

Abstract
FABACs (fatty acid–bile acid conjugates) are synthetic molecules that are designed to treat a range of lipid disorders. The compounds prevent cholesterol gallstone formation and diet-induced fatty liver, and increase reverse cholesterol transport in rodents. The aim of the present study was to investigate the effect of FABACs on cholesterol efflux in human cells. Aramchol (3β-arachidylamido-7α,12α,5β-cholan-24-oic acid) increased cholesterol efflux from human skin fibroblasts in a dose-dependent manner in the absence of known efflux mediators such as apoA-I (apolipoprotein A-I), but had little effect on phospholipid efflux. An LXR (liver X receptor) agonist strongly increased Aramchol-induced cholesterol efflux; however, in ABCA1 (ATP-binding cassette transporter A1)-deficient cells from Tangier disease patients, the Aramchol effect was absent, indicating that activity of ABCA1 was required. Aramchol did not affect ABCA1 expression, but plasma membrane levels of the transporter increased 2-fold. Aramchol is the first small molecule that induces ABCA1-dependent cholesterol efflux without affecting transcriptional control. These findings may explain the beneficial effect of the compound on atherosclerosis.
Keywords: ATP-binding cassette transporter A1 (ABCA1), Aramchol, atherosclerosis, cholesterol efflux, fatty acid–bile acid conjugate (FABAC), reverse cholesterol transport
Abbreviations: ABCA1, ATP-binding cassette transporter A1; apoA-I, apolipoprotein A-I; Aramchol, 3β-arachidylamido-7α,12α,5β-cholan-24-oic acid; BCA, bicinchoninic acid; DMEM, Dulbecco's modified Eagle's medium; FABAC, fatty acid–bile acid conjugate; HDL, high-density lipoprotein; LDH, lactate dehydrogenase; LXR, liver X receptor; NHS, N-hydroxysuccinimide; PBST, PBS containing 0.05% (w/v) Tween 20; RCT, reverse cholesterol transport
INTRODUCTION
FABACs (fatty acid–bile acid conjugates) are a new class of molecules originally synthesized with the aim of treating of cholesterol gallstone disease [1]. Indeed, FABACs, and particularly Aramchol (3β-arachidylamido-7α,12α,5β-cholan-24-oic acid or arachidyl-amido-cholanoic acid), were shown to retard cholesterol crystallization [1] and to dissolve preformed cholesterol crystals in model bile solutions [2,3], as well as in native human gall-bladder bile ex vivo [2,3]. Aramchol, when administered intragastrically, was absorbed and prevented the formation of biliary cholesterol crystals in inbred mice, as well as in hamsters [1]. In addition, it prevented gallstone formation in inbred mice. Furthermore, dissolution of pre-existing cholesterol crystals, as well as stones [3], was observed in vivo. Interestingly, Aramchol did not influence biliary lipid or bile salt concentrations in a variety of animal models [4]. In addition to the beneficial effect on gallstone disease, Aramchol was shown to decrease diet-induced fatty liver, to reduce atherosclerosis in C57Bl6 mice and increase faecal bile salt and neutral sterol secretion [4–7].
An important factor in the aetiology of atherosclerosis is the process of RCT (reverse cholesterol transport), i.e. the transport of excess cholesterol from peripheral cells to the liver followed by its secretion into bile. Activity of ABCA1 (ATP-binding cassette transporter A1) in the liver is considered to be a major factor in the control of this process (see [8,9] for reviews), although RCT also occurs in the absence of the protein [10]. Liver ABCA1 lipidates apoA-I (apolipoprotein A-I) with phospholipids and cholesterol, thus forming pre-β-HDL (high-density lipoprotein), which can take up cholesterol in the periphery, again via ABCA1. In several mouse models, overexpression of ABCA1 in the macrophage compartment has been shown to have a beneficial effect on atherosclerosis [11,12], and regulation of its expression is controlled by the nuclear receptors LXRα (liver X receptor α) and LXRβ [13].
The molecular mechanism by which ABCA1 mediates lipid efflux is not yet clear and is subject to intense investigation [14–21]. Since Aramchol has been shown to ameliorate atherosclerosis in mice and to increase faecal neutral sterol secretion [6,7], we speculated that this molecule could play an active role in cholesterol efflux from cells. Using human fibroblasts as a model system, we show in the present study results that support our hypothesis. Interestingly, the effects of Aramchol on fibroblasts depended strongly on the activity of the ABCA1 transporter.
MATERIALS AND METHODS
Materials
Cholesterol, phosphatidylcholine (from egg yolk), fatty-acid-free BSA, streptavidin–agarose and cholic acid (>98%) were purchased from Sigma Chemical Co. The different FABACs used in this study were a gift from Galmed (Tel Aviv, Israel). ApoA-I and HDL were purchased from Calbiochem. T090137 was from Cayman Chemical Co. (Ann Arbor, MI, U.S.A.). [1,2-3H]Cholesterol (38 μCi/mmol) and [methyl-3H]choline chloride (80 μCi/mmol) were purchased from Amersham Biosciences. Sulpho-NHS (N-hydroxysuccinimide)–long-chain biotin was from Pierce. Calpeptin was a gift from Dr E. A. Beuling (Academic Medical Centre, Amsterdam, The Netherlands). Monoclonal antibody raised against the C-terminal domain of human ABCA1 was a gift from Dr M. R. Hayden (University of British Columbia, Vancouver, Canada). Polyclonal antibody raised against the N-terminal sequence of Na+/K+-ATPase was a gift from Dr J. Koenderink (Catholic University of Nijmegen, Nijmegen, The Netherlands).
Aramchol preparation
Aramchol was prepared as described previously [1]. Arachidic acid (n-C20O2H40) was conjugated via an amide bond to the third position of cholic acid. The conjugation was in the β-configuration. The final product was purified by silica gel chromatography and characterized by 1H-NMR and MS. Aramchol was determined to be at least 98% pure by NMR.
Cell culture
Human skin fibroblasts from healthy subjects and from ABCA1 mutated (Tangier disease) patients were cultured in DMEM (Dulbecco's modified Eagle's medium; BioWhittaker, Verviers, Belgium) supplemented with 10% (v/v) fetal calf serum, 2 mM glutamine, 100 units/ml penicillin and 100 μg/ml streptomycin in a humidified 37 °C incubator in the presence of 10% CO2. The cells used for efflux experiments were seeded into 24-well plates in a 1:2 split ratio and were grown until confluent over 7 days.
Cell viability measurements
Cell toxicity caused by Aramchol was determined by performing LDH (lactate dehydrogenase) assays [22]. Aramchol was dissolved in ethanol and was added to serum-free medium. LDH leakage was determined after overnight incubation of the fibroblasts with different amounts of Aramchol (1–15 μg/ml). LDH leakage was expressed as the percentage of enzyme activity in the medium [=(activity in medium)/(activity in medium+activity in cells)×100]. Incubations were carried out in duplicate.
Cholesterol efflux assay
Cholesterol efflux is defined as the transfer rate of [3H]cholesterol from cells to culture medium. The assays were performed as described by van Wijland et al. [23]. In brief, cholesterol loading was performed by incubating fibroblasts for 20 h with DMEM supplemented with 30 μg/ml cholesterol, 0.5 μCi/ml of [3H]cholesterol and 0.2% fatty-acid-free BSA. After incubation, the cells were washed four times with PBS containing 0.2% fatty-acid-free BSA. Efflux was initiated by the addition of efflux medium consisting of DMEM supplemented with 0.2% fatty-acid-free BSA and different cholesterol acceptors. After 20–24 h of incubation in a humidified 37 °C incubator in the presence of 10% CO2, medium was collected and centrifuged at 10000 g for 5 min to remove cellular debris. Aliquots of the supernatant were taken to count the effluxed [3H]cholesterol. The remaining cell-associated [3H]cholesterol was determined after extraction for at least 30 min with propan-2-ol. Efflux was determined in the presence, or absence, of Aramchol. Aramchol was dissolved in ethanol and added to the medium at the concentrations indicated. The final concentration of ethanol in the efflux medium was not higher than 0.15% (v/v). To activate LXRα and LXRβ, 1 μM T090137 in DMSO (0.1%, v/v) was added to the efflux medium in some experiments. All control cells received an equivalent amount of DMSO and/or ethanol. The amount of [3H]cholesterol in medium and cellular extract was quantified by liquid-scintillation counting. The radioactivity released into the medium was expressed as the fraction of the total radioactive cholesterol that was present in each well.
Phospholipid efflux assay
Efflux of phospholipid from [3H]choline-labelled cells was assessed under the conditions described above for cholesterol efflux, with some modifications. Briefly, cells were cultured in (low-choline) Ham's F-10 medium (GIBCO®; Invitrogen) containing glutamine supplemented with 10% fetal calf serum, 100 units/ml of penicillin and 100 μg/ml of streptomycin. Loading with 30 μg/ml cholesterol was performed in the presence of 2 μCi/ml of [3H]choline chloride in Ham's F-10 medium supplemented with 0.2% fatty-acid-free BSA. After incubation with efflux medium, the medium was centrifuged for 5 min at 12000 g to remove any dissociated cells and a 200 μl aliquot was then extracted with chloroform/methanol as described by Bligh and Dyer [24]. The level of [3H]choline label in the chloroform phase of both medium and cells was measured by liquid-scintillation counting.
Density-gradient ultracentrifugation
Cholesterol efflux was performed as described above. Efflux medium was separated on a KBr gradient by ultracentrifugation as described previously [25] with some modifications. In brief, 0.95 g of KBr (Merck) was mixed with 2 ml of efflux medium. On to this mixture, 2 ml of a KBr solution with a density of 1.26 g/ml was layered, followed by 5 ml of a KBr solution with a density of 1.1 g/ml and finalized by the addition of 4 ml of 50 mM Tris/HCl, pH 8.2. The tubes (14 mm×95 mm; Beckman), were centrifuged for 20 h at 35000 rev./min at 20 °C in a Beckman SW 40 rotor. A linear gradient was obtained with density limits of 1.3 g/ml and 1.045 g/ml. The tubes were divided into 12 fractions. [3H]Cholesterol in each of the fractions was measured by liquid-scintillation counting. BSA in each of the fractions was measured using the BCA (bicinchoninic acid) method [26]. ApoA-I levels were measured by ELISA.
Cellular ABCA1 content and cell-surface ABCA1 biotinylation
Biotinylation of cell-surface proteins was performed as described previously [27,28], with some modifications. Fibroblasts were loaded with cholesterol and the efflux was collected in acceptor-containing medium with, or without, 2 μg/ml Aramchol. Cells were put on ice and washed twice with ice-cold PBS. Cell-surface proteins were biotinylated using 0.5 mg/ml sulpho-NHS–long-chain biotin in Hanks balanced salt solution without Phenol Red, for 30 min at 4 °C on a platform incubator. Cells were washed twice with ice-cold quench buffer, pH 7.5 (50 mM Tris/HCl; 0.1 mM EDTA and 150 mM NaCl). Cells were scraped into 1.5 ml of ice-cold PBS. Cell pellets were obtained by centrifugation (20 min, 12000 g, 4 °C) and subsequently resuspended in 100 μl of lysis buffer [25 mM Hepes, pH 7.5, 5 mM MgCl2, 5 mM EDTA, 2 mM PMSF and Complete™ protease inhibitor cocktail (Roche)]. Cell lysates were obtained by four cycles of freezing (−80 °C) and thawing (37 °C) of the cell suspensions. Protein concentrations were determined by BCA assay [26]. A proportion (10 μg) of the cell protein was set aside to examine total cellular ABCA1 content. Total cellular protein (60 μg in a final volume of 60 μl in lysis buffer) was added to 30 μl of streptavidin–agarose (Sigma) and was incubated overnight on a platform incubator at 4 °C. The streptavidin–agarose was pelleted by centrifugation at 12000 g for 5 min and washed three times with lysis buffer. Loading dye (20 μl) was added to the streptavidin–agarose pellet (the volume of the pellet was approx. 20 μl) and was incubated at 37 °C for 30 min. A proportion (6.7 μl) was used for Western blot analysis.
ABCA1 stabilization
Cholesterol-loaded fibroblasts were washed, and then incubated for 24 h with DMEM/BSA efflux medium containing calpeptin (25 μM) or Aramchol (2 μg/ml) or both. Proteins were harvested and ABCA1 protein levels were subsequently determined by Western blotting.
Western blotting
Lysis buffer containing 25 mM Hepes, pH 7.5, 5 mM MgCl2, 5 mM EDTA, 2 mM PMSF and Complete™ protease inhibitor was added to the cell pellets. Cell lysates were obtained by four cycles of freezing (−80 °C) and thawing (37 °C) of the cell suspensions. The protein concentrations of the lysates were determined using the BCA assay [26]. Equal amounts of protein (10 μg) were separated on SDS/PAGE (6% gels) and electrophoretically transferred on to nitrocellulose membranes (Schleicher and Schuell, 's-Hertogenbosch, The Netherlands). Membranes were probed with using a mouse monoclonal anti-ABCA1 antibody, diluted 1:1000 [30], followed by immunoreactivity detection by Lumi-Light Western blotting substrate (Roche). In selected cases, the membranes were stripped for 30 min by incubation in buffer containing 100 mM 2-mercaptoethanol, 2% (w/v) SDS and 62.5 mM Tris/HCl, pH 6.7, at 50 °C, followed by washing and probing with a rabbit anti-Na+/K+-ATPase antibody. Protein abundance was calculated by densitometry using LumiAnalyst 3.1 software (Roche).
Analytical procedures
Aramchol was measured in cellular extracts and culture medium with a LC (liquid chromatography)-tandem MS method exactly as described in [31]. Human apoA1 concentrations were determined by indirect sandwich ELISA. Flat-bottom Maxi-Sorp microtitre plates (Nunc) were coated overnight at 4 °C with 100 μl of rabbit anti-(human apoA-I) polyclonal antibody (Calbiochem), diluted 1:400 in 100 mM NaHCO3, pH 8.3. Coated wells were then blocked during 2 h at room temperature with 300 μl of PBS containing 0.05% (w/v) Tween 20 (PBST), and supplemented with 5% (w/v) non-fat dried milk. Samples were diluted in a range from 1:10 to 1:5000 in PBST, and 100 μl aliquots were added to the wells and incubated for 2 h at room temperature (20 °C). Plates were then incubated for 1 h at room temperature with 100 μl of 1:1000 diluted mouse anti-(human apoA-I) monoclonal antibody (Calbiochem), and finally incubated for 1 h at room temperature with 100 μl of 1:2000 diluted rabbit anti-(mouse IgG) conjugated to horseradish peroxidase (DakoCytomation), both in PBST supplemented with 2% (w/v) non-fat dried milk. Extensive automated washing with PBST was performed in between all antibody incubation steps. Colorimetric reactions were induced by 100 μl of tetramethylbenzidine substrate solution (Sigma). Reactions were stopped after 10 min by the addition of 100 μl of 2 M H2SO4. Absorbances were read at 450 nm using a microtitre spectrophotometer (Molecular Devices). For quantification purposes, a series of diluted human apoA-I standards (Sigma) was included on each plate, and diluted (e.g. 1:300000) human plasma served as positive control.
Statistical analysis
Data are shown as means±S.D. Statistical significance of differences was evaluated using Student's t test or Mann–Whitney U test. Significance was set at P<0.05.
RESULTS
Aramchol stimulated cholesterol but not phospholipid efflux
Figure 1(A) shows that cholesterol efflux rates from human fibroblasts mediated by 2 μg/ml Aramchol or 5 μg/ml apoA-I in the presence of 2 mg/ml BSA and also for BSA alone, were linear up to 20 h. At a concentration of 2 μg/ml, Aramchol was less potent than apoA-I. Figure 1(B) depicts a dose–response curve for Aramchol-induced cholesterol efflux. A hyperbolic response was observed. Since Aramchol at concentrations higher than 10 μg/ml induced toxicity in fibroblasts, as evident by a slight increase of LDH release, we performed all subsequent experiments at a concentration of 2 μg/ml which gives just suboptimal efflux and no toxicity. The presence of BSA in the medium was required for optimal efflux conditions. In the absence of BSA, as shown in Figure 2(A), both basal and Aramchol-induced efflux were lower. One could argue that contaminating apoA-I or other apolipoproteins in the BSA preparation may have been involved in the efflux observed. We could not demonstrate the presence of apoA-I or apoE in our BSA preparations at a detection limit of 0.2 μg/ml. To investigate whether one of the constituents of the Aramchol conjugate, i.e. cholic acid or fatty acid, would induce a similar efflux, we determined the effects of equimolar concentrations cholic acid and C16, C20 and C22 fatty acids. No effect on efflux was observed (results not shown). Interestingly, Aramchol had very little effect on efflux of choline-phospholipids: a 20% increase in efflux was considered insignificant compared with the 280% increase induced by apoA-I (Figure 2B).
Figure 1. Cholesterol efflux from human fibroblasts induced by Aramchol, apoA-I and BSA.

(A) Cultured human fibroblasts were loaded with [3H]cholesterol, and subsequent efflux of cholesterol in response to 2 μg/ml Aramchol, 2 mg/ml BSA or 5 μg/ml apoA-I, was determined, as described in the Materials and methods section. At the time points indicated, incubations were terminated. (B) Cholesterol efflux from human fibroblasts was performed using increasing concentrations of Aramchol in the presence of 2 mg/ml BSA. Efflux activity by the cells is given as percentage of the total radioactivity present in the cells at the onset of the experiment. The different incubations were carried out in triplicate and data are presented as means±S.D.
Figure 2. Effect of Aramchol on cholesterol and phospholipid efflux.

(A) Efflux from human cholesterol-loaded fibroblasts took place in the presence of 2 μg/ml Aramchol with or without 2 mg/ml BSA. (B) Choline-phospholipid efflux was induced by Aramchol (2 μg/ml) or apoA-I (5 μg/ml) in the presence of 2 mg/ml BSA. Efflux was determined after a 20 h incubation. 2 mg/ml BSA alone was used as a control. Three separate experiments were carried out in triplicate. Results are presented as means±S.D.
Effect of FABACs with different fatty-acid chain lengths
In previous studies on the effect of FABACs on gallstone disease and cholesterol crystallization, Aramchol was shown to be the most potent member of the family [4]. We therefore tested a range of molecules differing in the length of the fatty acid moiety in the cholesterol efflux assays. As shown in Figure 3, at a concentration of 2 μg/ml, all FABACs stimulated efflux, but Aramchol (C20) was by far the most effective.
Figure 3. Comparison of the potency of different FABACs in efflux induction.

Efflux from human cholesterol-loaded fibroblasts was performed in the presence of FABACs (2 μg/ml), which have a constant cholate moiety, but different acyl chain length. BSA was used as a control. Experiments were carried out in triplicate and results are presented as means±S.D.
The mechanism of Aramchol-induced efflux
ApoA-I-dependent cholesterol efflux has been shown to be fully dependent on the activity of ABCA1 [32,33]. To test the role of ABCA1 in Aramchol-induced efflux, we studied cholesterol efflux in response to Aramchol or apoA-I in fibroblasts derived from control subjects and Tangier disease patients. As shown in Figure 4, Aramchol failed to elicit cholesterol efflux from ABCA1-deficient fibroblasts, indicating that the protein is essential for the Aramchol-induced effect. Confirming earlier results [8,13], efflux to apoA-I is abrogated in Tangier disease fibroblasts, efflux to HDL decreased but methyl-β-cyclodextrin invokes normal cholesterol efflux. To substantiate further the relationship between Aramchol-mediated efflux and ABCA1, fibroblasts were incubated with the LXR agonist T0901317, the results of which are shown in Figure 5. T0901317 strongly increased both apoA-I- and Aramchol-induced efflux. To investigate whether Aramchol and apoA-I compete for the same site on the fibroblast cell membrane, competition assays were performed. Figure 6(A) shows the effect of increasing concentrations of Aramchol on a fixed amount of 5 μg/ml apoA-I. Aramchol did not increase efflux, and at the higher concentrations actually decreased efflux slightly, by approx. 20%. A similar experiment was performed using a constant level of HDL (50 μg). Aramchol did not inhibit HDL-induced efflux; in fact, HDL efflux slightly increased in response to increasing Aramchol (Figure 6A). To investigate further the interaction between Aramchol and apoA-I, the apolipoprotein was titrated on a fixed suboptimal concentration of 1 μg/ml Aramchol. At 0.25 μg/ml apoA-I, Aramchol addition stimulated efflux. The effect was, however, not additive and turned to slight inhibition at concentrations of apoA-I higher than 0.5 μg/ml, when compared with vehicle alone (Figure 6B).
Figure 4. ABCA1 mutations abrogate Aramchol induced cholesterol efflux.

Cholesterol-loaded fibroblasts from a control and two patients with Tangier disease were incubated with either 2 mg/ml BSA or 2 μg/ml Aramchol. Experiments were carried out in triplicate and results are presented as means±S.D.
Figure 5. The LXR agonist T090137 stimulates efflux mediated by Aramchol and apoA-I.

Fibroblasts were labelled with a trace amount of [3H]cholesterol in the presence or absence of 1 μM T0901317, and efflux in response to 2 mg/ml BSA, 2 μg/ml Aramchol or 5 μg/ml apoA-I was determined. Results are presented as means±S.D. (n=3).
Figure 6. Competition between Aramchol and apoA-I- or HDL-induced efflux.

(A) Efflux from cholesterol-loaded fibroblasts was performed in the presence of 5 μg/ml apoA-I or 50 μg/ml HDL. Increasing amounts of Aramchol were added, as indicated. (B) Efflux from cholesterol-loaded fibroblasts was performed in the presence of 2 μg/ml Aramchol. Increasing amounts of apoA-I were added, as indicated. Experiments were carried out in triplicate and results are presented as means±S.D.
Buoyant density of effluxed cholesterol
To explore the underlying mechanisms of Aramchol-induced cholesterol efflux, carriers of cholesterol in the efflux medium were separated by density-gradient ultracentrifugation. Figure 7(A) shows that, in the presence of apoA-I and Aramchol, cholesterol efflux appeared in two peaks, one at low density (1.055 g/ml) and one at high density (1.26 g/ml). In the control incubation with BSA as acceptor, no cholesterol was found at low density (Figure 7A). Total protein was also measured in the fractions as well as Aramchol and apoA-I in the gradients where these compounds were used as acceptors. Both total protein (predominantly BSA) and apoA-I could only be detected in the high-density fractions (Figure 7B). Aramchol was determined in pooled low- and high-density fractions. The compound was present exclusively in the high-density fraction (results not shown). Separate experiments in which choline-containing phospholipids were labelled with [3H]choline revealed a similar distribution, with approx. 50% of the choline label appearing in the low-density peak (results not shown).
Figure 7. Protein, apoA-I and cholesterol distribution in density-gradient fractions of efflux medium.

(A) Cholesterol-loaded fibroblasts were incubated with 2 mg/ml BSA with or without 5 μg/ml apoA-I or 2 μg/ml Aramchol and subjected to the efflux protocol. Efflux media were separated on a KBr density gradient by ultracentrifugation. A linear gradient was obtained, which was separated into 12 fractions. Fraction number (nr) 1 had a density of 1.045 g/ml and fraction 12 had a density of 1.3 g/ml. Results are presented as means±S.D. for three separate series of experiments. [3H]Cholesterol was determined by direct liquid-scintillation counting of the fractions. (B) Cholesterol-loaded fibroblasts were incubated with 2 mg/ml BSA with or without 5 μg/ml apoA-I and subjected to the efflux protocol, but in the absence of [3H]cholesterol. Efflux media were separated on a KBr density gradient by ultracentrifugation as described in (A). Total (Tot.) protein was measured in the fractions from the BSA-only gradient and apoA-I in the gradient where the protein was present. Three separate experiments were carried out and results are presented as means±S.D.
Distribution of Aramchol between cells and medium
To investigate the potential uptake of the hydrophobic molecule, the distribution of Aramchol between cells and medium was measured. The results of the extracted media and cells followed by LC–tandem MS measurements are summarized in Table 1. After efflux in the presence of BSA, 91% of Aramchol was found in the medium. Upon addition of apoA-I or HDL, 94% and 97% of the Aramchol was found in the medium respectively, suggesting that this hydrophobic compound does not accumulate in cells. Apparently, fibroblasts do not possess a transporter for Aramchol, and the compound cannot traverse the membrane bilayer spontaneously or the cells may contain an efficient efflux pump. Consequently, Aramchol might exert its activity outside the cell, possibly through binding to albumin.
Table 1. Aramchol distribution in cells and medium after efflux.
Cultured human fibroblasts were loaded with cholesterol and subsequently efflux of cholesterol in response to 2 mg/ml BSA, 5 μg/ml apoA-I or 50 μg/ml HDL was determined in the presence of 2 μg/ml aramchol. Subsequently, aramchol was measured in cells and supernatant by LC–tandem MS as described in the Materials and methods section. Results are means±S.D. (n=6).
| Treatment | Percentage in medium (mean±S.D.) | Recovery (%) |
|---|---|---|
| BSA | 91±0.7 | 95 |
| BSA, apoA-I | 94±0.3 | 93 |
| BSA, HDL | 97±2.1 | 99.6 |
Aramchol affects ABCA1 protein levels
The results obtained in the present study suggest that Aramchol affects cholesterol efflux in an ABCA1-dependent manner. We wondered whether Aramchol was also able to influence ABCA1 mRNA expression and/or protein levels in fibroblasts. RNA and protein were isolated from fibroblasts after cholesterol efflux in the presence, or absence, of 2 μg/ml Aramchol. No differences in mRNA expression level were observed when cells were incubated with Aramchol (results not shown). ABCA1 protein concentration, however, increased approx. 2-fold when efflux took place in the presence of Aramchol, compared with untreated cells (Figure 8A). To address the question of whether the observed increase is, indeed, accounted for by plasma membrane ABCA1, cell-surface proteins were biotinylated and isolated using streptavidin–agarose. Total cellular ABCA1 protein and ABCA1 protein isolated from the cell surface were analysed by Western blotting. As shown in Figure 8(B), the increase in ABCA1 protein level caused by Aramchol could be assigned to the plasma membrane fraction.
Figure 8. ABCA1 protein (A) and plasma membrane (B) levels in the presence or absence of Aramchol; effect of calpain (C).
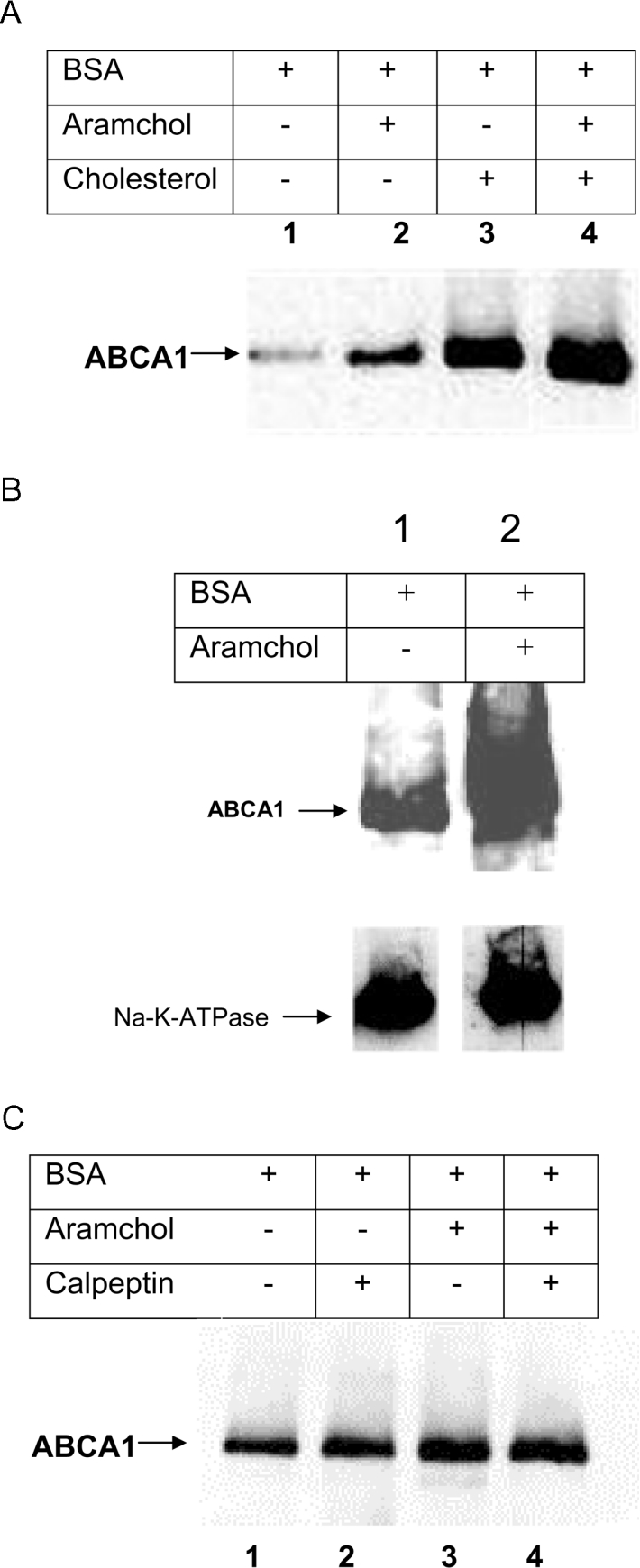
(A) Efflux from fibroblasts was measured in the presence, or absence of Aramchol (2 μg/ml). Subsequently, fibroblasts were harvested and protein extracts were subjected to Western blotting. Lane 1, no additions; lane 2, Aramchol (2 μg/ml); lane 3, cholesterol loading; lane 4, cholesterol loading plus 2 μg/ml Aramchol. (B) Cholesterol-loaded fibroblasts were incubated with, or without, Aramchol (2 μg/ml) and subjected to the efflux protocol. Subsequently the cells were put on ice and membrane proteins were biotinylated as described in the Materials and methods section. Biotinylated membrane fractions were isolated by streptavidin-affinity column chromatography. ABCA1 in these fractions was visualized by Western blotting. Lane 1, BSA; lane 2, BSA plus 2 μg/ml Aramchol. The blots were probed using anti-ABCA1 antibody, and after stripping with anti-Na+/K+-ATPase antibody to control for aspecific effects and equal loading of membrane protein. (C) Efflux from cholesterol-loaded fibroblasts was performed in the presence or absence of Aramchol (2 μg/ml) and with or without calpeptin (25 μM). After the incubations, cells were harvested and ABCA1 was visualized by Western blotting as described in the Materials and methods section. Lane 1, BSA; lane 2, BSA plus calpeptin; lane 3, BSA plus 2 μg/ml Aramchol; lane 4, BSA plus 2 μg/ml Aramchol plus calpeptin. Representative experiments are shown, and three independent experiments were carried out for all conditions.
ABCA1 stabilization by Aramchol
Aramchol is able to stabilize ABCA1 protein. To investigate the underlying mechanism, we examined whether this effect could be due to inhibition of calpain. Calpain is a thiol protease capable of digesting ABCA1 [34]. Calpeptin is a specific inhibitor of calpain [35]. Indeed, as shown in Figure 8(C), calpeptin enhances ABCA1 when cells were incubated in the presence of BSA. However, calpeptin did not result in an increase in ABCA1 protein levels when added to Aramchol-containing medium.
DISCUSSION
Aramchol represents a new group of synthetic molecules (FABACs) that, besides their ability to prevent and dissolve gallstones, were shown to ameliorate atherosclerosis and increase RCT. The major finding of the present study is that Aramchol promotes cholesterol efflux in an ABCA1-dependent manner. The molecular mechanism by which ABCA1 mediates transport of cholesterol and phospholipids is unclear and is subject to intense investigation [14–21,36–44]. ABCA1 has been suggested to induce a differential release of cholesterol and phospholipids [45,46] as opposed to coupled transport of both lipids. The role of raft-like domains in the plasma membrane is also a controversial subject area [18,45]. Conclusive data that are able to differentiate between the different mechanisms have not yet been produced (for a discussion, see [16]). To our knowledge, ABCA1-dependent efflux mediated by a non-apolipoprotein acceptor has not yet been reported. The fact that Aramchol induces efflux invalidates earlier observations suggesting that direct interaction of an apolipoprotein with ABCA1 is essential for the efflux process. Whether Aramchol interacts directly with ABCA1 is unclear. In the presence of saturating concentrations of apoA-I, a slight, but significant, inhibition of efflux was observed in response to increasing concentrations of Aramchol. At suboptimal concentrations of apoA-I, Aramchol stimulated efflux, but the effect was not additive. This may be explained by competition for the same site at the membrane. To explain the lack of a synergistic effect on efflux in the higher concentration range of both Aramchol and apoA-I, we hypothesize that Aramchol has a high affinity for the efflux site but a low capacity.
Liu et al. [47] reported that cholesterol efflux under the influence of apoA-I occurs in different types of particles. Intriguingly, most of the cholesterol was incorporated in large vesicles that did not contain apoA-I. In the present study we have confirmed these results by employing density-gradient ultracentrifugation to separate the different types of particles. Although the resolution of this method is not as clear as that of gel-permeation chromatography, the recovery (≈80%) is much better. Analysis of apoA-I efflux medium showed that less than half of the amount of cholesterol is present in the high-density fractions where apoA-I is present. The remainder was found in the low-density fraction, in the presence of choline-phospholipid presumably in the form of membrane vesicles, which is similar to the results reported previously by Liu et al. [47]. Surprisingly, Aramchol-induced efflux showed almost the same distribution pattern for cholesterol in density fractions of medium as apoA-I. No detectable Aramchol was present in the low-density fraction. We speculate that the cholesterol was solubilized in raft-derived lipid, as has recently been suggested by Duong et al. [48].
The chain length of the fatty acid moiety of the FABAC molecule influenced the potency of efflux induction. The C20-containing compound was the most effective, which may be due to an influence on the special domains in the membrane created by ABCA1 activity. Marguet and Chimini [16] proposed that ABCA1, by flipping phosphatidylserine, produces locally unstable membrane domains through which lipids can be taken up via microsolubilization. The C20 FABAC Aramchol may lower activation energy further for lipid uptake from these domains. In contrast with what has been observed for apoA-I, Aramchol induced very little choline-phospholipid efflux. Apparently, the compound has little affinity for phospholipids. Aramchol did not influence levels of ABCA1 mRNA; however, increased ABCA1 protein levels were observed, which were accessible to biotinylation, indicating localization on the plasma membrane. Elegant work of Vaughan and Oram [49] showed that overexpression of ABCA1 in the absence of apolipoproteins redistributed membrane cholesterol to cell-surface domains, which are accessible to cholesterol oxidase, suggesting the possibility that Aramchol extracts cholesterol from these domains. Stabilization studies with calpeptin and Aramchol suggest that these compounds may compete in inhibiting ABCA1 protein turnover. We hypothesize that the increase in ABCA1 protein level can be ascribed to inhibition of ABCA1 degradation, which has been similarly suggested for apoA-I [34].
In conclusion, in the present study, we have shown that small molecules such as FABACs induce ABCA1-dependent cholesterol efflux in an apolipoprotein-independent manner. Stimulation of RCT may explain the amelioration of atherosclerosis in mouse models induced by Aramchol as well as the increase in faecal neutral sterol secretion invoked by the compound [6,7].
Acknowledgments
We thank Ing Henk Overmars for excellent technical assistance. This study was supported in part by a ‘Meelmeyergrant’ from the Academic Medical Centre in Amsterdam and by grants 902-23-193 and 912-02-063 from The Netherlands Organization for Scientific Research.
References
- 1.Gilat T., Somjen G. J., Mazur Y., Leikin-Frenkel A., Rosenberg R., Halpern Z., Konikoff F. Fatty acid bile acid conjugates (FABACs) – new molecules for the prevention of cholesterol crystallisation in bile. Gut. 2001;48:75–79. doi: 10.1136/gut.48.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gilat T., Leikin-Frenkel A., Goldiner L., Laufer H., Halpern Z., Konikoff F. M. Arachidyl amido cholanoic acid (Aramchol) is a cholesterol solubilizer and prevents the formation of cholesterol gallstones in inbred mice. Lipids. 2001;36:1135–1140. doi: 10.1007/s11745-001-0824-3. [DOI] [PubMed] [Google Scholar]
- 3.Gilat T., Leikin-Frenkel A., Goldiner I., Halpern Z., Konikoff F. M. Dissolution of cholesterol gallstones in mice by the oral administration of a fatty acid bile acid conjugate. Hepatology. 2002;35:597–600. doi: 10.1053/jhep.2002.31868. [DOI] [PubMed] [Google Scholar]
- 4.Konikoff F. M., Leikin-Frenkel A., Goldiner I., Michowitz M., Brezowski E., Harats D., Gilat T. Biliary and systemic effects of fatty acid bile acid conjugates. Eur. J. Gastroenterol. Hepatol. 2003;15:649–655. doi: 10.1097/00042737-200306000-00012. [DOI] [PubMed] [Google Scholar]
- 5.Gilat T., Leikin-Frenkel A., Goldiner I., Juhel C., Lafont H., Gobbi D., Konikoff F. M. Prevention of diet-induced fatty liver in experimental animals by the oral administration of a fatty acid bile acid conjugate (FABAC) Hepatology. 2003;38:436–442. doi: 10.1053/jhep.2003.50348. [DOI] [PubMed] [Google Scholar]
- 6.Gonen A., Shaish A., Leikin-Frenkel A., Gilat T., Harats D. Fatty acid bile acid conjugates inhibit atherosclerosis in the C57BL/6 mouse model. Pathobiology. 2002;70:215–218. doi: 10.1159/000069332. [DOI] [PubMed] [Google Scholar]
- 7.Leikin-Frenkel A., Weinbroum A. A., Leikin-Gobbi D., Krupitzky L., Goldiner I., Shafat L., Gilat T., Konikoff F. M. Faecal sterol output is increased by arachidyl amido cholanoic acid (Aramchol) in rats. Biochem. Soc. Trans. 2004;32:131–133. doi: 10.1042/bst0320131. [DOI] [PubMed] [Google Scholar]
- 8.Attie A. D., Kastelein J. P., Hayden M. R. Pivotal role of ABCA1 in reverse cholesterol transport influencing HDL levels and susceptibility to atherosclerosis. J. Lipid Res. 2001;42:1717–1726. [PubMed] [Google Scholar]
- 9.Oram J. F. ABCA1 as a new therapeutic target for treating cardiovascular disease. Drug News Perspect. 2002;15:24–28. doi: 10.1358/dnp.2002.15.1.840027. [DOI] [PubMed] [Google Scholar]
- 10.Groen A. K., Bloks V. W., Bandsma R. H., Ottenhoff R., Chimini G., Kuipers F. Hepatobiliary cholesterol transport is not impaired in Abca1-null mice lacking HDL. J. Clin. Invest. 2001;108:843–850. doi: 10.1172/JCI12473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aiello R. J., Brees D., Bourassa P. A., Royer L., Lindsey S., Coskran T., Haghpassand M., Francone O. L. Increased atherosclerosis in hyperlipidemic mice with inactivation of ABCA1 in macrophages. Arterioscler. Thromb. Vasc. Biol. 2002;22:630–637. doi: 10.1161/01.atv.0000014804.35824.da. [DOI] [PubMed] [Google Scholar]
- 12.van Eck M., Bos I. S., Kaminski W. E., Orso E., Rothe G., Twisk J., Bottcher A., Van Amersfoort E. S., Christiansen-Weber T. A., Fung-Leung W. P., et al. Leukocyte ABCA1 controls susceptibility to atherosclerosis and macrophage recruitment into tissues. Proc. Natl. Acad. Sci. U.S.A. 2002;99:6298–6303. doi: 10.1073/pnas.092327399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Repa J. J., Mangelsdorf D. J. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu. Rev. Cell Dev. Biol. 2000;16:459–481. doi: 10.1146/annurev.cellbio.16.1.459. [DOI] [PubMed] [Google Scholar]
- 14.Hamon Y., Chambenoit O., Chimini G. ABCA1 and the engulfment of apoptotic cells. Biochim. Biophys. Acta. 2002;1585:64–71. doi: 10.1016/s1388-1981(02)00325-6. [DOI] [PubMed] [Google Scholar]
- 15.Hovingh G. K., Van Wijland M. J., Brownlie A., Bisoendial R. B., Hayden M. R., Kastelein J. J., Groen A. K. The role of the ABCA1 transporter and cholesterol efflux in familial hypoalphalipoproteinemia. J. Lipid Res. 2003;44:1251–1255. doi: 10.1194/jlr.M300080-JLR200. [DOI] [PubMed] [Google Scholar]
- 16.Marguet D., Chimini G. The ABCA1 transporter and ApoA-I: obligate or facultative partners? Trends Cardiovasc. Med. 2002;12:294–298. doi: 10.1016/s1050-1738(02)00177-9. [DOI] [PubMed] [Google Scholar]
- 17.Murthy S., Born E., Mathur S. N., Field F. J. Liver X receptor-mediated increase in ABCA1 expression is attenuated by fatty acids in CaCo-2 cells: effect on cholesterol efflux to high-density lipoprotein. Biochem. J. 2003;377:545–552. doi: 10.1042/BJ20030903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oram J. F. HDL apolipoproteins and ABCA1: partners in the removal of excess cellular cholesterol. Arterioscler. Thromb. Vasc. Biol. 2003;23:720–727. doi: 10.1161/01.ATV.0000054662.44688.9A. [DOI] [PubMed] [Google Scholar]
- 19.Alder-Baerens N., Muller P., Pohl A., Korte T., Hamon Y., Chimini G., Pomorski T., Herrmann A. Headgroup-specific exposure of phospholipids in ABCA1-expressing cells. J. Biol. Chem. 2005;280:26321–26329. doi: 10.1074/jbc.M413993200. [DOI] [PubMed] [Google Scholar]
- 20.Chen W., Wang N., Tall A. R. A PEST deletion mutant of ABCA1 shows impaired internalization and defective cholesterol efflux from late endosomes. J. Biol. Chem. 2005;280:29277–29281. doi: 10.1074/jbc.M505566200. [DOI] [PubMed] [Google Scholar]
- 21.Fitzgerald M. L., Okuhira K., Short G. F., III, Manning J. J., Bell S. A., Freeman M. W. ATP-binding cassette transporter A1 contains a novel C-terminal VFVNFA motif that is required for its cholesterol efflux and ApoA-I binding activities. J. Biol. Chem. 2004;279:48477–48485. doi: 10.1074/jbc.M409848200. [DOI] [PubMed] [Google Scholar]
- 22.Amador E., Dorfman L. E., Wacker W. E. Serum lactic dehyrogenase activity: an analytical assessment of current assays. Clin. Chem. 1963;12:391–399. [PubMed] [Google Scholar]
- 23.Van Wijland M. J., de Waart D. R., Groen A. K. Biliary anionic peptide fraction/calcium binding protein inhibits apolipoprotein A-I-mediated cholesterol efflux from cultured cells. Biochem. Biophys. Res. Commun. 2001;287:1–4. doi: 10.1006/bbrc.2001.5542. [DOI] [PubMed] [Google Scholar]
- 24.Bligh E. G., Dyer W. J. A rapid method of total lipid extraction and purification. Can. J. Med. Sci. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 25.Groen A. K., Ottenhoff R., Jansen P. L. M., van Marle J., Tytgat G. N. J. Effect of cholesterol nucleation promoting activity on cholesterol solubilization in model bile. J. Lipid Res. 1989;30:51–58. [PubMed] [Google Scholar]
- 26.Smith P. K., Krohn R. I., Hermanson G. T., Mallia A. K., Gartner F. H., Provenzano M. D., Fujimoto E. K., Goeke N. M., Olson B. J., Klenk D. C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 27.Witting S. R., Maiorano J. N., Davidson W. S. Ceramide enhances cholesterol efflux to apolipoprotein A-I by increasing the cell surface presence of ATP-binding cassette transporter A1. J. Biol. Chem. 2003;278:40121–40127. doi: 10.1074/jbc.M305193200. [DOI] [PubMed] [Google Scholar]
- 28.Haidar B., Denis M., Marcil M., Krimbou L., Genest J., Jr Apolipoprotein A-I activates cellular cAMP signaling through the ABCA1 transporter. J. Biol. Chem. 2004;279:9963–9969. doi: 10.1074/jbc.M313487200. [DOI] [PubMed] [Google Scholar]
- 29. Reference deleted.
- 30.Wellington C. L., Walker E. K., Suarez A., Kwok A., Bissada N., Singaraja R., Yang Y. Z., Zhang L. H., James E., Wilson J. E., et al. ABCA1 mRNA and protein distribution patterns predict multiple different roles and levels of regulation. Lab Invest. 2002;82:273–283. doi: 10.1038/labinvest.3780421. [DOI] [PubMed] [Google Scholar]
- 31.Goldiner I., Overmars H., Groen A. K., Kulik W. Method for the quantitative assay of fatty acid–bile acid conjugates by tandem mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 2003;795:35–40. doi: 10.1016/s1570-0232(03)00494-x. [DOI] [PubMed] [Google Scholar]
- 32.Lawn R. M., Wade D. P., Garvin M. R., Wang X., Schwartz K., Porter J. G., Seilhamer J. J., Vaughan A. M., Oram J. F. The Tangier disease gene product ABC1 controls the cellular apolipoprotein-mediated lipid removal pathway. J. Clin. Invest. 1999;104:R25–R31. doi: 10.1172/JCI8119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marcil M., Brooks-Wilson A., Clee S. M., Roomp K., Zhang L. H., Yu L., Collins J. A., van Dam M., Molhuizen H. O. F., Loubster O., et al. Mutations in the ABC1 gene in familial HDL deficiency with defective cholesterol efflux. Lancet. 1999;354:1341–1346. doi: 10.1016/s0140-6736(99)07026-9. [DOI] [PubMed] [Google Scholar]
- 34.Martinez L. O., Agerholm-Larsen B., Wang N., Chen W., Tall A. R. Phosphorylation of a pest sequence in ABCA1 promotes calpain degradation and is reversed by ApoA-I. J. Biol. Chem. 2003;278:37368–37374. doi: 10.1074/jbc.M307161200. [DOI] [PubMed] [Google Scholar]
- 35.Tsujinaka T., Kajiwara Y., Kambayashi J., Sakon M., Higuchi N., Tanaka T., Mori T. Synthesis of a new cell penetrating calpain inhibitor (calpeptin) Biochem. Biophys. Res. Commun. 1988;153:1201–1208. doi: 10.1016/s0006-291x(88)81355-x. [DOI] [PubMed] [Google Scholar]
- 36.Kiss R. S., Maric J., Marcel Y. L. Lipid efflux in human and mouse macrophagic cells: evidence for differential regulation of phospholipid and cholesterol efflux. J. Lipid Res. 2005;46:1877–1887. doi: 10.1194/jlr.M400482-JLR200. [DOI] [PubMed] [Google Scholar]
- 37.Nakamura K., Kennedy M. A., Baldan A., Bojanic D. D., Lyons K., Edwards P. A. Expression and regulation of multiple murine ATP-binding cassette transporter G1 mRNAs/isoforms that stimulate cellular cholesterol efflux to high density lipoprotein. J. Biol. Chem. 2004;279:45980–45989. doi: 10.1074/jbc.M408652200. [DOI] [PubMed] [Google Scholar]
- 38.Passarelli M., Tang C., McDonald T. O., O'Brien K. D., Gerrity R. G., Heinecke J. W., Oram J. F. Advanced glycation end product precursors Impair ABCA1-dependent cholesterol removal from cells. Diabetes. 2005;54:2198–2205. doi: 10.2337/diabetes.54.7.2198. [DOI] [PubMed] [Google Scholar]
- 39.Schmitz G., Langmann T. Transcriptional regulatory networks in lipid metabolism control ABCA1 expression. Biochim. Biophys. Acta. 2005;1735:1–19. doi: 10.1016/j.bbalip.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 40.Tsujita M., Wu C. A., Abe-Dohmae S., Usui S., Okazaki M., Yokoyama S. On the hepatic mechanism of HDL assembly by the ABCA1/apoA-I pathway. J. Lipid Res. 2005;46:154–162. doi: 10.1194/jlr.M400402-JLR200. [DOI] [PubMed] [Google Scholar]
- 41.Vedhachalam C., Liu L., Nickel M., Dhanasekaran P., Anantharamaiah G. M., Lund-Katz S., Rothblat G. H., Phillips M. C. Influence of ApoA-I structure on the ABCA1-mediated efflux of cellular lipids. J. Biol. Chem. 2004;279:49931–49939. doi: 10.1074/jbc.M406924200. [DOI] [PubMed] [Google Scholar]
- 42.Yamauchi Y., Chang C. C., Hayashi M., Abe-Dohmae S., Reid P. C., Chang T. Y., Yokoyama S. Intracellular cholesterol mobilization involved in the ABCA1/apolipoprotein-mediated assembly of high density lipoprotein in fibroblasts. J. Lipid Res. 2004;45:1943–1951. doi: 10.1194/jlr.M400264-JLR200. [DOI] [PubMed] [Google Scholar]
- 43.Yokoyama S. Assembly of high density lipoprotein by the ABCA1/apolipoprotein pathway. Curr. Opin. Lipidol. 2005;16:269–279. doi: 10.1097/01.mol.0000169346.15450.90. [DOI] [PubMed] [Google Scholar]
- 44.Zheng H., Kiss R. S., Franklin V., Wang M. D., Haidar B., Marcel Y. L. ApoA-I lipidation in primary mouse hepatocytes. Separate controls for phospholipid and cholesterol transfers. J. Biol. Chem. 2005;280:21612–21621. doi: 10.1074/jbc.M502200200. [DOI] [PubMed] [Google Scholar]
- 45.Wang N., Tall A. R. Regulation and mechanisms of ATP-binding cassette transporter A1-mediated cellular cholesterol efflux. Arterioscler. Thromb. Vasc. Biol. 2003;23:1178–1184. doi: 10.1161/01.ATV.0000075912.83860.26. [DOI] [PubMed] [Google Scholar]
- 46.Wang N., Silver D. L., Costet P., Tall A. R. Specific binding of ApoA-I, enhanced cholesterol efflux, and altered plasma membrane morphology in cells expressing ABC1. J. Biol. Chem. 2000;275:33053–33058. doi: 10.1074/jbc.M005438200. [DOI] [PubMed] [Google Scholar]
- 47.Liu L., Bortnick A. E., Nickel M., Dhanasekaran P., Subbaiah P. V., Lund-Katz S., Rothblat G. H., Phillips M. C. Effects of apolipoprotein A-I on ATP-binding cassette transporter A1-mediated efflux of macrophage phospholipid and cholesterol: formation of nascent high density lipoprotein particles. J. Biol. Chem. 2003;278:42976–42984. doi: 10.1074/jbc.M308420200. [DOI] [PubMed] [Google Scholar]
- 48.Duong P. T., Collins H. L., Nickel M., Lund-Katz S., Rothblat G. H., Phillips M. C. Characterization of nascent HDL particles and microparticles formed by ABCA1-mediated efflux of cellular lipids to ApoA-I. J. Lipid Res. 2006;47:832–843. doi: 10.1194/jlr.M500531-JLR200. [DOI] [PubMed] [Google Scholar]
- 49.Vaughan A. M., Oram J. F. ABCA1 redistributes membrane cholesterol independent of apolipoprotein interactions. J. Lipid Res. 2003;44:1373–1380. doi: 10.1194/jlr.M300078-JLR200. [DOI] [PubMed] [Google Scholar]