr/UCr ratios of 53+/−16, vs. 23+/−17 in littermate controls. Lama5neo/neo mice uniformly died before 4 weeks of age, with renal failure as the likely cause of death. Cysts were not found in liver (commonly observed in human ADPKD) or pancreas (rarely noted in human ADPKD) (data not shown).
Tubular ultrastructure
Transmission electron microscopic analysis of large cysts from 3 week-old mice (Figure 3) revealed flattened epithelial cells that were often devoid of characteristic tubular cell features, such as a dense brush border. The underlying BMs were of uniform density and thickened. At advanced stages, cyst BMs typically overlay deposits of fibrillar collagen, which was abundant in the expanded interstitium. When early cysts from P1 mice were examined, however, BMs appeared normal (Figure 3), indicating that BM formation per se was not perturbed in Lama5neo/neo mice. These data also suggest that BM thickening represents secondary changes associated with cyst enlargement and is not a primary abnormality.
Figure 3. Ultrastructural analysis of cyst walls.
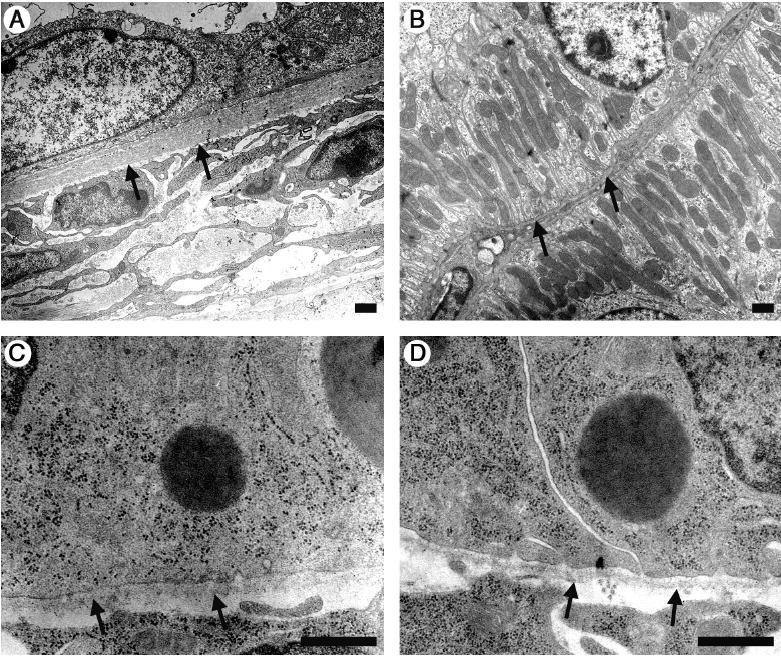
(A) Electron microscopic analysis of a cyst wall from a P23 Lama5neo/neo kidney shows flattened epithelial cells and a markedly thickened BM compared to the control (B). At P1 tubular BM ultrastructure was similar in mutants (C) and control (D). Arrows: BM. Scale bars: 800 nm.
Cell polarity, proliferation, and apoptosis
In PKD, tubular epithelial cells frequently exhibit alterations in polarity. However, here we did not observe significant changes in the localization of normally polarized cell markers, such as E-cadherin, β-catenin, and aquaporin-2 (data not shown). Alterations in proliferation and apoptosis are also frequently observed in PKD. Here, proliferation was increased, as more tubular epithelial cells in Lama5neo/neo mice exhibited BrdU uptake at P20, most notably in cystic epithelia (Figure 4A). Proliferation was also noted to a lesser extent in the interstitial compartment. As assessed by TUNEL assay, there was a focal increase in apoptosis, primarily in cystic tubular epithelia (Figure 4C).
Figure 4. Proliferation and apoptosis assays.
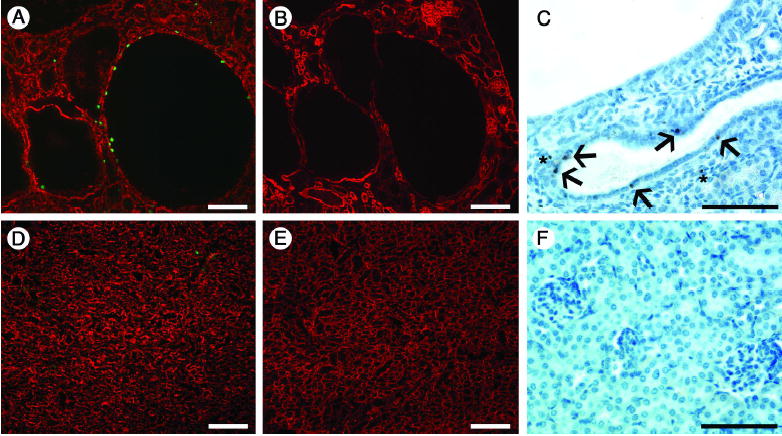
(A and D) Assay for BrdU (green), counterstained with anti-laminin-111 to label basement membranes (red), shows proliferating cells in cyst-lining epithelia (A). BrdU-positive cells were also occasionally observed in the interstitium and in normal tubules in mutants (A) but were rarely found in control (D). (B and E) Serial sections with respect to A and D show staining for laminin α5, which is focally decreased in cysts (B) but more homogenously expressed in control (E). (C and F) TUNEL staining at P20. Apoptotic cells were present in cyst-lining epithelia (arrows in C), as well as interstitium (asterisks), but were absent in controls (F). Scale bars: 100 μm.
Alterations in laminin α5 protein and mRNA
We investigated how the PGKneo insertion affected laminin α5 protein levels by immunohistochemistry and western blotting. Accumulation of laminin α5 was consistently reduced overall in Lama5neo/neo kidney BMs (Figure 2D,E). This was observed as early as E17.5 (data not shown). Moreover, the pattern of laminin α5 immunoreactivity was not uniform. In mutants, some tubular BMs stained normally, whereas others showed little to no expression. In addition, most cysts exhibited little or no staining for laminin α5 (Figures 2, 4), suggesting that the deficiency in laminin α5 protein is associated with cyst formation. Western blot of total kidney BM preparations reflected the immunofluorescence results; laminin α5 protein was greatly reduced compared with littermate controls at both P4 and P23 (Figure 5A). Lung, intestine, and heart showed uniform decreases in laminin α5 protein by immunofluorescence, but they have manifested no detectable defects (data not shown).
Figure 5. Laminin α5 protein and RNA analyses.
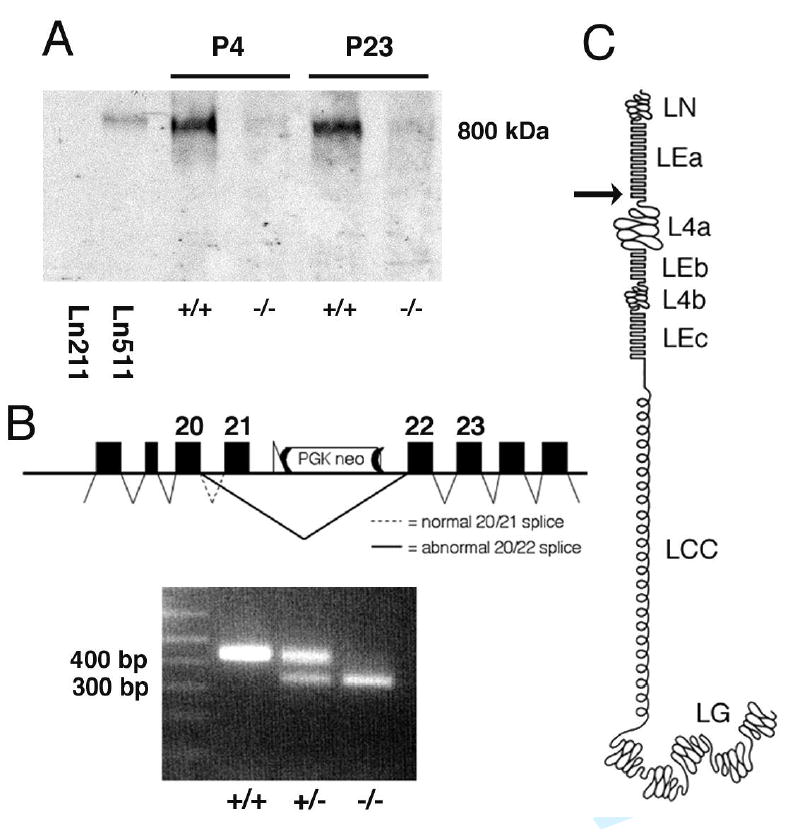
(A) Western blot analysis of non-reduced, BM-enriched samples probed with antibody to laminin α5. The bands at ~800 kDa are consistent with the size of trimeric laminins. Levels are greatly reduced in Lama5neo/neo kidney (−/−) at both P4 and P23 compared with controls (+/+), reflecting the immunofluorescence results. Mouse laminin-511 (Ln511) also reacts, whereas human laminin-211 (Ln211) does not. (B) RT-PCR from kidney RNA with primers designed to amplify cDNA from exons 20–23 of Lama5 produces an amplicon of the expected size (432 bp) in wild-type and Lama5+/neo, but it is barely detectable in Lama5neo/neo. In addition, both Lama5+/neo and Lama5neo/neo kidney exhibit a smaller band of ~300bp that is the predominant amplicon in the latter and results from splicing exon 20 directly to 22, causing a frameshift, as shown in the schematic. (C) Schematic of laminin α5 domain structure. The arrow indicates the site of truncation.
Regarding the mechanism leading to the reduced levels of laminin α5 in BMs, we hypothesized that the PGKneo cassette in intron 21 was disrupting gene transcription and/or RNA splicing, and we used RT-PCR analysis to investigate this. When RT-PCR was performed with primers designed to amplify cDNA from exons 20–23 of Lama5, an amplicon of the expected size (432 bp) was obtained from both wild-type and Lama5+/neo kidney RNA (Figure 5B). In addition, a smaller and more intense band (~300 bp) was detected in Lama5+/neo and Lama5neo/neo kidneys (Figure 5B). Sequencing of the ~300 bp amplicon revealed that it represented the product of splicing exon 20 directly to exon 22 (Figure 5B). The skipped exon 21 is 100 bp, resulting in a shift of the reading frame for the remaining 59 exons (of 80 total) downstream. Any protein translated from this mutant mRNA would contain the first 833 amino acids (of 3718 total), comprising only domains LN and most of LEa (Figure 5C). Furthermore, quantitative real time RT-PCR utilizing primers designed to amplify cDNA from exons 21–22, which are both present only in properly spliced mRNA, showed a variable 2- to 8-fold decrease in full length Lama5 mRNA in Lama5neo/neo mice compared with controls.
We also considered the possibility that the PGKneo cassette was causing PKD by altering the expression of a nearby gene (21). We therefore searched the Ensembl Mouse database (http://www.ensembl.org/Mus_musculus/index.html) to identify genes neighboring Lama5 on chromosome 2. The 5′ end of Adrm1, which encodes adhesion regulating molecule-1, lies 25 kb 3′ of the PGKneo insertion, in a practically overlapping tail-to-tail orientation with Lama5. We performed RT-PCR for Adrm1 and obtained equivalent bands from wild-type and Lama5neo/neo kidney RNA (data not shown), suggesting that disruption of Adrm1 expression by PGKneo is unlikely.
Detection of truncated laminin α5
As discussed above, the reading frame shift could lead to production of a truncated protein containing α5 domains LN and LEa. This truncated protein should be incapable of assembling with β and γ chains because it would lack the laminin coiled-coil (LCC) domain (Figure 5C). Using an antibody specific to α5LN/LEa, we detected laminin α5 in BMs in all kidneys tested, but in Lama5+/neo and Lama5neo/neo kidney we also detected intracellular punctate staining that was absent from wild-type (Figure 2F and data not shown). Antibody to α5LEb/L4b did not show this punctate staining (Figure 2G). We conclude that a truncated protein is produced from the aberrantly spliced mRNA, but it is unlikely to incorporate into BMs. In addition, because the heterozygous mice are normal, the truncated protein itself does not cause PKD. Taken together, these results are consistent with the cystic transformation of tubules being a consequence of greatly reduced laminin α5 protein in some tubular BMs.
Glomerular Defects
Laminin α5 is necessary for GBM formation (7). By immunofluorescence, expression of laminin α5 appeared less perturbed in glomeruli than in tubules (Figures 2, 4). A variety of non-specific changes in glomeruli consistent with advanced renal failure were apparent by light microscopy, including ischemic atrophy of the glomerular tuft, occasional globally sclerosed glomeruli, and hypertrophy of the glomerular tuft (Figure 6 and data not shown). GBM thickening and foot process effacement were present in some glomeruli at late stages, but relatively normal areas of glomerular capillary wall were also present (Figure 6 and data not shown). These findings indicate that a decrease in laminin α5 protein does not prevent assembly of the GBM, but the presence of proteinuria within a few days of birth (as discussed above) suggests that a breach in the glomerular barrier exists.
Figure 6. Glomerular histology and ultrastructure.
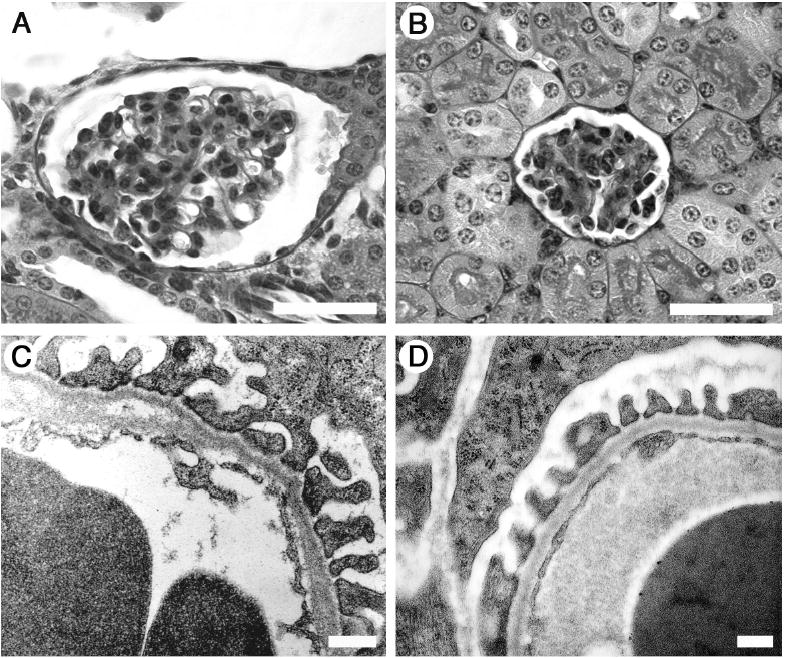
At 3 weeks, Lama5neo/neo glomeruli (A) in general appear somewhat enlarged compared to control littermates (B), but are otherwise relatively normal by light microscopy. Lama5neo/neo GBM, however, displays focal abnormalities including thickening of the lamina rara interna and splitting (arrows), though podocyte foot process architecture appears relatively normal (C), similar to control (D). Scale bars in A and B: 40 μm; in C and D: 800 nm.
Aberrant expression of laminin-332 and integrin α6β4
Laminin-332 (α3β3γ2, formerly laminin-5) is not normally detected in mature nephron BMs, but has previously been found in tubular BMs in human PKD (18). Similarly, Lama5neo/neo mice showed laminin-332 deposition in some (Figure 7A,B) but not all (for example, the cysts shown in Figure 4A and B lacked laminin-332) cyst basement membranes. Laminin-332 expression was observed as early as P2 and therefore is likely a compensatory response to the deficiency in laminin α5 rather than simply a secondary result of cystogenesis.
Figure 7. Characterization of cysts and cilia.
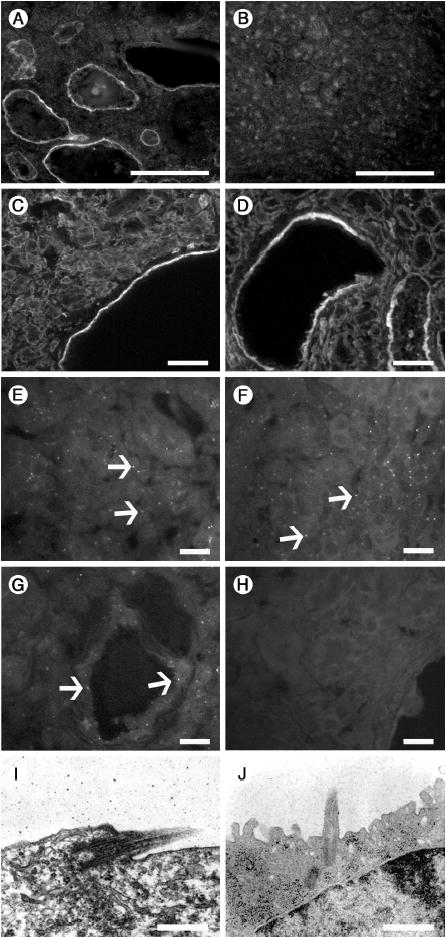
(A–D) Ectopic laminin-332 and increased integrin α6β4 expression is associated with cysts. Laminin-332 is deposited in cyst BMs at 3 weeks of age (A), but is absent from controls (B). Integrins α6 (C) and β4 (D), which comprise the major receptor for laminin-332, are variably upregulated in cyst epithelia in Lama5neo/neo kidneys. Scale bars: 100 μm. (E–J) Primary cilia are present in Lama5neo/neo kidneys. In Lama5neo/neo kidney, antibody to acetylated α-tubulin identifies cilia in normal (E) as well as cystic (G) tubules in Lama5neo/neo, similar to wild-type (F). (H) Negative control with no primary antibody. (I and J) Ultrastucture of a primary cilium within a cyst (I) and in wild-type (J). Scale bars in: A–D, 100 μm; E–H, 20 μm; I–J, 800 nm.
The known cell surface receptors for laminins include integrins, dystroglycan, and the Lutheran glycoprotein (1). By immunofluorescence, the expression patterns of integrin α3, Lutheran, and α-dystroglycan were unaltered in Lama5neo/neo kidneys (data not shown). Integrin α6β4 is regarded as the preferred receptor for laminin-332, although it may also mediate cell adhesion via laminin α5-containing isoforms (22). Both α6 and β4 integrins were variably elevated in cyst-lining epithelia in Lama5neo/neo mice (Figure 7C,D). Thus, aberrant expression of laminin-332 in BMs is accompanied by increased expression of its preferred receptor.
Cilia are present in cyst epithelial cells
Most forms of PKD have been related to defects in the structure or function of primary cilia (23). We therefore considered the possibility that the reduction in laminin α5 led to a secondary defect in assembly of cilia, perhaps due to altered epithelial cell phenotype. Immunostaining for acetylated α-tubulin as a marker for primary cilia was unchanged in Lama5neo/neo mice (Figure 7E-G), and cilia were detectable by transmission electron microscopy (Figure 7I-J). However, we cannot be sure that these cilia function properly.
Discussion
We have shown previously that laminin α5 is required for vascularization of glomeruli and that its COOH-terminal LG domain mediates binding of mesangial cells to the GBM so that they can maintain capillary loop structure (24, 7). Here we show that laminin α5 is also involved in maintaining homeostasis of the tubular compartment of the nephron. The kidneys of Lama5neo/neo mice display remarkable similarities to human PKD, as well as to PKD animal models. As in other PKD models, we found dysregulated proliferation and apoptosis. The unusually rapid loss of renal function in Lama5neo/neo mice likely results from glomerular disease superimposed on the cystic transformation of tubules.
This is the first demonstration that a primary defect in an extracellular matrix component can cause PKD. The insertion of a PGKneo cassette in an intron of Lama5 generated a hypomorphic allele resulting from aberrant RNA splicing and reduced full length mRNA and protein levels. Because BMs assembled normally, and early cystic tubules exhibited apparently normal BMs, we conclude that there exists a role for laminin α5 in maintaining epithelial homeostasis beyond a purely structural role in the extracellular matrix.
Selectable markers inserted in non-coding regions have been previously shown to affect gene expression, both at the DNA and RNA levels (25–28). Here, our RT-PCR results show that the presence of PGKneo in intron 21 of the ~50 kb primary Lama5 transcript impairs splicing and results in frequent skipping of exon 21. In addition, the transcription termination signal in the PGKneo cassette likely reduces the level of transcripts that include the downstream exons, whether they are correctly spliced or not. Interestingly, the tubular segments that appear to express normal levels of α5 also express the truncated form, whereas other segments express neither (Figure 2). This suggests that there may be cell type-specific effects on the ability to read through the PGKneo cassette so that splicing and polyadenylation of RNA can occur and protein can be synthesized, whether truncated or full length.
Most of the developmental defects in laminin α5 null mice are associated with breakdown or discontinuity of BMs (9, 7), but we did not observe overtly defective tubular BMs in Lama5neo/neo mice. It is possible that adhesion of tubular epithelia to BMs with decreased laminin α5 content is altered, leading to phenotypic changes. Consistent with this hypothesis, cystic epithelia from the murine cpk model of PKD exhibit increased integrin-mediated adhesion to type I collagen, type IV collagen, and laminin (29), representing an inherently altered behavior. Likewise, a potential explanation for our findings is that signaling cascades mediated by integrins are altered as a consequence of the reduction in laminin α5 content and/or the ectopic deposition of laminin-332, a trimer not normally found in nephron BMs but that has been reported in human PKD (18).
The great majority of PKD models involve mutations in genes relating to the structure and function of primary cilia (23). How could reduced levels of a BM protein produce a similar phenotype? When the primary structure of PC1 was first described, it was speculated to function as a receptor for ECM molecules (19, 20, 30, 31). Although the precise localization of PC1 is still not without controversy, there is evidence that it is expressed on basolateral surfaces, as well as apically in association with cilia (32). Recombinant PC1 peptides have been shown to bind laminin in vitro (20), suggesting the possibility that PC1 might normally interact with laminin α5 at the basal cell surface.
Several reports have cited an increase in EGF signaling in PKD and its animal models (32). The N-terminal arm of most laminin chains contains multiple EGF-like repeats interrupted by globular domains. Fragments of laminin-111 containing EGF-like repeats increase cell proliferation by a mechanism which likely involves the EGF receptor (33), and laminin-332 fragments have been shown to bind the EGF receptor and stimulate cell migration (34). The truncated α5 protein likely produced in Lama5neo/neo kidney would contain most of domain LEa, which consists of EGF-like repeats. Although highly speculative, it is possible that such a truncated product would be secreted and potentially stimulate EGF receptors and promote cystogenesis in Lama5neo/neo kidneys. Alternatively, proteolytic processing of laminin-332 (35) may be a source of biologically active laminin EGF-like fragments. On the other hand, aberrant signaling from laminin-332 through its receptor detected at elevated levels in cysts, integrin α6β4, could be responsible for the cystic transformation.
Aberrant expression of laminins is a recurring theme in kidney disorders involving BM proteins. For example, mice lacking the laminin β2 chain, a component of laminin-521 that is crucial for GBM function, abnormally retain the laminin β1 chain in the GBM (36). Mice, dogs, and humans lacking the α3α4α5 collagen IV network and exhibiting features of Alport syndrome display ectopic deposition of laminin α1 and/or α2 in the GBM (37–41). An unresolved issue in these cases is the extent to which abnormal expression of the ectopic laminin isoform contributes to disease pathogenesis. Likewise, it is not clear whether aberrant expression of laminin-332 directly causes cystogenesis, as the protein is found in some but not all cysts.
Cilia-mediated signaling is hypothesized to influence cell polarity and maintenance of the differentiated epithelial cell phenotype. Our results provide the first genetic evidence in vivo that cell/matrix interactions are also involved in maintaining tubular epithelial cell homeostasis. It is possible that a downstream function of cilia-mediated signaling is to regulate interactions with the BM, or, conversely, that cell/matrix interactions influence cell phenotype and the response to cilia-mediated signaling. Disturbing either the ciliary or the cell/matrix aspect can then result in PKD. Investigations are underway in our laboratory to explore these possibilities.
Methods
Generation and genotyping of mutant mice
Mice carrying the Lama5neo locus (Figure 1) were generated from two independently targeted embryonic stem cell clones as previously described (10). This locus carries a PGKneo cassette flanked by frt recombination sites inserted into a KpnI site in intron 21 of Lama5. Primers F14 (5′-ACCATGGGTATCCCGACTGTCACG-3′) and R15 (5′-TCGGGTCACAGGTTGTATGCAAGG-3′) were used in tail DNA PCR reactions to identify mice carrying the targeted locus. Animal studies were approved by the Washington University Animal Studies Committee.
Histology and electron microscopy
Tissues were fixed in 10% buffered formalin (DiRuscio and Associates) and embedded in paraffin. 3 μm sections were cut and stained with hematoxylin and eosin. For electron microscopy, tissues were fixed in 2.5% glutaraldehyde/2% paraformaldehyde/0.1M sodium cacodylate (pH=7.4), post-fixed in aqueous 1.25% osmium tetroxide, dehydrated through an ethanol series, embedded in Polybed, and sectioned on a Reichert Ultracut. Ultrathin sections were stained with 4% uranyl acetate and lead citrate and visualized under a Jeol CX-100 transmission electron microscope.
Antibodies and immunofluorescence
Rabbit antibodies 8948 and 1586 to mouse laminin α5 recognize epitopes in domains LEb and L4b (formerly IIIb and IVa; encoded by exons COOH-terminal to the insertion) and have been previously described (4). Rabbit anti-mouse laminin α5 domains LN/LEa (formerly VI/V; encoded by exons NH2-terminal to the insertion) was a gift from Takako Sasaki and the late Rupert Timpl, Martinsried, Germany (42). Rabbit anti-human laminin-332 (α3β3γ2; formerly laminin-5) was a gift from Peter Marinkovich, Stanford University (35). Rabbit anti-mouse laminin-111 was purchased from Sigma. Rat anti-integrin α6 and β4 were purchased from BD Biosciences. Mouse anti-acetylated tubulin (6–11B-1) was purchased from Sigma. Nephron segment identification was determined by staining with rat anti-mouse cytokeratin 8 (TROMA-1; Developmental Studies Hybridoma Bank) to mark collecting ducts, rabbit anti-Tamm-Horsfall protein (Biomedical Technologies) to mark thick ascending limb of the loop of Henle, and FITC-conjugated Lotus tetragonolobus lectin (Vector Laboratories) to label proximal tubules.
Tissues were embedded in OCT and frozen in 2-methylbutane cooled with a dry-ice/ethanol bath. 7 μm cryosections were fixed in 2% paraformaldehyde in PBS. Blocking was performed with 1% goat serum and 1% BSA in PBS. Primary antibodies were applied for one hour at room temperature or overnight at 4C. Appropriate Cy3- or FITC-conjugated goat anti-rabbit or goat anti-rat secondary antibodies (ICN/Cappel Biomedicals) were applied for 30 min to 1 h. Images were visualized under a Nikon E800 epifluorescence microscope and captured with a Diagnostics Instruments Spot 2 CCD camera.
RT-PCR and DNA sequencing
Tissues were flash frozen in liquid nitrogen or placed in RNA Later (Qiagen) prior to freezing. Following homogenization, RNA was extracted with Tri Reagent (Molecular Research Center) and treated with DNase utilizing the DNA-Free kit (Ambion). 3 μg total RNA was reverse transcribed with Superscript III (Invitrogen) using random hexamers as primers. A 1.5 μL aliquot of this reaction was amplified with KlentaqLA DNA polymerase (Clontech) for subsequent sequencing. For real time quantitative RT-PCR, cDNA was amplified utilizing Sybr Green I PCR Master Mix (Applied Biosystems) with the addition of uracil DNA glycosylase (Invitrogen) to prevent the reamplification of carryover PCR products. Amplicons were generated and analyzed with the GeneAmp 5700 Sequence Detection System (Applied Biosytems). Primers utilized for laminin α5 were: sense, 5;-ACCCAAGGACCCACCTGTAG-3′ and antisense, 5′-TCATGTGTGCGTAGCCTCTC-3′. Primers for GAPDH were: sense TGGCAAAGTGGAGATTGTTGCC ; antisense, AAGATGGTGATGGGCTTCCCG. Quantification of Lama5 gene expression was determined by the standard curve method with GAPDH as an endogenous control. A two-step PCR protocol was followed with a melting temperature of 95° C and annealing temperature of 60° C for 40 cycles.
PCR products generated with primer sets designed to amplify cDNA flanking the PGKneo insertion (exons 20–23) were gel purified, cloned into a TA vector using the TOPO Cloning kit (Invitrogen), and sequenced utilizing the BigDye 3.1 cycle sequencing kit (Applied Biosystems). Amplification primers were: sense primer in exon 20, 5′-TTCTTTGGCCTGGATTATGC-3′; antisense primer in exon 23, 5′-GGTACAGCTGGAAAGCTTGC-3′.
Western blot analysis
Kidneys were flash frozen in liquid nitrogen. BM-enriched extracts were prepared as described (4). Samples of 15 μg total protein were subjected to 3% SDS-PAGE under non-reducing conditions. Western blotting was performed with antibody 1586. Purified mouse laminin-511 and human laminin-211 were included as positive and negative controls, respectively.
Acknowledgments
We thank Peter Marinkovich, Michael Dipersio, Takako Sasaki, and the late Rupert Timpl for antibodies; Jeannette Cunningham, Steve Cok, Casey Moulson, Gloriosa Go, Jennifer Richardson, and Dan Martin for assistance; Renate Lewis and Joshua Sanes for facilitating ES cell targeting; and the Mouse Genetics Core for care of mice. This work was funded by grants from the NIH (R01DK064687 and R01GM060432) to JHM. JHM is an Established Investigator of the American Heart Association. MBS was supported by NIH T32DK062705. SJH was supported by a National Kidney Foundation Research Fellowship. Portions of this work were presented at the 2004 Annual Meeting of the American Society of Nephrology in St. Louis, MO.
References
- 1.Miner JH, Yurchenco PD. Laminin functions in tissue morphogenesis. Annu Rev Cell Dev Biol. 2004;20:255–284. doi: 10.1146/annurev.cellbio.20.010403.094555. [DOI] [PubMed] [Google Scholar]
- 2.Miner JH, Lewis RM, Sanes JR. Molecular cloning of a novel laminin chain, α5, and widespread expression in adult mouse tissues. J Biol Chem. 1995;270:28523–28526. doi: 10.1074/jbc.270.48.28523. [DOI] [PubMed] [Google Scholar]
- 3.Sorokin LM, Pausch F, Durbeej M, Ekblom P. Differential expression of five laminin alpha (1–5) chains in developing and adult mouse kidney. Dev Dyn. 1997;210:446–462. doi: 10.1002/(SICI)1097-0177(199712)210:4<446::AID-AJA8>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 4.Miner JH, Patton BL, Lentz SI, Gilbert DJ, Snider WD, Jenkins NA, Copeland NG, Sanes JR. The laminin alpha chains: expression, developmental transitions, and chromosomal locations of alpha1–5, identification of heterotrimeric laminins 8–11, and cloning of a novel alpha3 isoform. J Cell Biol. 1997;137:685–701. doi: 10.1083/jcb.137.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aumailley M, Bruckner-Tuderman L, Carter WG, Deutzmann R, Edgar D, Ekblom P, Engel J, Engvall E, Hohenester E, Jones JC, Kleinman HK, Marinkovich MP, Martin GR, Mayer U, Meneguzzi G, Miner JH, Miyazaki K, Patarroyo M, Paulsson M, Quaranta V, Sanes JR, Sasaki T, Sekiguchi K, Sorokin LM, Talts JF, Tryggvason K, Uitto J, Virtanen I, von der Mark K, Wewer UM, Yamada Y, Yurchenco PD. A simplified laminin nomenclature. Matrix Biol. 2005;24:326–332. doi: 10.1016/j.matbio.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 6.Miner JH. Renal basement membrane components. Kidney Int. 1999;56:2016–2024. doi: 10.1046/j.1523-1755.1999.00785.x. [DOI] [PubMed] [Google Scholar]
- 7.Miner JH, Li C. Defective glomerulogenesis in the absence of laminin α5 demonstrates a developmental role for the kidney glomerular basement membrane. Dev Biol. 2000;217:278–289. doi: 10.1006/dbio.1999.9546. [DOI] [PubMed] [Google Scholar]
- 8.Nguyen NM, Miner JH, Pierce RA, Senior RM. Laminin alpha5 is required for lobar septation and visceral pleural basement membrane formation in the developing mouse lung. Dev Biol. 2002;246:231–244. doi: 10.1006/dbio.2002.0658. [DOI] [PubMed] [Google Scholar]
- 9.Miner JH, Cunningham J, Sanes JR. Roles for laminin in embryogenesis: Exencephaly, syndactyly, and placentopathy in mice lacking the laminin α5 chain. J Cell Biol. 1998;143:1713–1723. doi: 10.1083/jcb.143.6.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nguyen NM, Kelley DG, Schlueter JA, Meyer MJ, Senior RM, Miner JH. Epithelial laminin alpha5 is necessary for distal epithelial cell maturation, VEGF production, and alveolization in the developing murine lung. Dev Biol. 2005;282:111–125. doi: 10.1016/j.ydbio.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 11.Coles EG, Gammill LS, Miner JH, Bronner-Fraser M. Abnormalities in neural crest cell migration in laminin alpha5 mutant mice. Dev Biol. 2006;289:218–228. doi: 10.1016/j.ydbio.2005.10.031. [DOI] [PubMed] [Google Scholar]
- 12.Wilson PD, Hreniuk D, Gabow PA. Abnormal extracellular matrix and excessive growth of human adult polycystic kidney disease epithelia. J Cell Physiol. 1992;150:360–369. doi: 10.1002/jcp.1041500220. [DOI] [PubMed] [Google Scholar]
- 13.Cuppage FE, Huseman RA, Chapman A, Grantham JJ. Ultrastructure and function of cysts from human adult polycystic kidneys. Kidney Int. 1980;17:372–381. doi: 10.1038/ki.1980.43. [DOI] [PubMed] [Google Scholar]
- 14.Katz SK, Hakki A, Miller AS, Finkelstein SD. Ultrastructural tubular basement membrane lesions in adult polycystic kidney disease. Ann Clin Lab Sci. 1989;19:352–359. [PubMed] [Google Scholar]
- 15.Wilson PD, Falkenstein D. The pathology of human renal cystic disease. Curr Top Pathol. 1995;88:1–50. doi: 10.1007/978-3-642-79517-6_1. [DOI] [PubMed] [Google Scholar]
- 16.Carone FA, Jin H, Nakamura S, Kanwar YS. Decreased synthesis and delayed processing of sulfated glycoproteins by cells from human polycystic kidneys. Lab Invest. 1993;68:413–418. [PubMed] [Google Scholar]
- 17.Carone FA, Makino H, Kanwar YS. Basement membrane antigens in renal polycystic disease. Am J Pathol. 1988;130:466–471. [PMC free article] [PubMed] [Google Scholar]
- 18.Joly D, Morel V, Hummel A, Ruello A, Nusbaum P, Patey N, Noel LH, Rousselle P, Knebelmann B. Beta4 integrin and laminin 5 are aberrantly expressed in polycystic kidney disease: role in increased cell adhesion and migration. Am J Pathol. 2003;163:1791–1800. doi: 10.1016/s0002-9440(10)63539-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millan JL, Gamble V, Harris PC. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet. 1995;10:151–160. doi: 10.1038/ng0695-151. [DOI] [PubMed] [Google Scholar]
- 20.Malhas AN, Abuknesha RA, Price RG. Interaction of the leucine-rich repeats of polycystin-1 with extracellular matrix proteins: possible role in cell proliferation. J Am Soc Nephrol. 2002;13:19–26. doi: 10.1681/ASN.V13119. [DOI] [PubMed] [Google Scholar]
- 21.Olson EN, Arnold HH, Rigby PW, Wold BJ. Know your neighbors: three phenotypes in null mutants of the myogenic bHLH gene MRF4. Cell. 1996;85:1–4. doi: 10.1016/s0092-8674(00)81073-9. [DOI] [PubMed] [Google Scholar]
- 22.Kikkawa Y, Sanzen N, Fujiwara H, Sonnenberg A, Sekiguchi K. Integrin binding specificity of laminin-10/11: laminin-10/11 are recognized by alpha3beta1, alpha6beta1 and alpha6beta4 integrins. J Cell Sci. 2000;113:869–876. doi: 10.1242/jcs.113.5.869. [DOI] [PubMed] [Google Scholar]
- 23.Watnick TJ, Germino GG. From cilia to cyst. Nat Genet. 2003;34:355–356. doi: 10.1038/ng0803-355. [DOI] [PubMed] [Google Scholar]
- 24.Kikkawa Y, Virtanen I, Miner JH. Mesangial cells organize the glomerular capillaries by adhering to the G domain of laminin alpha5 in the glomerular basement membrane. J Cell Biol. 2003;161:187–196. doi: 10.1083/jcb.200211121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 26.Meyers EN, Lewandoski M, Martin GR. An Fgf8 mutant allelic series generated by Cre-and Flp-mediated recombination. Nat Genet. 1998;18:136–141. doi: 10.1038/ng0298-136. [DOI] [PubMed] [Google Scholar]
- 27.McDevitt MA, Shivdasani RA, Fujiwara Y, Yang H, Orkin SH. A "knockdown" mutation created by cis-element gene targeting reveals the dependence of erythroid cell maturation on the level of transcription factor GATA-1. Proc Natl Acad Sci USA. 1997;94:6781–6785. doi: 10.1073/pnas.94.13.6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scacheri PC, Crabtree JS, Novotny EA, Garrett-Beal L, Chen A, Edgemon KA, Marx SJ, Spiegel AM, Chandrasekharappa SC, Collins FS. Bidirectional transcriptional activity of PGK-neomycin and unexpected embryonic lethality in heterozygote chimeric knockout mice. Genesis. 2001;30:259–263. doi: 10.1002/gene.1072. [DOI] [PubMed] [Google Scholar]
- 29.van Adelsberg J. Murine polycystic kidney epithelial cell lines have increased integrin-mediated adhesion to collagen. Am J Physiol. 1994;267:F1082–F1093. doi: 10.1152/ajprenal.1994.267.6.F1082. [DOI] [PubMed] [Google Scholar]
- 30.Weston BS, Bagneris C, Price RG, Stirling JL. The polycystin-1 C-type lectin domain binds carbohydrate in a calcium-dependent manner, and interacts with extracellular matrix proteins in vitro. Biochim Biophys Acta. 2001;1536:161–176. doi: 10.1016/s0925-4439(01)00046-1. [DOI] [PubMed] [Google Scholar]
- 31.Consortium IP. Polycystic kidney disease: the complete structure of the PKD1 gene and its protein. Cell. 1995;81:289–298. doi: 10.1016/0092-8674(95)90339-9. [DOI] [PubMed] [Google Scholar]
- 32.Wilson PD. Polycystic kidney disease. N Engl J Med. 2004;350:151–164. doi: 10.1056/NEJMra022161. [DOI] [PubMed] [Google Scholar]
- 33.Panayotou G, End P, Aumailley M, Timpl R, Engel J. Domains of laminin with growth-factor activity. Cell. 1989;56:93–101. doi: 10.1016/0092-8674(89)90987-2. [DOI] [PubMed] [Google Scholar]
- 34.Schenk S, Hintermann E, Bilban M, Koshikawa N, Hojilla C, Khokha R, Quaranta V. Binding to EGF receptor of a laminin-5 EGF-like fragment liberated during MMP-dependent mammary gland involution. J Cell Biol. 2003;161:197–209. doi: 10.1083/jcb.200208145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marinkovich MP, Lunstrum GP, Burgeson RE. The anchoring filament protein kalinin is synthesized and secreted as a high molecular weight precursor. J Biol Chem. 1992;267:17900–17906. [PubMed] [Google Scholar]
- 36.Noakes PG, Miner JH, Gautam M, Cunningham JM, Sanes JR, Merlie JP. The renal glomerulus of mice lacking s-laminin/laminin β2: nephrosis despite molecular compensation by laminin β1. Nat Genet. 1995;10:400–406. doi: 10.1038/ng0895-400. [DOI] [PubMed] [Google Scholar]
- 37.Abrahamson DR, Prettyman AC, Robert B, St John PL. Laminin-1 reexpression in Alport mouse glomerular basement membranes. Kidney Int. 2003;63:826–834. doi: 10.1046/j.1523-1755.2003.00800.x. [DOI] [PubMed] [Google Scholar]
- 38.Cosgrove D, Rodgers K, Meehan D, Miller C, Bovard K, Gilroy A, Gardner H, Kotelianski V, Gotwals P, Amatucci A, Kalluri R. Integrin alpha1beta1 and transforming growth factor-beta1 play distinct roles in Alport glomerular pathogenesis and serve as dual targets for metabolic therapy. Am J Pathol. 2000;157:1649–1659. doi: 10.1016/s0002-9440(10)64802-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kashtan CE, Kim Y, Lees GE, Thorner PS, Virtanen I, Miner JH. Abnormal glomerular basement membrane laminins in murine, canine and human Alport syndrome: aberrant laminin alpha2 deposition is species-independent. J Amer Soc Nephrol. 2001;12:252–260. doi: 10.1681/ASN.V122252. [DOI] [PubMed] [Google Scholar]
- 40.Miner JH. Of laminins and delamination in Alport syndrome. Kidney Int. 2003;63:1158–1159. doi: 10.1046/j.1523-1755.2003.00841.x. [DOI] [PubMed] [Google Scholar]
- 41.Andrews KL, Mudd JL, Li C, Miner JH. Quantitative trait loci influence renal disease progression in a mouse model of Alport syndrome. Am J Pathol. 2002;160:721–730. doi: 10.1016/S0002-9440(10)64892-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garbe JH, Gohring W, Mann K, Timpl R, Sasaki T. Complete sequence, recombinant analysis and binding to laminins and sulphated ligands of the N-terminal domains of laminin alpha3B and alpha5 chains. Biochem J. 2002;362:213–221. doi: 10.1042/0264-6021:3620213. [DOI] [PMC free article] [PubMed] [Google Scholar]