

Abstract
P-glycoprotein (P-gp; ABCB1) actively transports a broad range of structurally unrelated compounds out of the cell. An important step in the transport cycle is coupling of drug binding with ATP hydrolysis. Drug substrates such as verapamil bind in a common drug-binding pocket at the interface between the TM (transmembrane) domains of P-gp and stimulate ATPase activity. In the present study, we used cysteine-scanning mutagenesis and reaction with an MTS (methanethiosulphonate) thiol-reactive analogue of verapamil (MTS-verapamil) to test whether the first TM segment [TM1 (TM segment 1)] forms part of the drug-binding pocket. One mutant, L65C, showed elevated ATPase activity (10.7-fold higher than an untreated control) after removal of unchanged MTS-verapamil. The elevated ATPase activity was due to covalent attachment of MTS-verapamil to Cys65 because treatment with dithiothreitol returned the ATPase activity to basal levels. Verapamil covalently attached to Cys65 appears to occupy the drug-binding pocket because verapamil protected mutant L65C from modification by MTS-verapamil. The ATPase activity of the MTS-verapamil-modified mutant L65C could not be further stimulated with verapamil, calcein acetoxymethyl ester or demecolcine. The ATPase activity could be inhibited by cyclosporin A but not by trans-(E)-flupentixol. These results suggest that TM1 contributes to the drug-binding pocket.
Keywords: cysteine-scanning mutagenesis, drug transport, drug-binding pocket, P-glycoprotein, protein modification, transmembrane segment 1 (TM1)
Abbreviations: ABC transporter, ATP-binding-cassette transporter; BHK cell, baby hamster kidney cell; calcein-AM, calcein acetoxymethyl ester; DTT, dithiothreitol; HEK-293 cell, human embryonic kidney 293 cell; M4M, 1,4-butanediyl bismethanethiosulphonate; M8M, 3,6-dioxaoctane-1,8-diyl bismethanethiosulphonate; M11M, 3,6,9-trioxaundecane-1,11-diyl bismethanethiosulphonate; M17M, 3,6,9,12,15-pentaoxaheptadecane-1,17-diyl bismethanethiosulphonate; MTS, methanethiosulphonate; NBD, nucleotide-binding domain; P-gp, P-glycoprotein; PXR, pregnane X receptor; TBS, Tris-buffered saline; TM, transmembrane; TM1 (etc.), TM segment 1 (etc.); TMD, TM domain; TMD1, N-terminal TMD containing TM1–TM6; TMD2, C-terminal TMD containing TM7–TM12
INTRODUCTION
The human multidrug resistance P-gp (P-glycoprotein; ABCB1) is an ATP-dependent transporter that catalyses the efflux of hydrophobic molecules out of the cell [1]. The physiological role of P-gp is unknown because studies on knockout mice show that it is not essential for viability or reproduction but the mice demonstrate hypersensitivity to cytotoxic compounds [2]. The protein is expressed in relatively high levels in the apical membranes of epithelial cells of intestine, kidney, liver and blood–brain/testes barriers [3] and its presence in these organs suggests that it may protect us from cytotoxic compounds in our diets and environment. P-gp is clinically important because of its potential to undermine cancer and AIDS/HIV chemotherapies [4].
P-gp is a 170 kDa plasma membrane protein and is one of 48 members of the ABC transporter (ATP-binding-cassette transporter) family found in humans [5]. It has 1280 amino acids that are organized as two similar halves (43% amino acid identity) and are joined by a linker region [6]. Each half has a TMD [TM (transmembrane) domain] containing six TM segments followed by a hydrophilic NBD (nucleotide-binding domain) [7].
The minimum functional unit in P-gp is a monomer [8], although the two halves of the molecule do not have to be covalently linked for function [9]. The interface between the two halves of the molecule is critical for function because the ATP-binding sites are located between the NBDs [10,11]. Although both NBDs can hydrolyse ATP, inhibition at either site inhibits the activity of the protein [12,13]. P-gp may hydrolyse ATP by an alternating mechanism [13].
Similarly, the drug-binding pocket is located between the TMDs of P-gp [14–18]. A deletion mutant that lacked both NBDs retained the ability to bind drug substrates [9]. It has been proposed that P-gp may have up to four distinct drug-binding sites [19–22] and that each substrate has a distinct binding site. One way to reconcile the ability of P-gp to bind and transport numerous structurally unrelated compounds is to propose a common drug-binding pocket at the interface between the TMDs and that substrates bind through a ‘substrate-induced fit’ mechanism [17,23]. In this mechanism, a substrate would generate its own binding site by using various combinations of residues from different TMs. The number and types of residues involved in binding the substrate would determine its affinity for the substrate. Therefore binding of a relatively large molecule such as cyclosporin A most likely involves many more residues than binding of a relatively small molecule such as verapamil, and may account for P-gp having a higher affinity for cyclosporin than verapamil [24]. Also, structurally different substrates could share the same residues during drug binding.
To determine the mechanism of P-gp, it is important to identify TMs that contribute residues to the drug-binding pocket and to understand how substrate binding is coupled with ATP hydrolysis. Recently, we revised the TM packing model of P-gp based on results from cross-linking studies [25–27] and by comparison with the crystal structure of the bacterial ABC transporter MsbA [28]. In the revised model, TM1 (TM segment 1) is close to TM4–TM6 that are predicted to line the drug-binding pocket. Another observation indicating that TM1 may lie close to TM6 is that mutations in TM6 can alter the protease sensitivity of the extracellular end of TM1 [29]. Recently, it was shown that ATP hydrolysis promoted interactions between the extracellular ends to TM1 and TM11 [30]. Since residues in TM11 have been shown to contribute to substrate binding [15,31], interactions between TM1 and TM11 during ATP hydrolysis suggested that TM1 could also contribute to drug binding.
In the present study, we used cysteine-scanning mutagenesis and reaction with a thiol-reactive analogue of verapamil, MTS (methanethiosulphonate)-verapamil, to test for activation of ATP hydrolysis after covalent modification of residues in TM1.
MATERIALS AND METHODS
Construction of mutants
Cysteine-less (Cys-less) P-gp was constructed by replacing the seven endogenous cysteine residues at positions 137, 431, 717, 956, 1074, 1125 and 1227 with alanine residues [7]. None of the cysteine residues are essential for function [7]. Single cysteine residues were then introduced at each position in TM1 (residues 51–71) in the Cys-less P-gp as described previously [32]. All of the mutants contained a His10 tag at the C-terminal end to facilitate purification of the mutant P-gp by nickel-chelate chromatography [33]. A series of double cysteine mutants containing L65C in TM1 with another cysteine in TMD2 (C-terminal TMD containing TM7–TM12) predicted to line the drug-binding pocket [34] (i.e. F942C or T945C in TM11 and L975C, V981C, V982C, G984C or A985C in TM12) were also constructed for cross-linking analysis. A His-tagged mutant containing two cysteine residues L65C(TM1)/I306C(TM5) was also constructed.
Expression of mutants
The mutant P-gps were transiently expressed in HEK-293 cells (human embryonic kidney 293 cells) as described previously [33]. The mutant L65C was also stably expressed in BHK cells (baby hamster kidney cells). Briefly, BHK cells were transfected with the His-tagged mutant L65C cDNA in pMT21 as described previously [35]. P-gp-expressing cells were selected on 2 nM vinblastine and P-gp expression was confirmed by subjecting whole cell lysates to immunoblot analysis with rabbit polyclonal antibody against P-gp [36] followed by enhanced chemiluminescence (Pierce, Rockford, IL, U.S.A.). BHK or HEK-293 cells expressing mutant L65C were also grown in the presence of 10 μM cyclosporin A for 24 h because it acts as a pharmacological/specific chemical chaperone to increase the yield of mature enzyme [37]. To prevent potential interference of cyclosporin A in subsequent labelling or disulphide cross-linking studies, the cyclosporin A-treated cells were grown in the absence of cyclosporin A for 24 h before harvest.
Reaction with MTS-verapamil and measurement of ATPase activity
HEK-293 or BHK cells expressing His-tagged mutant L65C from 20 (10 cm diameter) plates were washed three times with PBS (10 mM sodium phosphate, pH 7.4, and 150 mM NaCl) and then suspended in a total volume of 1.5 ml of TBS (Tris-buffered saline; 10 mM Tris/HCl, pH 8.0, and 150 mM NaCl). The cells were solubilized by addition of an equal volume of TBS containing 2% (w/v) n-dodecyl-β-D-maltoside. Insoluble material was removed by centrifugation at 16000 g for 15 min at 4 °C. DNA was removed from the supernatant by passage through a miniprep plasmid DNA spin column (Qiagen, Mississauga, ON, Canada). Half of the supernatant (1.3 ml) was then incubated with the desired concentration of MTS-verapamil (0–1 mM) for 10 min at 20 °C, while the remaining sample served as an untreated control. In the drug protection studies, the solubilized material was pre-incubated with 3 mM rhodamine B or 2 mM verapamil (saturating concentrations) for 10 min at 20 °C prior to labelling with MTS-verapamil. The samples were then cooled in an ice bath, followed by addition of 0.15 ml of 3 M NaCl and 0.05 ml of 1 M imidazole (pH 7.0). His-tagged P-gp was then isolated by nickel-chelate chromatography as described previously [33]. The recovery of P-gp was monitored by immunoblot analysis with rabbit anti-P-gp polyclonal antibody [36].
A sample of the isolated His-tagged P-gp was mixed with an equal volume of 10 mg/ml sheep brain phosphatidylethanolamine (Type II-S; Sigma) or 10 mg/ml Escherichia coli lipids (Avanti Polar Lipids, Alabaster, AL, U.S.A.) that had been washed and suspended in TBS. The sample was sonicated and ATPase activity was measured in the absence of drug substrate or in the presence of saturating levels of calcein-AM (calcein acetoxymethyl ester; 0.6 mM), demecolcine (3 mM), verapamil (1 mM), cyclosporin A (0.15 mM) or trans-(E)-flupentixol (0.6 mM) [38]. The samples were incubated for 30 min at 37 °C and the amount of inorganic phosphate released was determined [39].
To determine the characteristics of the single cysteine P-gp mutants that were not exposed to MTS-verapamil, the mutant P-gps were assayed for drug-stimulated ATPase activity in the presence of various concentrations of verapamil (1–1000 μM), vinblastine (0.6–60 μM) or colchicine (0.1–10 mM).
Disulphide cross-linking analysis
Mutants L65C, F942C, T945C, L975C, V981C, V982C, G984C, A985C, L65C/F942C, L65C/T945C, L65C/975C, L65C/V981C, L65C/V982C, L65C/G984C and L65C/A985C were transiently expressed in HEK-293 cells. The cells were harvested and washed three times with PBS (pH 7.4) and the membranes were prepared as described previously [25]. The membranes were suspended in TBS. A sample of the membrane was then treated with zero-length cross-linker (1 mM copper phenanthroline) or with 0.2 mM of the homobifunctional MTS cross-linkers with spacer arms of various lengths [M4M (1,4-butanediyl bismethanethiosulphonate; 7.8 Å spacer arm; 1 Å=0.1 nm), M8M (3,6-dioxaoctane-1,8-diyl bismethanethiosulphonate; 13 Å), M11M (3,6,9-trioxaundecane-1,11-diyl bismethanethiosulphonate; 16.9 Å) or M17M (3,6,9,12,15-pentaoxaheptadecane-1,17-diyl bismethanethiosulphonate; 24.7 Å)] (Toronto Research Chemicals, Toronto, ON, Canada) for 15 min at 4 °C [16]. The reactions were stopped by addition of 2×SDS sample buffer [125 mM Tris/HCl, pH 6.8, 20% (v/v) glycerol and 4% (w/v) SDS] containing 50 mM EDTA and no reducing agent. The reaction mixtures were then subjected to SDS/PAGE (7.5% gel) and immunoblot analysis with a rabbit polyclonal antibody against P-gp [36]. Intramolecular disulphide cross-linking between TMD1 (N-terminal TMD containing TM1–TM6) and TMD2 can be detected because the cross-linked product migrates with a slower mobility on SDS/polyacrylamide gels [38].
RESULTS
P-gp contains 12 TM segments (Figure 1A). To identify TMs that line the drug-binding pocket, we previously used cysteine-scanning mutagenesis followed by reaction with thiol-reactive analogues of drug substrates such as verapamil and Rhodamine to determine those residues which when modified resulted in inhibition of ATPase activity. The rationale in those studies was that drug binding stimulated ATPase activity and therefore covalent modification of residues in the drug-binding pocket should inhibit the ATPase activity. The results from such studies showed that TM4–TM6 of TMD1 and TM9–TM12 of TMD2 line the drug-binding pocket [15,17,32].
Figure 1. Schematic models of P-gp.

(A) The 12 TMs of P-gp are shown as numbered cylinders. The branched lines represent glycosylation sites. The locations of residues L65C in TM1, I306C in TM5 and F343C in TM6 are shown. The ATPase activities of these mutants were permanently activated after modification with thiol-reactive drug substrate analogues. (B) Predicted packing of the TM segments of P-gp. The common drug-binding pocket is at the interface between TMD1 and TMD2.
In our revised P-gp model for TM packing, TM1 is placed close to TM5 and TM6 (Figure 1B). This model is based on results from disulphide cross-linking studies [26,27] and comparison with the crystal structure of the bacterial transporter, MsbA [28]. Although TM1 is now predicted to contribute to the drug-binding pocket, we did not see any evidence that TM1 was important for drug binding in previous cysteine-scanning mutagenesis and thiol-reactivity studies in assays that were designed to detect for inhibition of ATPase activity [15,17,32]. It was later found, however, that covalent attachment of a drug substrate to P-gp did not always inhibit drug-stimulated ATPase activity. For example, modification of Cys306 in TM5 with MTS-verapamil [40] or Cys343 in TM6 with MTS-Rhodamine [41] permanently activated the ATPase activity of P-gp. The covalently-attached molecule (Figure 2A) appeared to mimic P-gp interactions with normal verapamil or Rhodamine since maximal drug-stimulated ATPase activities were obtained after covalent modification and labelling of P-gp by MTS-verapamil or MTS-Rhodamine could be blocked by the presence of verapamil or Rhodamine respectively [40,41].
Figure 2. Reaction and ATPase activity of mutant L65C treated with MTS-verapamil.

(A) Schematic reaction of a thiol group with MTS-verapamil and attachment of verapamil to the protein via a disulphide bond. (B) His-tagged Cys-less or mutants M51C–V71C P-gps were expressed in HEK-293 cells and solubilized with n-dodecyl-β-D-maltoside. Insoluble material was removed by centrifugation and the supernatants were treated with or without 1 mM MTS-verapamil. His-tagged P-gps were then isolated by nickel-chelate chromatography. Equivalent amounts of P-gp were mixed with lipid, sonicated and assayed for ATPase activity in the absence of drug substrate. The fold stimulation is the ratio of activity of a sample treated with MTS-verapamil to that in an untreated sample. Each value is the average of three different experiments. ND, not determined because of low expression. (C) His-tagged mutant L65C expressed in HEK-293 cells was solubilized with n-dodecyl-β-D-maltoside, treated with various concentrations of MTS-verapamil and isolated by nickel-chelate chromatography. The ATPase activities of the isolated protein were determined as described above.
Therefore we tested whether labelling of any cysteine residue introduced into TM1 by MTS-verapamil could also result in permanent activation of ATPase activity. MTS-verapamil was selected for the study because verapamil is one of the most potent activators of P-gp ATPase activity [31]. Accordingly, we constructed cysteine mutants that contained a cysteine residue at positions 51–71 of TM1 of the His-tagged Cys-less P-gp and transiently expressed the mutants in HEK-293 cells. A potential complicating factor in our earlier study to screen for mutants showing permanent activation after reaction with MTS-verapamil was that the presence of cyclosporin A in the growth media could inhibit labelling [40]. Therefore the cells were grown in the absence of cyclosporin A for 24 h prior to harvest. The single cysteine mutants were first isolated by nickel-chelate chromatography and ATPase activity was determined in the presence of various concentrations of verapamil, colchicine or vinblastine. Colchicine and vinblastine were included because they are classic substrates of P-gp [1]. Table 1 shows that all mutants, except for mutants H61C and G64C, showed characteristics similar to that of the Cys-less parent. Mutants H61C and G64C showed approx. 4–5-fold reduction in apparent affinity for verapamil and had smaller reductions in affinity for colchicine. Mutant H61C also exhibited a reduction in the apparent affinity for vinblastine. The activities of mutants V52C, G54C and G62C were not determined because of very low expression [32].
Table 1. Drug-stimulated ATPase activity of TM1 cysteine mutants.
ND, not determined because of very low expression.
| Verapamil | Colchicine | Vinblastine | ||||
|---|---|---|---|---|---|---|
| Mutant | Vmax (%)* | S50 (μM)† | Vmax (%) | S50 (μM) | Vmax (%) | S50 (μM) |
| M51C | 101 | 11.0±0.6 | 96 | 391±36 | 94 | 2.4±0.2 |
| V52C | ND | ND | ND | ND | ND | ND |
| V53C | 104 | 12.0±0.2 | 101 | 389±30 | 102 | 2.2±0.1 |
| G54C | ND | ND | ND | ND | ND | ND |
| T55C | 114 | 10.3±1.1 | 95 | 418±22 | 91 | 2.2±0.1 |
| L56C | 103 | 12.2±0.3 | 87 | 440±41 | 95 | 2.5±0.2 |
| A57C | 108 | 11.3±0.3 | 98 | 377±34 | 92 | 2.4±0.2 |
| A58C | 90 | 12.5±0.2 | 94 | 434±20 | 95 | 2.6±0.3 |
| I59C | 115 | 11.2±0.8 | 95 | 380±33 | 114 | 2.5±0.2 |
| I60C | 102 | 11.1±0.7 | 91 | 408±18 | 110 | 2.5±0.2 |
| H61C | 97 | 54.0±5.0 | 61 | 912±86 | 105 | 5.4±0.4 |
| G62C | ND | ND | ND | ND | ND | ND |
| A63C | 114 | 10.5±1.2 | 99 | 362±42 | 105 | 2.0±0.3 |
| G64C | 106 | 45.0±6.0 | 88 | 613±55 | 60 | 2.4±0.1 |
| L65C | 72 | 9.3±1.1 | 112 | 368±32 | 78 | 2.0±0.2 |
| P66C | 95 | 13.0±0.5 | 86 | 480±39 | 97 | 2.8±0.4 |
| L67C | 101 | 12.3±0.3 | 106 | 423±21 | 100 | 2.3±0.1 |
| M68C | 119 | 9.7±1.1 | 105 | 365±32 | 92 | 2.3±0.2 |
| M69C | 107 | 11.8±0.6 | 110 | 431±25 | 108 | 2.2±0.1 |
| L70C | 94 | 11.4±0.7 | 90 | 413±18 | 98 | 2.3±0.1 |
| V71C | 106 | 11.9±0.3 | 90 | 370±27 | 102 | 2.5±0.5 |
| Cys-less | 100 | 12.0±1.0 | 100 | 412±48 | 100 | 2.2±0.3 |
* Maximum activity relative to that of Cys-less P-gp.
† Substrate concentration at 50% maximal activity.
To test for interaction of the TM1 single cysteine mutants with MTS-verapamil, cells expressing each mutant were then solubilized with n-dodecyl-β-D-maltoside. The insoluble material was removed and the extracts were treated with or without 1 mM MTS-verapamil. His-tagged P-gp was then isolated by nickel-chelate chromatography, a step that also removed unchanged MTS-verapamil. Treatment of the P-gp mutants with MTS-verapamil did not affect their subsequent recovery by nickel-chelate chromatography (results not shown). The isolated P-gps were then mixed with lipid, sonicated and ATPase activity was determined. Figure 2(B) shows that Cys-less P-gp treated with 1 mM MTS-verapamil did not exhibit enhanced ATPase activity compared with an untreated sample. We found that the activity of only one cysteine mutant in TM1 (L65C) was permanently activated after treatment with MTS-verapamil (Figure 2B). Mutant L65C showed a 10.7-fold increase in activity after modification with 0.3–1 mM MTS-verapamil compared with an untreated control. The half-maximal concentration for activation was 38 μM (Figure 2C). We previously showed that treatment of any of the TM1 cysteine mutants with MTS-verapamil did not inhibit verapamil-stimulated ATPase activity [15].
To test whether activation of mutant L65C by MTS-verapamil was due to covalent attachment of verapamil, we treated the modified mutant with 20 mM of the reducing agent DTT (dithiothreitol) and then measured ATPase activity. Figure 3 shows that the presence of DTT almost completely abolished the enhanced ATPase activity of mutant L65C. After treatment with DTT, the activity of MTS-verapamil-treated mutant L65C could still be stimulated 10-fold with 1 mM verapamil (Figure 3). These results show that attachment of MTS-verapamil to Cys65 mimics the interaction of P-gp with the drug substrate verapamil.
Figure 3. Effect of DTT on mutant L65C modified with MTS-verapamil.
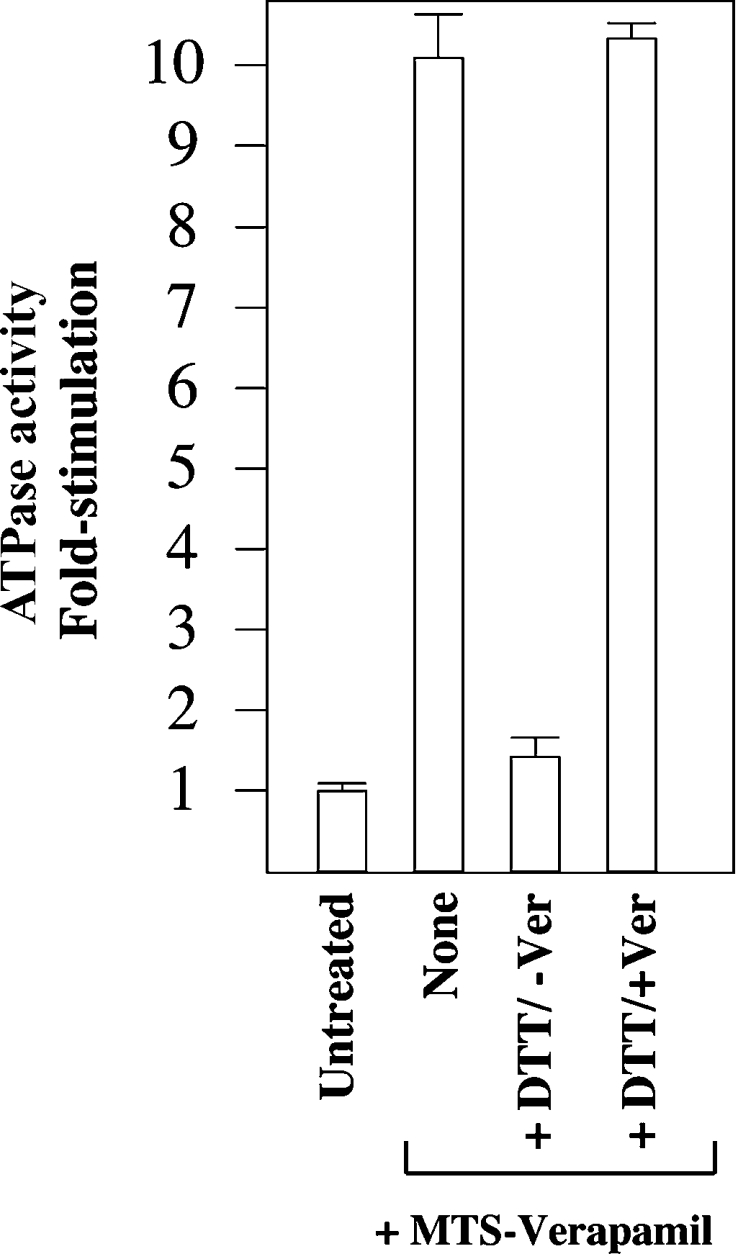
His-tagged mutant L65C was expressed in HEK-293 cells. The cells were solubilized with n-dodecyl-β-D-maltoside and then incubated in the absence (Untreated) or presence (+MTS-Verapamil) of 0.3 mM MTS-verapamil. The His-tagged proteins were isolated by nickel-chelate chromatography, mixed with an equal volume of lipid containing no (None) or 40 mM DTT (+DTT). After 5 min at 20 °C, the samples were incubated with an equal volume of 2×ATPase buffer (100 mM Tris/HCl, pH 7.5, 100 mM NaCl, 20 mM MgCl2, 10 mM ATP, 2 mM ouabain, 4 mM EGTA and 10 mM sodium azide) containing no (−Ver) or 2 mM verapamil (+Ver). The fold stimulation is the ratio of the activity of the treated sample to that of an untreated sample. Each value is the average of three different experiments.
We then tested whether labelling of mutant L65C with MTS-verapamil could be inhibited by the drug substrate verapamil. The rationale for these experiments was that if residue L65C contributed to binding of MTS-verapamil, then the presence of verapamil should protect mutant L65C from being modified. We also tested whether Rhodamine B had any effect on labelling of mutant L65C by MTS-verapamil. It was shown that verapamil and Rhodamine B interact with P-gp at separate sites in the drug-binding pocket [41]. In this study [41], it was found that the ATPase activity of mutant F343C in TM6 could be permanently activated by covalent attachment of MTS-Rhodamine. While the presence of bound Rhodamine prevented further stimulation of ATPase activity by drug substrates such as colchicine or calcein-AM, the activity could still be increased in the presence of verapamil [41]. Therefore Rhodamine and verapamil can bind simultaneously to P-gp.
Accordingly, BHK cells stably expressing mutant L65C were used for these studies because they do not show any variation in P-gp expression as observed with transient expression in HEK-293 cells. A potential problem with generating stable lines by selecting under very high drug substrate concentrations is that one could inadvertently select a cell line containing a different mutation in P-gp [42]. Therefore the BHK cells were selected using a relatively low concentration of vinblastine (2 nM) which inhibited growth of non-P-gp-expressing cells.
BHK cells stably expressing His-tagged L65C were harvested, solubilized with n-dodecyl-β-D-maltoside and then incubated with saturating levels of verapamil (2 mM) or Rhodamine B (3 mM) for 10 min at 20 °C. The samples were then reacted for 10 min at 20 °C in the absence or presence of 0.1 mM MTS-verapamil, as this was the minimum concentration that gave almost complete modification of mutant L65C (Figure 2C). The His-tagged mutant was then isolated by nickel-chelate chromatography that also removed the drug substrates (verapamil or Rhodamine B) and unchanged MTS-verapamil. Immunoblot analysis of the eluted samples showed that the presence of drug substrates did not affect the yield of P-gp–His10 (results not shown). The eluted samples were mixed with lipid, sonicated and assayed for ATPase activity. Figure 4 shows that the presence of verapamil reduced the labelling efficiency of mutant L65C by MTS-verapamil by more than 80%. In contrast, the presence of Rhodamine B during labelling with MTS-verapamil caused very little (<10%) reduction in labelling of mutant L65C (Figure 4). These results are consistent with the finding that verapamil and Rhodamine bind in different areas of the drug-binding pocket and that covalent attachment of MTS-verapamil to Cys65 leads to permanent occupation of the verapamil-binding site.
Figure 4. MTS-verapamil labelling of mutant L65C is inhibited by verapamil.
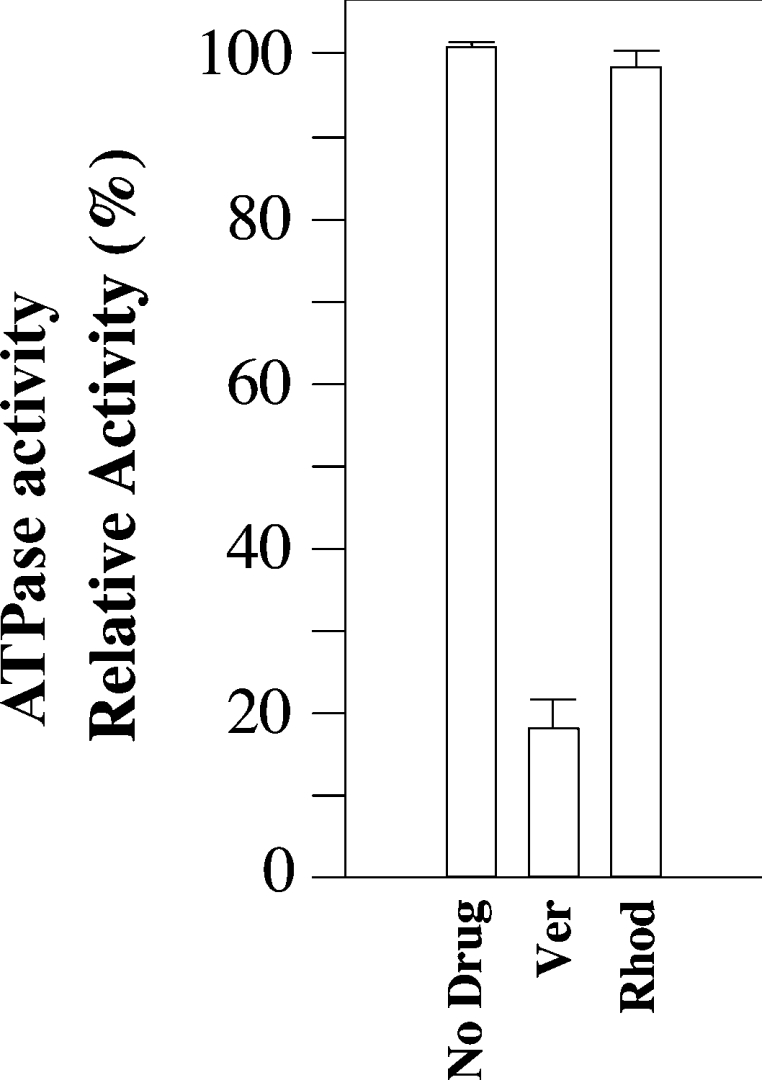
BHK cells stably expressing His-tagged mutant L65C were solubilized with n-dodecyl-β-D-maltoside. Insoluble material was removed by centrifugation. Equivalent amounts of supernatant were incubated for 10 min at 20 °C in the presence of no drug (No Drug), 2 mM verapamil (Ver) or 3 mM Rhodamine B (Rhod). The samples were then incubated with 0.1 mM MTS-verapamil for 10 min at 20 °C. His-tagged P-gps were isolated by nickel-chelate chromatography. Equivalent amounts of the isolated P-gps were mixed with lipid and assayed for ATP activity in the absence of drug substrate. The activities are expressed relative to a sample treated with MTS-verapamil in the absence of drug substrate. Each value is the average of three different experiments.
We then tested whether other drug substrates could still interact with mutant L65C that had been covalently labelled with MTS-verapamil. The rationale was that substrates will not affect the ATPase activity of MTS-verapamil-modified mutant L65C if they occupy the same binding site as verapamil or if their binding site significantly overlaps that of verapamil, but will further stimulate or inhibit the activity if their binding sites are different from that of verapamil. The drug substrates calcein-AM, demecolcine, cyclosporin A and trans-(E)-flupentixol were chosen for the present study because calcein-AM and demecolcine are more potent stimulators of P-gp ATPase activity than verapamil, whereas cyclosporin A and trans-(E)-flupentixol are potent inhibitors of activity [24]. BHK cells stably expressing His-tagged mutant L65C were solubilized with n-dodecyl-β-D-maltoside and then treated with or without 0.3 mM MTS-verapamil. His-tagged P-gp was then isolated by nickel-chelate chromatography, mixed with lipids from E. coli, sonicated and assayed for ATPase activity in the presence of saturating concentrations of calcein-AM (0.6 mM), demecolcine (3 mM), verapamil (1 mM), cyclosporin A (0.15 mM) or trans-(E)-flupentixol (0.6 mM). The composition of lipid surrounding P-gp has a great effect on the ATPase activity of P-gp [43]. The presence of sheep brain lipid gives relatively low levels of basal P-gp ATPase activity and high drug-stimulated ATPase activity when compared with activity in the presence of E. coli lipids. We used E. coli lipids because the higher basal ATPase activity made it easier for us to detect for inhibition of activity. Figure 5(A) shows that in unmodified mutant L65C, verapamil stimulated the ATPase activity 4.6-fold, whereas calcein-AM and demecolcine stimulated the ATPase activity 7.0- and 5.9-fold respectively. In contrast, cyclosporin A and trans-(E)-flupentixol inhibited the basal activity 0.85- and 0.7-fold respectively. When mutant L65C was modified with MTS-verapamil however, its basal activity was increased 4.6-fold compared with an untreated sample (Figure 5B). The activity of the modified mutant was not markedly changed by the presence of the calcein-AM, demecolcine or trans-(E)-flupentixol. The presence of cyclosporin A, however, reduced the ATPase activity of the modified mutant to basal levels. These results suggest that modification of mutant L65C by MTS-verapamil blocks interaction of P-gp with drug substrates calcein-AM, demecolcine and trans-(E)-flupentixol, but not its interaction with cyclosporin.
Figure 5. Effect of drug substrates and inhibitors on the ATPase activity of mutant L65C before and after labelling with MTS-verapamil.

BHK cells stably expressing His-tagged mutant L65C were solubilized with n-dodecyl-β-D-maltoside and then incubated in the absence (Untreated; A) or presence (+MTS-Verapamil; B) of 0.3 mM MTS-verapamil. P-gp was then isolated by nickel-chelate chromatography, mixed with E. coli lipids and ATPase activity was measured in the presence of no drug (No Drug), 0.6 mM calcein-AM (CAM), 3 mM demecolcine (Deme), 1 mM verapamil (Ver), 0.15 mM cyclosporin A (Cyclo) or 0.6 mM trans-(E)-flupentixol (T-Flu). The fold stimulation is the ratio of the activity with drug substrate to that without drug substrate. Each value is the average of triplicate determinations.
The characteristics of the mutant L65C after labelling with MTS-verapamil suggested that Cys65 lined the drug-binding pocket. Another method to test for this possibility is to test whether Cys65 can be cross-linked to cysteine residues in the TMs of TMD2 that were previously shown to be involved in drug binding [15,17]. Cross-linking between cysteines in TMD1 and TMD2 can be detected because the cross-linked P-gp migrates with lower mobility in SDS/polyacrylamide gels [25]. Accordingly, we constructed a series of double cysteine mutants that contained Cys65 in TMD1 and another cysteine in TMD2 [F942C(TM11), T945C(TM11), L975C(TM12), V981C(TM12), V982C(TM12), G984C(TM12) or A985C(TM12)] predicted to line the drug-binding pocket [34]. The mutants were transiently expressed in HEK-293 cells. Membranes were prepared and cross-linked with or without a zero-length cross-linker (1 mM copper phenanthroline) or with 0.2 mM of the homobifunctional MTS cross-linkers, M4M, M8M, M11M or M17M containing spacer arms of various lengths [16], for various times at 4 °C. Cross-linking was done at 4 °C to minimize thermal motion in the protein. The reactions were stopped by addition of SDS sample buffer containing 50 mM EDTA and no reducing agent and the samples were subjected to immunoblot analysis. Cross-linked product was not detected with copper phenanthroline (results not shown). Cross-linking, however, was observed in mutants L65C(TM1)/T945C(TM11), L65C(TM1)/V982C(TM12), L65C(TM1)/G984C(TM12) and L65C(TM1)/A985C(TM12) with the M8M, M11M and M17M cross-linkers. Figure 6(A) shows the results obtained with the M11M cross-linker. No cross-linked product was detected in the mutants L65C(TM1) (Figure 6A) or V982C(TM12) (Figure 6B) that were treated with the M11M cross-linkers. Similarly, no cross-linked products were detected when mutants T945C(TM11), G984C(TM12) and A985C(TM12) were treated with M11M (results not shown). Cross-linking with M11M was time-dependent as shown in mutant L65C(TM1)/V982C(TM12) (Figure 6B). Cross-linked product was detected within 4–8 min after addition of M11M cross-linker. These results are consistent with the prediction that Cys65 lines the drug-binding pocket.
Figure 6. Disulphide cross-linking of P-gp mutants.

(A) Membranes were prepared from HEK-293 cells (A) expressing mutants L65C, L65C/T945C, L65C/V982C, L65C/G984C or L65C/A985C. The membranes were then treated with 0.2 mM of the homobifunctional disulphide cross-linker, M11M, for 15 min at 4 °C. The reactions were stopped by addition of SDS sample buffer containing EDTA and subjected to immunoblot analysis on SDS/7.5% polyacrylamide gels. (B) Membranes prepared from HEK-293 cells expressing mutants L65C, V982C or L65C/V982C were treated with 0.2 mM M11M for various times at 4 °C. The reactions were stopped by addition of SDS sample buffer containing EDTA and subjected to immunoblot analysis on SDS/7.5% polyacrylamide gels. The positions of the cross-linked (X-link) and mature (170 kDa) P-gps are indicated.
We previously found that labelling of Ile306 (TM5) with MTS-verapamil also caused permanent activation of P-gp ATPase activity [40]. Therefore we tested whether it was possible to further stimulate P-gp ATPase activity by labelling the double cysteine mutant, L65C(TM1)/I306C(TM5). Accordingly, His-tagged mutant L65(TM1)/I306C(TM5) was expressed in HEK-293 cells. The cells were solubilized with n-dodecyl-β-D-maltoside and insoluble material was removed by centrifugation. Equivalent amounts of the solubilized material were treated with or without 1 mM MTS-verapamil at 20 °C for 10 min and P-gp was isolated by nickel-chelate chromatography. The isolated P-gps were mixed with lipid, sonicated and ATPase activity was determined. Mutant L65C(TM1)/I306C(TM5) not treated with MTS-verapamil showed an 8.8-fold stimulation of ATPase activity with verapamil. The mutant showed a small (1.5–2-fold) reduction in apparent affinity for verapamil (S50; 19.7±1.4 μM), vinblastine (2.9±0.2 μM) and colchicine (980±61 μM) compared with Cys-less P-gp (Table 1). Mutant L65C(TM1)/I306C(TM5) showed only a 2.8-fold increase in activity after treatment with MTS-verapamil (Figure 7), whereas mutant L65C showed >10-fold increase in activity (Figure 2B). Apparently, labelling of both Cys65 and Cys306 with MTS-verapamil reduced its activity when compared with labelling at one position. Labelling of mutant L65C(TM1)/I306C(TM5) with MTS-verapamil did not permanently disrupt the mutant P-gp because treatment of the MTS-treated L65C(TM1)/I306C(TM5) mutant with DTT resulted in a 7.8-fold increase in the verapamil-stimulated ATPase activity (Figure 7).
Figure 7. Effect of MTS-verapamil labelling of mutant L65C(TM1)/I306C(TM5).
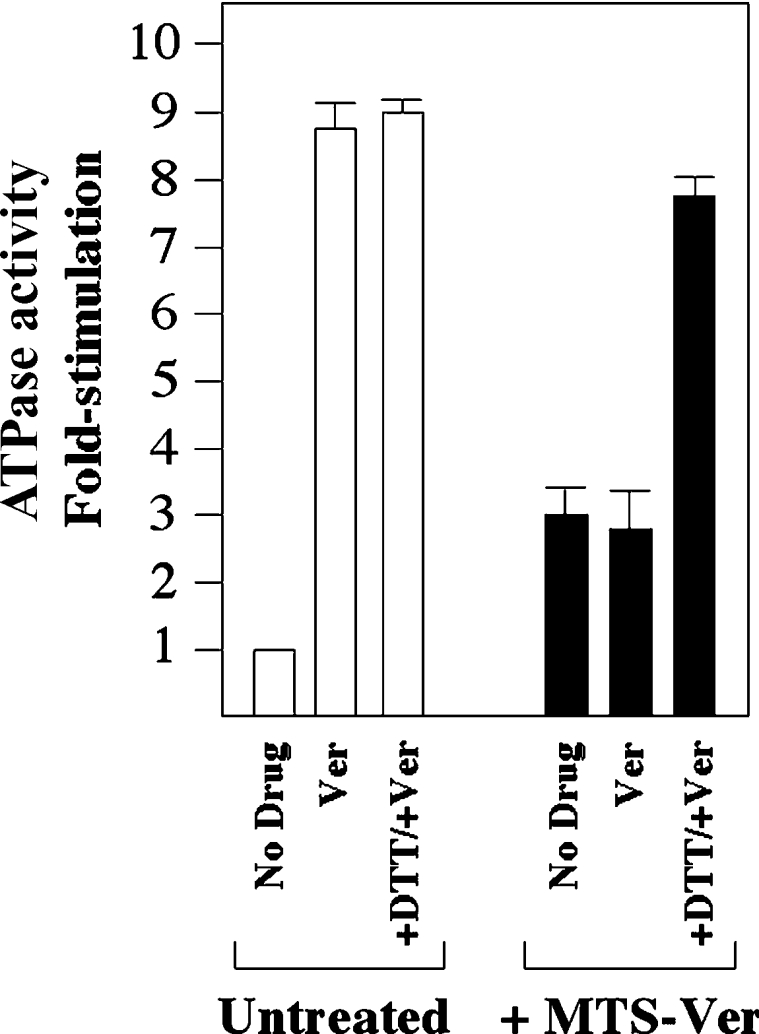
His-tagged mutant L65C(TM1)/I306C(TM5) was expressed in HEK-293 cells and solubilized with n-dodecyl-β-D-maltoside. Insoluble material was removed by centrifugation and equivalent amounts of supernatant were treated with (+MTS-Ver) or without (Untreated) 1 mM MTS-verapamil. P-gp was isolated by nickel-chelate chromatography, mixed with lipid and ATPase activity was determined in the presence or absence of 1 mM verapamil (Ver) or 10 mM DTT. The fold stimulation for the sample not treated with MTS-verapamil is the ratio of the activity in the presence of verapamil to that in the absence of verapamil. The fold stimulation of the MTS-verapamil-treated sample is the ratio of the activity to that of the activity of the untreated sample in the absence of verapamil.
DISCUSSION
Only one mutant in TM1, L65C, was able to adopt a conformation that permanently hydrolysed ATP after modification with MTS-verapamil. The increase in activity was due to disulphide linkage of verapamil to P-gp because reduction of the modified protein with DTT returned the activity of the protein to basal levels. Attachment of MTS-verapamil to Cys65 seems to mimic interaction of P-gp with verapamil because the ATPase activity of the MTS-verapamil-treated mutant L65C was very similar to that of untreated mutant L65C in the presence of saturating levels of verapamil. The results from the present study show that TM1 forms part of the drug-binding pocket.
There is supporting evidence that residues in TM1 are important for drug binding and for coupling ATPase activity to drug efflux. Mutational analysis of residues in TM1 showed that changes of His61, Gly64 and Leu65 to larger amino acids such as arginine altered the substrate specificity of human P-gp [44,45]. In all three cases, mutation to arginine increased the ability of the mutant P-gps to confer resistance to colchicine, but reduced their abilities to confer resistance to vinblastine [45]. Smaller changes in substrate specificity were observed when these residues were changed to smaller amino acids such as alanine or cysteine [44]. The authors of these studies then suggested that His61, Gly64 and Leu65 formed part of the recognition site for substrates of P-gp. Our results are consistent with these early studies. We also found that mutation of either His61 or Gly64 to cysteine altered the apparent affinity of P-gp for verapamil, colchicine or vinblastine (Table 1). Arrangement of the residues in TM1 in a cylindrical helix (Figure 8A) shows that residues that react with MTS-verapamil (L65C; the present study) show alterations in substrate specificity when mutated (H61C, G644C and L65C; [44,45]) or show ATP-dependent cross-linking (M68 and M69; [30]) and occupy one face of the helix. Similarly, mutational analysis of the CFTR (cystic fibrosis transmembrane conductance regulator) protein that functions as a chloride channel also implicated TM1 in forming part of the channel [46–48].
Figure 8. Arrangement of TM1 residues in a cylindrical helix and MTS-verapamil in the drug-binding pocket.

(A) The residues of TM1 are arranged in a cylindrical helix. Residues that show alterations in substrate specificity when mutated (H61C, G644C and L65C, circled) or show ATP-dependent cross-linking (Met68 and Met69, boxed) with residues in TM11 are shown as occupying one face of the helix. Residue Cys65 forms a covalent bond with MTS-verapamil and is shaded black. (B) Schematic diagram of the TM segments surrounding the drug-binding pocket. MTS-verapamil (ball and stick diagram) is bound to Cys306 in TM5 (upper panel) and to Cys65 in TM1 (lower panel) in different orientations.
The face of the TM1 helix occupied by Leu65 has also been implicated in P-gp conformational changes associated with ATP hydrolysis [30]. It was shown that cysteine residues introduced at positions 68 or 69 of TM1 could be cross-linked with oxidant to specific cysteine residues in TM11 during ATP hydrolysis but not in the presence of p[NH]ppA (adenosine 5′-[β,γ-imido]triphosphate). Since the present study has shown that L65C is involved in binding of verapamil, then cross-linking studies between TM1 and TM11 during ATP hydrolysis [30] would indicate that conformational changes in TM1 and TM11 probably contribute to the release of drug substrate during ATP hydrolysis. This may explain why drug substrate did not inhibit cross-linking between TM1 and TM11 during ATP hydrolysis. It is likely that the cross-linked products between TM1 and TM11 during ATP hydrolysis reflect a P-gp conformation in which the substrate has left the drug-binding pocket. Therefore TM1 appears to play a critical role in drug transport because of its importance in both drug binding and release of drug substrate during the transport cycle.
An important implication of the results of the present study is that verapamil can be covalently linked to cysteine residues in two different TMs [Cys65 in TM1 (the present study) and Cys306 in TM5 [40]] and still cause permanent activation of ATPase activity. Labelling of both sites was protected by verapamil. Since MTS-verapamil contains a rather short spacer arm of two carbons and a sulphur atom (Figure 2A), it is unlikely that verapamil attached to Cys65 in TM1 occupies the identical site in the drug-binding pocket as verapamil that is attached to Cys306 in TM6. This is supported by the findings that Cys65 and Cys306 that have been modified with MTS-verapamil have slightly different characteristics. While the ATPase activities of both mutants L65C and I306C modified by MTS-verapamil could not be further stimulated by calcein-AM or demecolcine, the inhibition of their activities were different. The ATPase activity of mutant I306C modified by MTS-verapamil could not be inhibited by cyclosporin A or trans-(E)-flupentixol [40], whereas that of MTS-verapamil-modified mutant L65C was inhibited only by cyclosporin A (Figure 5). This difference in sensitivity to inhibition by cyclosporin A suggests that both verapamil and cyclosporin A could bind simultaneously to mutant L65C, whereas covalent binding of verapamil to Cys306 prevents cyclosporin A from occupying its normal binding site. Therefore the bound verapamil molecules in mutants L65C(TM1) and I306C(TM5) have different orientations (Figure 8B), yet both are capable of permanently stimulating ATPase activity. It should be noted that not all orientations of covalently bound verapamil are capable of stimulating ATPase activity. In an earlier study, it was shown that modification of 15 other cysteine residues in the TMs by MTS-verapamil inhibited the ATPase activity of the enzyme [15].
The ability of a compound to bind to a protein in multiple orientations has also been observed in the human nuclear xenobiotic receptor protein, PXR (pregnane X receptor) [49]. The crystal structure of PXR complexed with ligand showed that the ligand could assume multiple orientations within a large binding pocket. The results from our studies suggest that the drug substrate could also assume multiple orientations in the drug-binding pocket. The ability of P-gp to accommodate verapamil molecules in different orientations may explain why R- and S-stereoisomers of verapamil show similar abilities to stimulate P-gp ATPase activity [50].
Labelling of mutant L65C (TM1) (the present study) or Ile306 (TM5) [40] with MTS-verapamil stimulated the ATPase activity of P-gp by more than 8–10-fold but labelling of the double cysteine mutant L65C(TM1)/I306C(TM5) with MTS-verapamil reduced the verapamil-stimulated ATPase activity of the enzyme (Figure 7). The presence of two verapamil molecules in the drug-binding pocket may inhibit conformational changes associated with ATP hydrolysis. Recently, it was shown that ATP hydrolysis promotes interactions between the extracellular ends of TM1 and TM11 [30]. Therefore the presence of a permanently bound verapamil molecule at I306C may interfere with movement between TM1 and TM11 during ATP hydrolysis (Figure 1B), resulting in inhibition of ATPase activity. The decrease in ATPase activity of mutant L65C(TM1)/I306C(TM5) after treatment with MTS-verapamil may explain why relatively high concentrations of verapamil (>1 mM) begin to inhibit ATPase activity. The presence of more than one verapamil molecule within the drug-binding pocket may start to jam the ‘scissor-like’ movement between the N- and C-terminal TM segments during the drug transport cycle [30].
In summary, the present study has shown that TM1 can contribute to the drug-binding pocket and that drug substrates can activate P-gp even when bound in different orientations.
Acknowledgments
This work was supported by grants from the National Cancer Institute of Canada through the Canadian Cancer Society and from the Canadian Institutes of Health Research. D.M.C. is the recipient of the Canada Research Chair in Membrane Biology.
References
- 1.Ambudkar S. V., Dey S., Hrycyna C. A., Ramachandra M., Pastan I., Gottesman M. M. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 1999;39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- 2.Schinkel A. H., Smit J. J., van Tellingen O., Beijnen J. H., Wagenaar E., van Deemter L., Mol C. A., van der Valk M. A., Robanus-Maandag E. C., te Riele H. P., et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell. 1994;77:491–502. doi: 10.1016/0092-8674(94)90212-7. [DOI] [PubMed] [Google Scholar]
- 3.Thiebaut F., Tsuruo T., Hamada H., Gottesman M. M., Pastan I., Willingham M. C. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc. Natl. Acad. Sci. U.S.A. 1987;84:7735–7738. doi: 10.1073/pnas.84.21.7735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee C. G., Gottesman M. M., Cardarelli C. O., Ramachandra M., Jeang K. T., Ambudkar S. V., Pastan I., Dey S. HIV-1 protease inhibitors are substrates for the MDR1 multidrug transporter. Biochemistry. 1998;37:3594–3601. doi: 10.1021/bi972709x. [DOI] [PubMed] [Google Scholar]
- 5.Dean M., Rzhetsky A., Allikmets R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001;11:1156–1166. doi: 10.1101/gr.184901. [DOI] [PubMed] [Google Scholar]
- 6.Chen C. J., Chin J. E., Ueda K., Clark D. P., Pastan I., Gottesman M. M., Roninson I. B. Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell. 1986;47:381–389. doi: 10.1016/0092-8674(86)90595-7. [DOI] [PubMed] [Google Scholar]
- 7.Loo T. W., Clarke D. M. Membrane topology of a cysteine-less mutant of human P-glycoprotein. J. Biol. Chem. 1995;270:843–848. doi: 10.1074/jbc.270.2.843. [DOI] [PubMed] [Google Scholar]
- 8.Loo T. W., Clarke D. M. The minimum functional unit of human P-glycoprotein appears to be a monomer. J. Biol. Chem. 1996;271:27488–27492. doi: 10.1074/jbc.271.44.27488. [DOI] [PubMed] [Google Scholar]
- 9.Loo T. W., Clarke D. M. The transmembrane domains of the human multidrug resistance P-glycoprotein are sufficient to mediate drug binding and trafficking to the cell surface. J. Biol. Chem. 1999;274:24759–24765. doi: 10.1074/jbc.274.35.24759. [DOI] [PubMed] [Google Scholar]
- 10.Loo T. W., Clarke D. M. Drug-stimulated ATPase activity of human P-glycoprotein is blocked by disulfide cross-linking between the nucleotide-binding sites. J. Biol. Chem. 2000;275:19435–19438. doi: 10.1074/jbc.C000222200. [DOI] [PubMed] [Google Scholar]
- 11.Loo T. W., Bartlett M. C., Clarke D. M. The ‘LSGGQ’ motif in each nucleotide-binding domain of human P-glycoprotein is adjacent to the opposing walker A sequence. J. Biol. Chem. 2002;277:41303–41306. doi: 10.1074/jbc.C200484200. [DOI] [PubMed] [Google Scholar]
- 12.Loo T. W., Clarke D. M. Covalent modification of human P-glycoprotein mutants containing a single cysteine in either nucleotide-binding fold abolishes drug- stimulated ATPase activity. J. Biol. Chem. 1995;270:22957–22961. doi: 10.1074/jbc.270.39.22957. [DOI] [PubMed] [Google Scholar]
- 13.Urbatsch I. L., Sankaran B., Bhagat S., Senior A. E. Both P-glycoprotein nucleotide-binding sites are catalytically active. J. Biol. Chem. 1995;270:26956–26961. doi: 10.1074/jbc.270.45.26956. [DOI] [PubMed] [Google Scholar]
- 14.Loo T. W., Clarke D. M. Identification of residues in the drug-binding site of human P-glycoprotein using a thiol-reactive substrate. J. Biol. Chem. 1997;272:31945–31948. doi: 10.1074/jbc.272.51.31945. [DOI] [PubMed] [Google Scholar]
- 15.Loo T. W., Clarke D. M. Defining the drug-binding site in the human multidrug resistance P-glycoprotein using MTS-verapamil. J. Biol. Chem. 2001;276:14972–14979. doi: 10.1074/jbc.M100407200. [DOI] [PubMed] [Google Scholar]
- 16.Loo T. W., Clarke D. M. Determining the dimensions of the drug-binding domain of human P-glycoprotein using thiol cross-linkers as molecular rulers. J. Biol. Chem. 2001;276:36877–36880. doi: 10.1074/jbc.C100467200. [DOI] [PubMed] [Google Scholar]
- 17.Loo T. W., Clarke D. M. Location of the rhodamine-binding site in the human multidrug resistance P-glycoprotein. J. Biol. Chem. 2002;277:44332–44338. doi: 10.1074/jbc.M208433200. [DOI] [PubMed] [Google Scholar]
- 18.Pleban K., Kopp S., Csaszar E., Peer M., Hrebicek T., Rizzi A., Ecker G. F., Chiba P. P-glycoprotein substrate binding domains are located at the transmembrane domain/transmembrane domain interfaces: a combined photoaffinity labeling–protein homology modeling approach. Mol. Pharmacol. 2005;67:365–374. doi: 10.1124/mol.104.006973. [DOI] [PubMed] [Google Scholar]
- 19.Dey S., Ramachandra M., Pastan I., Gottesman M. M., Ambudkar S. V. Evidence for two nonidentical drug-interaction sites in the human P-glycoprotein. Proc. Natl. Acad. Sci. U.S.A. 1997;94:10594–10599. doi: 10.1073/pnas.94.20.10594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pascaud C., Garrigos M., Orlowski S. Multidrug resistance transporter P-glycoprotein has distinct but interacting binding sites for cytotoxic drugs and reversing agents. Biochem. J. 1998;333:351–358. doi: 10.1042/bj3330351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shapiro A. B., Fox K., Lam P., Ling V. Stimulation of P-glycoprotein-mediated drug transport by prazosin and progesterone. Evidence for a third drug-binding site. Eur. J. Biochem. 1999;259:841–850. doi: 10.1046/j.1432-1327.1999.00098.x. [DOI] [PubMed] [Google Scholar]
- 22.Lugo M. R., Sharom F. J. Interaction of LDS-751 with P-glycoprotein and mapping of the location of the R drug binding site. Biochemistry. 2005;44:643–655. doi: 10.1021/bi0485326. [DOI] [PubMed] [Google Scholar]
- 23.Loo T. W., Bartlett M. C., Clarke D. M. Substrate-induced conformational changes in the transmembrane segments of human P-glycoprotein. Direct evidence for the substrate-induced fit mechanism for drug binding. J. Biol. Chem. 2003;278:13603–13606. doi: 10.1074/jbc.C300073200. [DOI] [PubMed] [Google Scholar]
- 24.Loo T. W., Bartlett M. C., Clarke D. M. Drug binding in human P-glycoprotein causes conformational changes in both nucleotide-binding domains. J. Biol. Chem. 2003;278:1575–1578. doi: 10.1074/jbc.M211307200. [DOI] [PubMed] [Google Scholar]
- 25.Loo T. W., Clarke D. M. The packing of the transmembrane segments of human multidrug resistance P-glycoprotein is revealed by disulfide cross-linking analysis. J. Biol. Chem. 2000;275:5253–5256. doi: 10.1074/jbc.275.8.5253. [DOI] [PubMed] [Google Scholar]
- 26.Loo T. W., Bartlett M. C., Clarke D. M. Disulfide cross-linking analysis shows that transmembrane segments 5 and 8 of human P-glycoprotein are close together on the cytoplasmic side of the membrane. J. Biol. Chem. 2004;279:7692–7697. doi: 10.1074/jbc.M311825200. [DOI] [PubMed] [Google Scholar]
- 27.Loo T. W., Bartlett M. C., Clarke D. M. Residues V133 and C137 in transmembrane segment 2 are close to residues A935 and G939 in transmembrane segment 11 of human P-glycoprotein. J. Biol. Chem. 2004;279:18232–18238. doi: 10.1074/jbc.M400229200. [DOI] [PubMed] [Google Scholar]
- 28.Chang G. Structure of MsbA from Vibrio cholera: a multidrug resistance ABC transporter homolog in a closed conformation. J. Mol. Biol. 2003;330:419–430. doi: 10.1016/s0022-2836(03)00587-4. [DOI] [PubMed] [Google Scholar]
- 29.Loo T. W., Clarke D. M. Quality control by proteases in the endoplasmic reticulum. Removal of a protease-sensitive site enhances expression of human P-glycoprotein. J. Biol. Chem. 1998;273:32373–32376. doi: 10.1074/jbc.273.49.32373. [DOI] [PubMed] [Google Scholar]
- 30.Loo T. W., Bartlett M. C., Clarke D. M. ATP hydrolysis promotes interactions between the extracellular ends of transmembrane segments 1 and 11 of human multidrug resistance P-glycoprotein. Biochemistry. 2005;44:10250–10258. doi: 10.1021/bi050705j. [DOI] [PubMed] [Google Scholar]
- 31.Loo T. W., Clarke D. M. Identification of residues in the drug-binding domain of human P-glycoprotein: analysis of transmembrane segment 11 by cysteine-scanning mutagenesis and inhibition by dibromobimane. J. Biol. Chem. 1999;274:35388–35392. doi: 10.1074/jbc.274.50.35388. [DOI] [PubMed] [Google Scholar]
- 32.Loo T. W., Clarke D. M. Identification of residues within the drug-binding domain of the human multidrug resistance P-glycoprotein by cysteine-scanning mutagenesis and reaction with dibromobimane. J. Biol. Chem. 2000;275:39272–39278. doi: 10.1074/jbc.M007741200. [DOI] [PubMed] [Google Scholar]
- 33.Loo T. W., Clarke D. M. Rapid purification of human P-glycoprotein mutants expressed transiently in HEK 293 cells by nickel-chelate chromatography and characterization of their drug-stimulated ATPase activities. J. Biol. Chem. 1995;270:21449–21452. doi: 10.1074/jbc.270.37.21449. [DOI] [PubMed] [Google Scholar]
- 34.Loo T. W., Clarke D. M. Do drug substrates enter the common drug-binding pocket of P-glycoprotein through ‘gates’? Biochem. Biophys. Res. Commun. 2005;329:419–422. doi: 10.1016/j.bbrc.2005.01.134. [DOI] [PubMed] [Google Scholar]
- 35.Loo T. W., Bartlett M. C., Clarke D. M. Rescue of ΔF508 and other misprocessed CFTR mutants by a novel quinazoline compound. Mol. Pharmacol. 2005;2:407–413. doi: 10.1021/mp0500521. [DOI] [PubMed] [Google Scholar]
- 36.Loo T. W., Clarke D. M. P-glycoprotein. Associations between domains and between domains and molecular chaperones. J. Biol. Chem. 1995;270:21839–21844. doi: 10.1074/jbc.270.37.21839. [DOI] [PubMed] [Google Scholar]
- 37.Loo T. W., Clarke D. M. Correction of defective protein kinesis of human P-glycoprotein mutants by substrates and modulators. J. Biol. Chem. 1997;272:709–712. doi: 10.1074/jbc.272.2.709. [DOI] [PubMed] [Google Scholar]
- 38.Loo T. W., Clarke D. M. Cross-linking of human multidrug resistance P-glycoprotein by the substrate, Tris-(2-maleimidoethyl)amine, is altered by ATP hydrolysis: evidence for rotation of a transmembrane helix. J. Biol. Chem. 2001;276:31800–31805. doi: 10.1074/jbc.M103498200. [DOI] [PubMed] [Google Scholar]
- 39.Chifflet S., Torriglia A., Chiesa R., Tolosa S. A method for the determination of inorganic phosphate in the presence of labile organic phosphate and high concentrations of protein: application to lens ATPases. Anal. Biochem. 1988;168:1–4. doi: 10.1016/0003-2697(88)90002-4. [DOI] [PubMed] [Google Scholar]
- 40.Loo T. W., Bartlett M. C., Clarke D. M. Permanent activation of the human P-glycoprotein by covalent modification of a residue in the drug-binding site. J. Biol. Chem. 2003;278:20449–20452. doi: 10.1074/jbc.C300154200. [DOI] [PubMed] [Google Scholar]
- 41.Loo T. W., Bartlett M. C., Clarke D. M. Methanethiosulfonate derivatives of rhodamine and verapamil activate human P-glycoprotein at different sites. J. Biol. Chem. 2003;278:50136–50141. doi: 10.1074/jbc.M310448200. [DOI] [PubMed] [Google Scholar]
- 42.Choi K. H., Chen C. J., Kriegler M., Roninson I. B. An altered pattern of cross-resistance in multidrug-resistant human cells results from spontaneous mutations in the mdr1 (P-glycoprotein) gene. Cell. 1988;53:519–529. doi: 10.1016/0092-8674(88)90568-5. [DOI] [PubMed] [Google Scholar]
- 43.Urbatsch I. L., Senior A. E. Effects of lipids on ATPase activity of purified Chinese hamster P-glycoprotein. Arch. Biochem. Biophys. 1995;316:135–140. doi: 10.1006/abbi.1995.1020. [DOI] [PubMed] [Google Scholar]
- 44.Taguchi Y., Kino K., Morishima M., Komano T., Kane S. E., Ueda K. Alteration of substrate specificity by mutations at the His61 position in predicted transmembrane domain 1 of human MDR1/P-glycoprotein. Biochemistry. 1997;36:8883–8889. doi: 10.1021/bi970553v. [DOI] [PubMed] [Google Scholar]
- 45.Taguchi Y., Morishima M., Komano T., Ueda K. Amino acid substitutions in the first transmembrane domain (TM1) of P-glycoprotein that alter substrate specificity. FEBS Lett. 1997;413:142–146. doi: 10.1016/s0014-5793(97)00899-5. [DOI] [PubMed] [Google Scholar]
- 46.Anderson M. P., Gregory R. J., Thompson S., Souza D. W., Paul S., Mulligan R. C., Smith A. E., Welsh M. J. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science. 1991;253:202–205. doi: 10.1126/science.1712984. [DOI] [PubMed] [Google Scholar]
- 47.Akabas M. H., Kaufmann C., Cook T. A., Archdeacon P. Amino acid residues lining the chloride channel of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 1994;269:14865–14868. [PubMed] [Google Scholar]
- 48.Ge N., Muise C. N., Gong X., Linsdell P. Direct comparison of the functional roles played by different transmembrane regions in the cystic fibrosis transmembrane conductance regulator chloride channel pore. J. Biol. Chem. 2004;279:55283–55289. doi: 10.1074/jbc.M411935200. [DOI] [PubMed] [Google Scholar]
- 49.Watkins R. E., Wisely G. B., Moore L. B., Collins J. L., Lambert M. H., Williams S. P., Willson T. M., Kliewer S. A., Redinbo M. R. The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science. 2001;292:2329–2333. doi: 10.1126/science.1060762. [DOI] [PubMed] [Google Scholar]
- 50.Loo T. W., Bartlett M. C., Clarke D. M. Simultaneous binding of two different drugs in the binding pocket of the human multidrug resistance P-glycoprotein. J. Biol. Chem. 2003;278:39706–39710. doi: 10.1074/jbc.M308559200. [DOI] [PubMed] [Google Scholar]