

Abstract
Antimycin A (antimycin), one of the first known and most potent inhibitors of the mitochondrial respiratory chain, binds to the quinone reduction site of the cytochrome bc1 complex. Structure-activity-relationship studies have shown that the N-formylamino-salicylamide group is responsible for most of the binding specificity, and suggested that a low pKa for the phenolic OH group and an intramolecular H-bond between that OH and the carbonyl O of the salicylamide linkage are important.
Two previous X-ray structures of antimycin bound to vertebrate bc1 complex gave conflicting results. A new structure reported here of the bovine mitochondrial bc1 complex at 2.28 Å resolution with antimycin bound, allows us for the first time to reliably describe the binding of antimycin and shows that the intramolecular hydrogen bond described in solution and in the small-molecule structure is replaced by one involving the NH rather than carbonyl O of the amide linkage, with rotation of the amide group relative to the aromatic ring. The phenolic OH and formylamino N form H-bonds with conserved Asp228 of cyt b, and the formylamino O H-bonds via a water molecule to Lys227. A strong density the right size and shape for a diatomic molecule is found between the other side of the dilactone ring and the αA helix.
Keywords: Keywords: cytochrome bc1, antimycin, respiratory chain, membrane protein complex, inhibitor binding site
Introduction
The cytochrome bc1 complex is an enzyme (E.C. 1.10.2.2, ubiquinol:cytochrome c oxidoreductase) that comprises the middle part of the mitochondrial respiratory chain. It is a multi-subunit membrane protein, with 10 or 11 protein chains in mitochondrial forms and 3 or more in bacterial complexes. It always contains the three redox subunits cytochrome b, cytochrome c1, and the iron-sulfur protein (ISP); which contain respectively two hemes B, heme C and a Rieske-type Fe2S2 iron-sulfur cluster. It catalyzes reversible electron transfer from ubiquinol to cytochrome c coupled to proton translocation across the inner mitochondrial membrane, probably by a mechanism like the “protonmotive Q cycle”1; 2; 3; 4.
The existence of inhibitors specifically binding to and inhibiting the two ubiquinone reaction sites was critical in the elucidation of the Q-cycle mechanism. One of the earliest known (for a review of early work see ref.5) and most potent (with a KD on the order of 30 pM5) of these is antimycin A (antimycin)b. Antimycin binds specifically to the quinone reduction site (Qi site) of the cytochrome bc1 complex. When bound, the UV-visible spectrum of the high-potential cytochrome b is shifted to the red and the fluorescence of antimycin is quenched, leading to the conclusion6 that antimycin binds near the heme bH.
Together with British Anti-Lewisite (BAL) or alkyl-hydroxynapthoquinone (HNQ), antimycin enabled the demonstration of two independent pathways for reduction of cytochrome b by ubiquinol, one sensitive to antimycin and the other blocked by BAL treatment or HNQ, in the “double-kill” experiment7.
When antimycin is bound, the bc1 complex exhibits an unexpected inverse relation between the redox poise of the high potential chain (iron-sulfur protein and cytochrome c1) and that of the b cytochromes, resulting in phenomena coined “oxidant-induced”8 and “reductant-controlled”9 reduction of b cytochromes. This is explained in the Q-cycle mechanism (Scheme 1) and in an earlier model10 by having the b cytochromes and the high-potential chain connected to each of two sequential one-electron steps in the oxidation of quinol at the antimycin insensitive (Qo) site. In the Q-cycle scheme antimycin blocks the reaction at the Qi site, at which cytochrome b equilibrates directly with the ubiquinone/ubiquinol couple, masking the oxidant-induced reduction in the absence of antimycin.
Generation of a semiquinone at the Qi site is expected from the Q-cycle mechanism due to sequential one-electron reduction of quinone there by successive turnovers of the Qo-site reaction. A semiquinone signal has been observed by epr spectroscopy and it is eliminated by antimycin11; 12, consistent with the predictions of the Q-cycle scheme. Thus antimycin could be considered a marker for the Qi site of the Q-cycle mechanism.
Antimycin dramatically increases the stability of the bovine bc1 complex in the presence of bile salt detergent taurocholate13, inhibiting the “cleavage” reaction quantitatively at stoichiometric concentrations. These and other observations6; 14; 15; 16 led to the conclusion that antimycin-binding induces a far-reaching conformational change in the cytochrome bc1 complex. Dithionite reduction of the complex results in a similar protection against cleavage13, and affects the apparent cooperativity of antimycin binding6 suggesting that redox state of cytochrome b may also be coupled to the conformational change. More recent indications of an antimycin-induced conformational change include an effect of antimycin on the sensitivity of the iron-sulfur protein to proteolytic cleavage17, and an apparent effect of antimycin on “half-the sites reactivity” of the Qo site18.
One model19 for the reaction mechanism of the bc1 complex invokes conformational coupling between events at the Qi site, where antimycin binds, and the Qo site, where the bifurcated transfer of electrons from ubiquinol to cytochrome b and the iron-sulfur protein must be enforced to maintain proton-pumping efficiency. After different positions of the ISP ectodomain were observed in crystals and proposed to be involved in the catalytic cycle20, it was suggested21; 22 that such conformational coupling might prevent the ISP from returning to the Qo site until the second electron had passed to heme bH and the Qi site, ensuring bifurcation.
Despite the circumstantial evidence for a conformational change involving the Qi site, no significant conformational change in the N-side or transmembrane domains of cytochrome b has been observed in the crystallographic structures. With the initial chicken bc1 structures, we reported23 an upper limit of 1A in the absence of stigmatellin (and 0.5 Å in its presence) for the maximum difference of C-α atom positions (residues 2–380) between crystals with and without antimycin. Gao et al.24 reported rmsd 0.33 A for all but one of the 378 residues modeled; comparison of structures 1ntk and 1ntz shows the largest deviations to be 2.2Å for residue 2 and 1.4 Å for residue 267.
In potentiometric titrations it has often been observed that the cytochrome b662 (b560 in bacteria) species attributed to heme bH titrates heterogeneously25; 26; 27; 28; 29; 30, with part showing a midpoint potential (Em) around 150 mv and the rest around 50 mv (in bacterial chromatophores at pH 7). In the presence of antimycin only the low potential component is observed30; 31. In the presence of funiculosin there is only one component, with Em near that of the high potential component30; 32. If the system is poised so that bH is partly reduced, addition of antimycin results in oxidation of cytochrome b25; 26. These phenomena have been explained as due to the mechanism by which cyt bH equilibrates with the Q pool via the Qi site26; 27; 31; 33, or alternatively as due to redox interaction between the cyt b heme and quinone species at the Qi 29; 30 in which the redox state of one component affects the midpoint potential of the other. If the latter explanation is correct, the possibility that antimycin and funiculosin mimic different redox states of ubiquinone at the Qi site seems attractive.
Thus there remains a large body of experimental observation concerning antimycin and the reaction at the Qi site that is not very well explained at present. In the process of unraveling the details of the reaction, and in explaining these diverse observations, it would be very helpful to know how antimycin, funiculosin, and ubiquinone bind to the site. In addition, the Qi site of fungal and other plant pathogens is an important target for crop protection agents 34; 35. The site is of potential significance in treatment of human disease if species-specific inhibitors can be designed. Thus antimycin has been the subject of numerous structure-activity-relationship studies aimed at understanding the mechanism of the enzyme and at developing powerful new crop protection agents16; 36; 37; 38; 39; 40; 41.
Antimycin (Figure 1a) has a head group consisting of 3-formylamino salicylate, amidified to a dilactone ring consisting of L-threonine (whose amino group is amidified to the salicylate moiety) and a 2-alkyl, 3,4-dihydroxyvalerate. It is the 4-hydroxy group of the latter that participates in the dilactone, and the 3-OH is esterified by a branched carboxylic acid (acyl side chain). There is heterogeneity in the 2-alkyl group (alkyl side chain) and in the acyl side chain, which at least in antimycin A1 has recently been shown to consist mainly of 2-methyl butanoate42 rather than isovalerate (3-methyl butanoate) as deduced earlier43; 44. High resolution chromatography has resolved commercial antimycin samples into as many as ten different compounds45.
Figure 1.
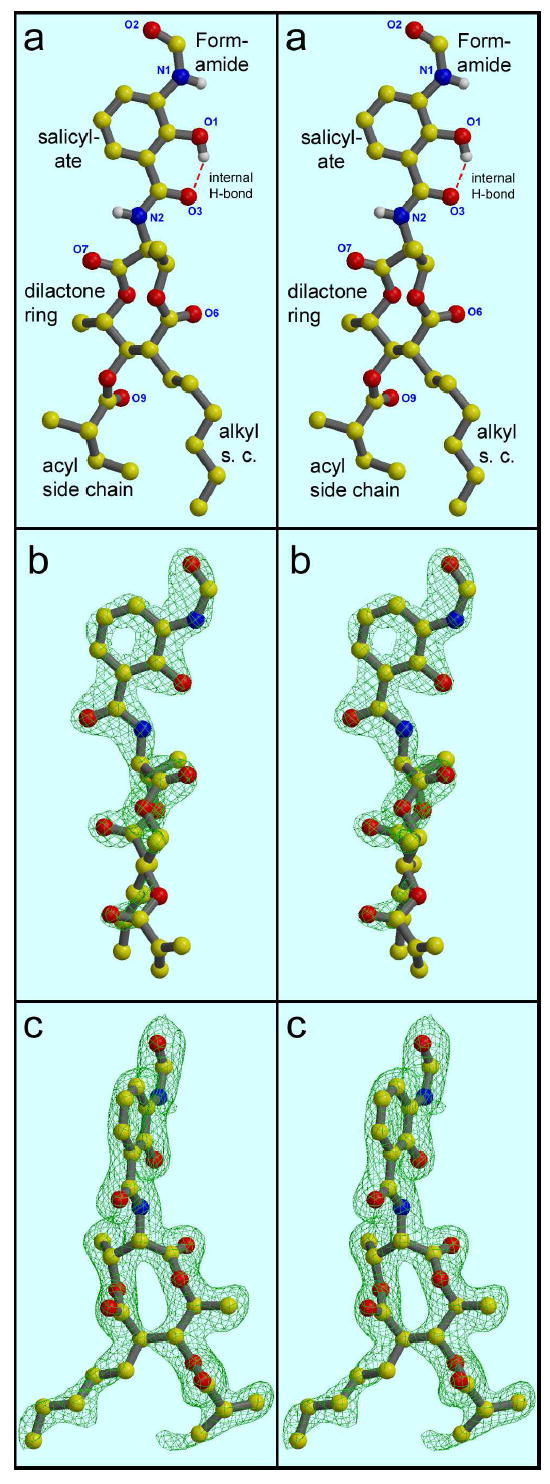
The structure of antimycin (stereo views). A, From the small-molecule crystal structure (42 ; coordinates from the Cambridge Structure Database, CCDC # 125007). Hydrogen atoms have been removed from the carbon atoms for clarity. B, from the structure 1PPJ, with the FSA ring and amide group in the plane of the picture. C, as B but rotated 75° to view the dilactone ring nearly face-on. The electron density in parts B and C is a 2Fo-Fc map contoured at 2.1 σ (B) or 0.9 σ (C) from structure 1PPJ.
Early structure-activity-relationship studies have led to the conclusions that the N-formylamino-salicyl group is responsible for most of the binding specificity, and to the importance of a low pKa for the phenolic OH group16. The dilactone ring and substituents can be replaced by a long-chain fatty amine with retention of tight (μM) binding and inhibition. More recent studies have examined stereospecificity of the dilactone37 and probed with substituents at various positions on the salicylamide group38; 40. Conclusions of the latter studies include the importance of the phenolic OH and formylamino groups and an intramolecular H-bond between the phenolic OH and the carbonyl O of the amide linkage by which the rest of the molecule is connected to the 3-formylaminosalicylic acid.
Antimycin was instrumental in locating the Qi site in the first crystal structure of a bc1 complex46, but no coordinates for antimycin were deposited in the Protein Data Bank (PDB). Since then two structures have been made available with coordinates for antimycin, PDB entries 3BCC (chicken) and 1NTK (bovine). The low resolution of the former structure made it impossible to discern details required for a rigorous description of antimycin binding. Structure 1NTK was processed at higher resolution (2.6 Å), however the work presented here shows that it, too, has errors in the details of binding.
In this work we introduce two new crystal structures of the bovine mitochondrial bc1 complex with stigmatellin at the Qo site. PDB Entry 1PP9 (2.23 Å) has no Qi-site ligand added, while 1PPJ (2.28 Å) was co-crystallized with antimycin. This allows us for the first time to reliably describe the binding mode of antimycin at the level of detail required to begin to understand its diverse effects on the bc1 complex.
Results
Resolution and quality of the structures
The diffraction was somewhat anisotropic, as judged by the “falloff” analysis in the program TRUNCATE and by anisotropic scaling during refinement in CNS which gave a B tensor with diagonal elements −15.3, 0.6, 14.7 Å2 for 1PP9 and −12.5, 3.8, 8.6 Å2 for 1PPJ. The data reduction and refinement programs we used have no provision for an ellipsoidal resolution cutoff, so to avoid losing any useful data in the well-ordered directions we used a resolution cutoff of 2.07 for 1PP9 and 2.0 for 1PPJ in the initial data reduction. In the final refinement for deposition and calculation of refinement statistics (Table 1b), a resolution limit of 2.1 Å was used for both structures. This should not be taken as the resolution of the structure, however, as the data in the outer shells was quite weak. A more objective measure of the resolution of a diffraction dataset47 is given by the “optical” resolution as calculated by the program SFCHECK48 . However the optical resolution is defined differently (how close two features can be and still be resolved by the data, rather than as a dmin cutoff), so they are not directly comparable. A sparse random survey of structures deposited with data during 2002 showed (unpublished work of E. Tung) that in the range from 1.2 to 3.0 Å the optical resolution Ropt was related to reported resolution cutoff dmin by the expression (Ropt = 0.42 + 0.59dmin). The datasets for structures 1PP9 and 1PPJ have optical resolution 1.72 and 1.75. By the above relation this is the type of resolution to be expected from the average structure using a resolution cutoff of 2.23 or 2.28 Å.
Table 1.
Statistics from the structure determination process.
| Protein Database entry: | 1PP9 | 1PPJ |
|---|---|---|
| Inhibitors co-crystallized: | stigmatellin | stigmatellin, antimycin |
| A. DATA REDUCTION | ||
| Unit Cell Dimensions | 139.12 × 171.06 × 227.20 Å | 128.53 × 168.75 × 231.53 Å |
| Solvent content | 58.2 % | 54.7 % |
| VM | 2.97 | 2.74 |
| X-ray wavelength a: | 0.99200 | 0.97977, 1.0000, 1.1000, 1.1808 |
| Unique Reflections | 312369 | 285923 |
| Resolution Range, Å a : | 50 - 2.1 (2.18 - 2.10) | 250. - 2.100 (2.15 - 2.10) |
| “Optical Resolution” b | 1.72 Å | 1.75 Å |
| Completeness : | 97.2% (83%) | 98.1% ( 94.3 %) |
| Data Redundancy: | 5.9 | 5.630 |
| R Merge on I : | 0.12 (>1.0) | 0.149 ( 0.879) |
| <I/σI> | 10.9 (1.037) | 18.6890 (2.819) |
| B. REFINEMENT: | ||
| Resolution | 24.99 - 2.10 (2.15-2.10) | 93.53 - 2.10 (2.15 - 2.10) |
| Data Cutoff (σF) | 0.0 | 0.0 |
| Completeness | 97.3 (91.9) | 97.7% ( 90.3%) |
| # Reflections | 305496 (19066) | 285060 (16565) |
| R Value | 0.250 (0.40) | 0.224 (0.33) |
| Free R Value | 0.287 (0.40) | 0.260 (0.38) |
| Number Of Atoms Used | ||
| Protein Atoms | 31493 | 31181 |
| Heterogen Atoms | 1005 | 962 |
| Solvent Atoms | 1461 | 1406 |
| B Values | ||
| From Wilson Plot | 27.3 Å2 | 33.50 Å2 |
| Mean atomic B Value | 46.9 Å2 | 50.20 Å2 |
| Anisotropic B11, B22, B33 | 15.35, -0.55, -14.81 Å2 | 12.34, -3.71, -8.63 Å2 |
| ESD,- (cross-validated)c | ||
| From Luzzati Plotc | 0.32 Å (0.39 Å) | 0.28 Å ( 0.35 Å) |
| From Sigmaac | 0.43 Å (0.47 Å) | 0.33 Å (0.38 Å) |
| Rms Deviations From Ideality | ||
| Bond Lengths | 0.007 Å | 0.006 Å |
| Bond Angles | 1.5° | 1.4° |
| Dihedral Angles | 21.8° | 21.8° |
| Improper Angles | 1.02° | 0.94° |
| C. VALIDATION: | ||
| Residues in “Most Favored” region of Ramachandran | 92.1% | 92.4% |
| Residues in Ramachandran “disallowed” region | 0.2% | 0.2% |
| Bad Contacts/100 residues | 0.5 | 0.3 |
| Overall G-factor (ProCheck): | 0.4 | 0.4 |
| Real-space R-value | 0.155 | 0.148 |
| Real-space Correlation Coeff. | 0.909 | 0.921 |
While this is only marginally higher resolution than the best yeast or bovine bc1 structures previously available, we think the quality of the structures is significantly higher. This is due to the presence of a dimer in the asymmetric unit, which for the same solvent content doubles the number of unique reflections at a given resolution. Because non-crystallographic symmetry was quite good for most of the protein, the use of NCS-restraints resulted in effectively doubling the data/parameters ratio with consequent improvement in the refinement process. In addition, while making the final model we had the benefit of using all the previously deposited structures for comparison and evaluation, which we gratefully acknowledge.
At the current state of refinement (Table 1b) the free-R factor is approximately 0.40 in the shell around 2.1 Å for 1PP9, and below 0.4 at 2.0 Å for 1PPJ, suggesting the datasets actually contain some useful information to these resolutions. Analysis of the structuresc by PROCHECK 49; 50 show them to be within the norm or better on all measures as compared to 2.0 Å structures. These structures are the first cytochrome bc1 structures to achieve greater than 90% of the residues in the allowed regions (A, B, L) of the Ramachandran plot, as expected for real proteins based on analysis of structures solved at better than 2 Å with R-factors below 20%50. Overall real-space R-factors are 0.155 and 0.148, and real-space correlation coefficients are 0.909 and 0.921 for structures 1PP9 and 1PPJ, respectively (EDS website, http://fsrv1.bmc.uu.se). Representative electron density from well-ordered regions in crystal 1PPJ are shown in stereo pairs of Figure 2 as well as in the figures documenting the mode of antimycin binding (Figures 1, 5, and 6).
Figure 2.
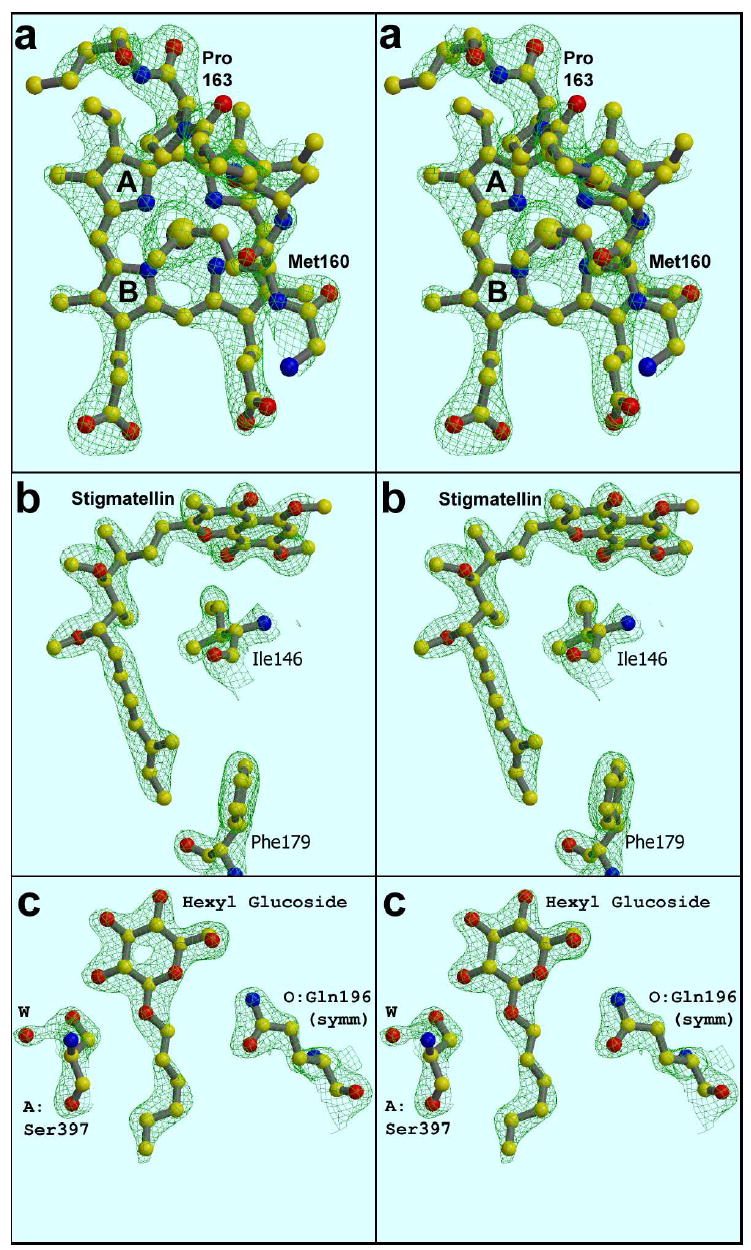
Representative density in well-ordered parts of the structure. (a). Heme-ligand met160, showing chirality about the Sδ atom. (b). Stigmatellin (c) hexyl glucoside molecule sandwiched in a crystal contact. The maps are 2Fo-Fc, calculated from data between 15 and 2.1 Å, sharpened with B −20, and contoured at 2.3 σ (a), 2.0 σ (b), or 1.8 σ (c); from structures 1PP9 (a) or 1PPJ (b), (c).
Figure 5.
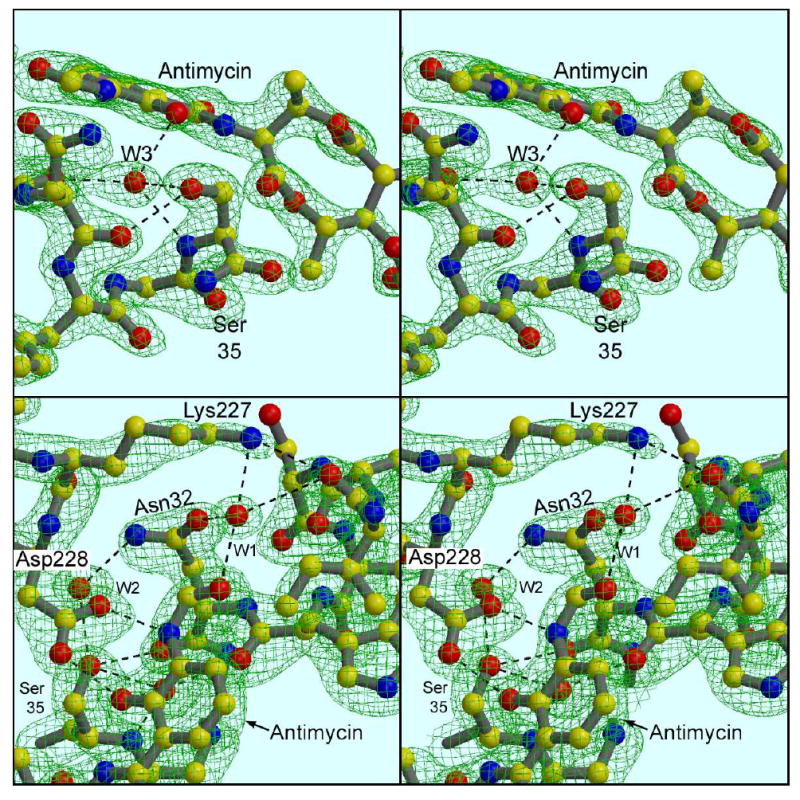
Ser35 carbonyl O faces away from antimycin, and Lys227 interacts with antimycin through a water molecule. (a) Ser35 and vicinity: a 2Fo-Fc map contoured at 1.5 σ. Asp228 has been removed for clarity, and W2 is not shown. (b) Lys227, Asp228, and vicinity: Antimycin is in the front and lower part of the figure, with its formylamino oxygen at the center. Water W1 bridges between the formylamino oxygen and Lys227Nζ at the top of the figure. W1 also makes H-bonds with Oγ1 of Ser32 (left) and the carbonyl oxygen of C27 (right). Also visible in this figure, two-point H-bonding of Asp228 to antimycin. In the background is Asn32 with H-bonds stabilizing W1 and W2, and a bond from the latter atom to Ser35. The intercalated water W3 is behind antimycin, barely visible through the salicyl ring, with H-bond to Ser35 indicated. The map is the same as in (a).
Figure 6.
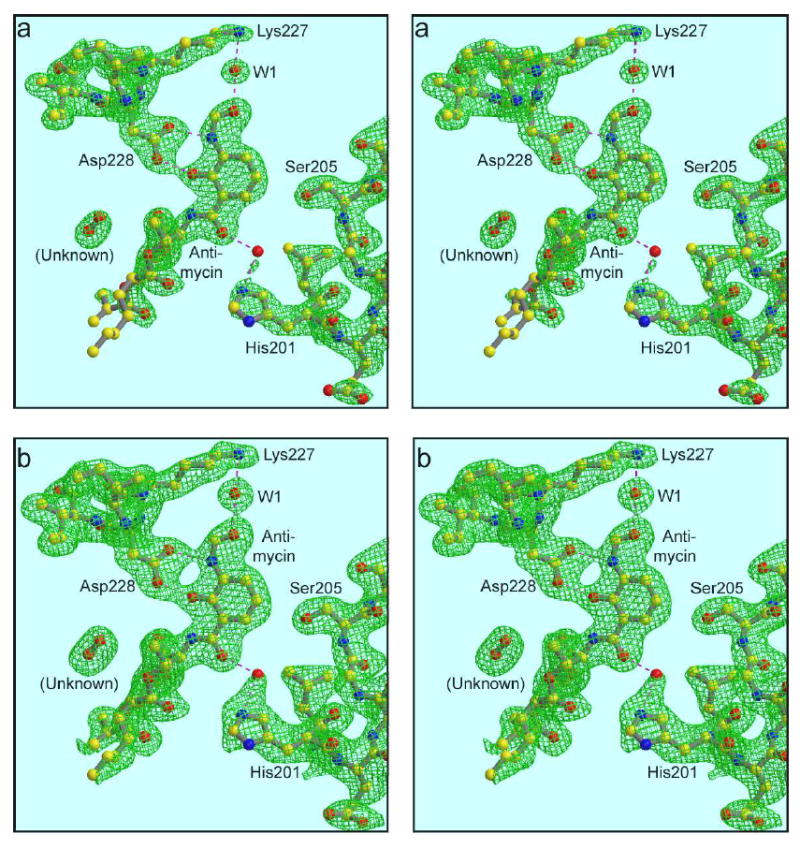
His201 and Ser205. The view is nearly the same as Figure 5b, showing antimycin, Lys227, Asp228 and water 1. Residues behind those have been removed for clarity, and the C-terminus of helix D containing His201 and Ser205 is shown. Two different density levels are used to elucidate the interaction of His201 with antimycin and the possible involvement of a water molecule. The maps are 2Fo-Fc, contoured at 1.5 σ in (a) and 1.0 σ in (b). Also shown is an unknown molecule modeled as dioxygen (see text).
Still, the current structures are disordered in a few sections, and so for some features it will be best to look at structures from other crystal forms. In such areas where the structure is not completely determined by the data, the electron density has been interpreted liberally to provide our best guess of the actual arrangement. To avoid over-interpretation of the structure and possible erroneous conclusions concerning features not described in the text, it is important to compare all the available structures, and to examine the electron density on which the feature is based. To facilitate independent evaluation of structural features by others, the original data (structure factor amplitudes) for the structures have been deposited with the Protein Data Bank.
Overall Structure
The overall structure of the eukaryotic bc1 complex has been described previously20; 46; 51; 52. The transmembraneous region is made up of 26 transmembrane helices, with each monomer contributing 13: eight from cyt b and one each from subunits 7, 10 and 11 plus the transmembrane anchor helices of the ISP and cyt c1. The redox-active ectodomains of the ISP and cyt c1 together with subunit 8 (acidic “hinge protein”) make up the membrane extrinsic portion on the external or “P” side of the membrane, while the two largest subunits (“core” proteins53) and subunit 6 make up the extrinsic part on the “N” side. Subunit 11 is peripherally bound to the transmembrane domain46 and readily dissociable after solubilization in DM. It is not present in this crystal form.
Table 2 lists the number of residues modeled for each subunit of each monomer of the two structures discussed here. It also defines the chain letters for the 10 subunits in each of two monomers: Chains A to J are subunits 1 to 10 of the “first” monomer, while N to W are the corresponding subunits in the second monomer. The hemes and iron-sulfur clusters are numbered starting at 501 in the same chain to which they are linked. Water molecules are numbered starting at 1, ligands present at the same place on both monomers are labeled starting at 2001 for monomer 1 and 3001 for monomer 2, and ligands without symmetry mates are numbered starting at 4001.
Table 2. Model completeness by subunit.
For each subunit is given the actual number of residues present in the complex based on sequence, and the number of residues modeled in each monomer of the two structures presented here. The chain letters assigned to each subunit in each monomer are also indicated. Major differences are due to the lack of subunit 11 in the crystals and disorder of the first 30 residues of subunit 10 in chain J of 1PPJ.
| modeled in structure: | ||||||
|---|---|---|---|---|---|---|
| 1PP9 | 1PPJ | |||||
| Subunit | number of residues actual | monomer #1 | monomer #2 | monomer #1 | monomer #2 | |
| 1 | “core”1 | 446 | A 442 | N442 | A 441 | N 441 |
| 2 | “core”2 | 439 | B 423 | O424 | B 424 | O 423 |
| 3 | cyt b | 379 | C365 | P 370 | C 365 | P 365 |
| 4 | cyt c1 | 241 | D241 | Q241 | D 241 | Q 241 |
| 5 | ISP | 196 | E 196 | R196 | E 196 | R 196 |
| 6 | 110 | F 99 | S 99 | F 99 | S 99 | |
| 7 | 81 | G 73 | T 74 | G 73 | T 74 | |
| 8 | “hinge” | 78 | H 66 | U 66 | H 66 | U 66 |
| 9 | signal | 78 | I 42 | V 42 | I 43 | V 43 |
| 10 | 62 | J 62 | W 62 | J 32 | W 61 | |
| 11 | 56 | K 0 | X 0 | K 0 | X 0 | |
| sum | 2166 | 2009 | 2016 | 1980 | 2009 | |
The 11-subunit bovine bc1 complex contains 2166 residues per monomer (ref 54, Table 2), and the 10-subunit preparation used here has 2110 of these. Due to omission of disordered areas, the final structures contains about 2009 residues in each monomer, or 95% of the residues present. The model is lacking the first 17 residues of subunit 2, the first 14 residues of cytochrome b, the first 11 of subunit 6, the first 12 of subunit 8, about half of subunit 9, and smaller sections elsewhere. Monomer 1 of 1PPJ has fewer residues because it is lacking the first 29 residues of subunit 10, which were disordered. Poorly ordered residues that are likely to have mistakes in the current model include the interdomain linkers of subunits 1 and 2 (A/N 223–230, B/O 230–233, ) ; E:79–80 E:178–190; and F109–110.
The entire stigmatellin molecule is well ordered in the current structures, with all but two atoms (the methoxy carbon C5A and final carbon of the tail) covered by 2Fo-Fc density at a contour level of 2.0 σ (Figure 2b). The stereochemistry of the 4 chiral centers and the planarity at the isoprenoid unit are clear, and are consistent with what is known from chemical investigations55.
Modeling of the lipids and detergents in these structures is not yet complete, and will be described in a later paper. At present there are five phospholipids in 1PP9 and four in 1PPJ. One of the best ordered (residues 2007 and 3007; with phosphate H-bonding Tyr103 and Tyr104 of cyt b) is in the position of one of the lipids in the chicken bc1 structures (e.g. 2BCC) and is also conserved in the yeast bc1 complex (1KB9, 1P84). Phospholipids 2006 and 3006 correspond to the “interhelical lipid” described in the yeast bc1 complex52, at the coming-together of transmembrane helices from subunits 3, 4, 5, and 10. As described also by Iwata56 there are two cardiolipin molecules in the bovine complex where one was modeled in yeast (1KB9).
Six hexyl glucoside (HG) molecules have been modeled in 1PP9, and nine in 1PPJ. For the most part these are poorly ordered and may be misidentified, however in 1PPJ there is one hexyl glucoside that is exquisitely defined by the density (Figure 2c). The hexose ring is pinned in a crystal contact between helix αMd of chain A (at the level of 393-394) and the imidazole ring of O:His192 in a symmetry-related dimer, presumably accounting for the good order. In addition there are H-bonds from O2 of the sugar to A:Ser397 (shown) and from O6 to A:Glu394. This well-ordered detergent is seen in all crystals examined so far that have cell edge a = 128 Å, but in the looser lattice of 1PP9 this contact does not occur and the detergent is disordered. In one crystal with cell edge a ~120 Å (not shown) this detergent is absent and O:His192 of the sym-related molecule packs directly against helix αM of chain A . Thus the detergent seems to be the “shim” which accounts for the frequent occurrence of the a=128 Å cell edge after partial dehydration of the crystals.
As expected in the presence of stigmatellin, the ISP is in the proximal or “b” position, with a hydrogen bond between His161 and stigmatellin, which is bound in cyt. b. The significance of this H-bond has been described in a recent note57. The methionine axial heme ligand in cyt c1 has “R” chirality at the Sδ atom, as in the chicken 1BCC or yeast 1EZV structures (Figure 2a). The heme planes of heme bH, heme bL, and cytochrome c1 are at angles of 25 – 26°, 5 – 6°, and 14 – 16° to the membrane normal, respectively. The orientation of the hemes about their pseudo-two fold axis is unambiguous and is the same as originally modeled in the chicken structure 1BCC. Cis- peptide linkages are present at the peptide bonds involving Pro222, Pro436 (cyt b) and Pro74 (cyt c1) as the (i+1)th residue. The assignment is unambiguous for these three residues, and has been verified in cross-validated SigmaA-weighted Fo-Fc omit maps calculated for 1PPJ omitting residues in a sphere of radius 3 Å around the residue and calculated between 93.5 and 2.2 Å resolutionc. Pro21 in subunit 2 is also modeled as a cis peptide, but the position of His20 is not well defined by the density so this is likely to be in error. No other cis-peptide linkages were found.
Heme-binding helix bundle and heme bH
As deduced from sequence analysis58; 59; 60 and described in previous structures20; 46; 52, cytochrome b is primarily α-helical, with eight transmembrane helices labeled A - H and four “surface” helices labeled αa (before Helix A), αcd1 and αcd2 (Between helices C and D), and αef (between helices E and F). There is one small β-sheet consisting of two antiparallel strands from the linker regions before helices A and E, which will be described below in connection with the antimycin site.
The two hemes are located within a four-helix bundle consisting of helices A, B, C, and D; with the high potential heme (heme bH) toward the N side and low potential heme (heme bL) toward the P side of the membrane. Both hemes have bis-histidyl ligation, with the histidine ligands provided by helices B and D (His83, 97, 182, and 196 in the bovine sequence). In addition there are four conserved glycines in helices A and C where the heme ring makes a close contact (Gly34, 48, 116, and 130).
Heme bH, with axial ligands His97 and His196, is distinctly curved: pyrrole ringse A and C bend toward the His97 side while rings B and D are nearly in a straight line with the iron (forming the axis of curvature). Pyrrole rings B and D lie along the axis of the four-helix bundle with rings A and C on the sides, inserting between the helices that comprise the bundle. Ring A, exposed between helices A and D, contributes to the antimycin binding site (below).
As reported20; 52 the propionate on the A ring is bent sharply back toward the axial ligand His97, making an ion-pair with the guanidino group of Arg-100 (bovine sequence numbering). We can see now (Figure 3) that this ion pair involves only one of the carboxylate oxygens and NH1 of the guanidino group of R100 (distance 2.8 Å), but that the propionate in addition binds two very well-ordered water molecules (Figure 3 and Table 3). The same propionate oxygen that ion-pairs with R100 has a second bond (2.8 Å) to an entity modeled as water W4, whose other ligands are the other (D) propionate and Ser205 Oγ. The other oxygen of the A propionate is separated by 3.3 Å from the NH2 atom of Arg100, but makes a very strong (2.44 Å) bond with another stable water molecule W5 which bridges between this propionate and the Nδ atom of the heme axial ligand His97. This water also makes bonds with the carbonyl oxygen of Trp30 and backbone N of C33 in the A helix. This rigid framework presumably serves to fix the plane of the heme-ligand histidine, and may be partly responsible for the heme curvature mentioned above. The sharply bent A propionate arm also forms one surface of the Qi binding site (see below), and may be on the path for electron transfer between heme bH and quinone at that site. The other propionate, on the D pyrrole ring of heme bH, H-bonds with one carboxylate oxygen to the side chains of Ser106 and Trp31, and with the other to the backbone nitrogen of Asn206 and to the water molecule W4 mentioned above. This arrangement of the propionates, Arg100, the two waters, and their ligands is the same in the presence or absence of antimycin, and is seen also in the yeast bc1 structures (e.g. 1P84), so it is likely to be a static arrangement. However if at some point in the reaction cycle the A propionate could be released to straighten out, it would put the carboxylate in the Qi site, as a possible ligand for a quinone species there, as well as modulating the charge density near the heme iron and curvature of the macrocycle.
Figure 3.
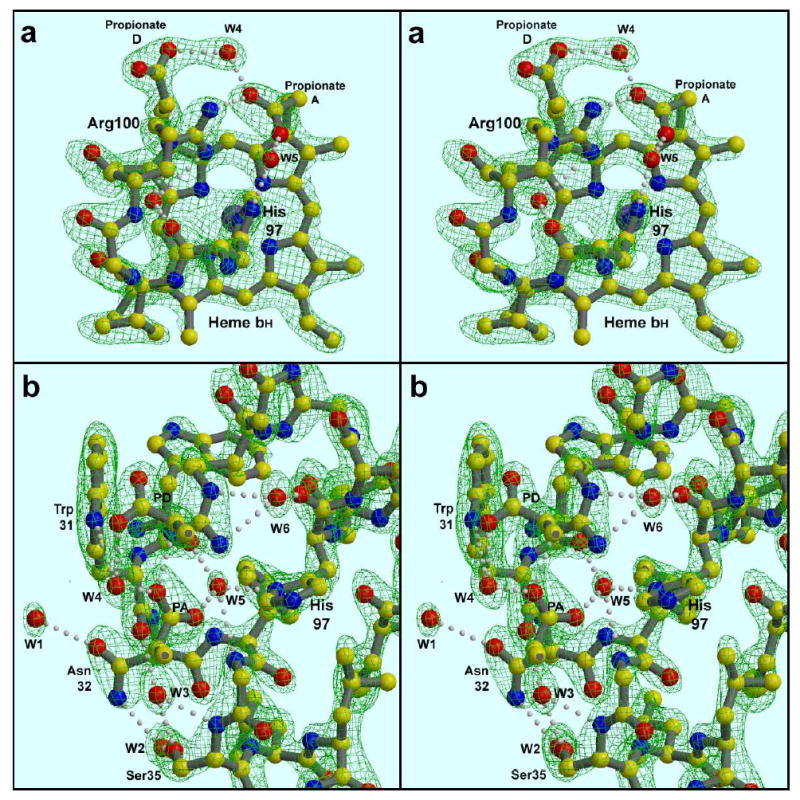
H-bonding around the high potential cyt b heme. (a)- Heme bH viewed from helix B. Water W4 bridges between the two heme propionate side chains, while W5 bridges between a propionate and the heme axial ligand His97. Arg100 interacts directly with this “bent” propionate, and via unlabeled water molecule W6 with the carbonyl O of His97. W5 is also H-bonding with backbone atoms of helix A, which has been removed from this view for clarity. (b). - The same region viewed from the heme position, looking toward helices A and B. The heme is removed except the two propionate side chains. Intercalation of waters W3 and W5 in the helical backbone of Helix A can be seen. Waters W1-3 are discussed in the text in connection with antimycin binding. The map is a 2Fo-Fc map calculated from data between 15 and 2.1 Å, sharpened with B −20, and contoured at 1.8 σ (a) or 1.7 σ (b).
Table 3. Potential H-bonding partners for six highly-ordered water molecules in the region of heme bH and the Qi site.
For each of the waters, O and N atoms within 3.5 Å are listed, together with the interatomic distance in each of the two monomers (chains C and P). The B-values for the waters in each monomer are also given.
| Water res. | H-bond Partner | Distance | B-value | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Label | (C, | P) | Res. | Num | Atom | C | P | C | P |
| W1 | 119 | 959 | ** Antimycin | O2 | 2.6 | 2.6 | 44 | 38 | |
| ** LYS | 227 | NZ | 2.6 | 2.6 | |||||
| ** SER | 28 | O | 3.1 | 2.9 | |||||
| ** ASN | 32 | OD1 | 3.2 | 3.3 | |||||
| W2 | 1008 | 214 | ** SER | 35 | OG | 2.7 | 3.0 | 29 | 32 |
| ** ASN | 32 | ND2 | 2.8 | 2.8 | |||||
| ** ASP | 228 | O | 3.0 | 2.7 | |||||
| W3 | 222 | 28 | Antimycin | N1 | 3.2 | 3.2 | 25 | 32 | |
| ** Antimycin | O1 | 2.9 | 2.8 | ||||||
| ** TRP | 31 | O | 2.7 | 2.7 | |||||
| ASN | 32 | O | 3.2 | 3.2 | |||||
| ** SER | 35 | N | 2.8 | 2.8 | |||||
| ** SER | 35 | OG | 3.0 | 2.9 | |||||
| W4 | 168 | 109 | ** SER | 205 | OG | 2.5 | 2.5 | 27 | 31 |
| ** HEM | 502 | O2A | 2.8 | 2.8 | |||||
| ** HEM | 502 | O1D | 2.7 | 2.7 | |||||
| W5 | 2 | 1 | ** HEM | 502 | O1A | 2.5 | 2.6 | 33 | 30 |
| ** TRP | 30 | O | 2.8 | 2.7 | |||||
| ** HIS | 97 | ND1 | 2.9 | 2.8 | |||||
| ** PHE | 33 | N | 3.1 | 3.3 | |||||
| ** GLY | 34 | N | 3.1 | 3.4 | |||||
| ARG | 100 | NH1 | 3.5 | 3.4 | |||||
| ARG | 100 | NH2 | 3.5 | 3.2 | |||||
| W6 | 108 | 35 | ** HIS | 97 | O | 2.6 | 2.7 | 31 | 32 |
| ** ARG | 100 | NE | 3.2 | 3.1 | |||||
| ** ARG | 100 | NH2 | 3.2 | 3.0 | |||||
| ** GLY | 101 | N | 3.3 | 3.4 | |||||
- potential hydrogen bond
Ser205, one of the ligands for strongly ordered water W4, is replaced by Asn221 in Rb. sphaeroides. It has been proposed that the Oδ1 atom of Asn221 occupies the position of W4 bridging between the two heme propionates, positioning the Nδ2 atom to serve as a ligand for quinone in the bacterial complex33.
Molecular configuration of bound antimycin
The antimycin in structure 1PPJ is very well ordered, with average B-factors in the two monomers of 41.5 and 43.6 Å2, barely above that for the backbone of cytochrome b (39.0, 40.9) and lower than the average b-factor for the structure (50.2 Å2). The electron density is correspondingly good, and there is little ambiguity in the placement of any of the atoms except the tips of the alkyl and acyl side chains. The degree of order is greatest on the formylaminosalicylamide portion, which is well defined in 2Fo-Fc maps contoured at 2.1 σ (Figure 1b), and decreases through the dilactone ring and into the alkyl and acyl side chains at the other end. At 1.5 σ (not shown) the acyl chain is visible through C3 and shows the methyl branch to be at the 2 position as reported42 rather than the 3 position as previously believed, and at 0.9 σ (Figure 1c) there is weak density for C4 in one monomer, tentatively modeled in Figure 4b and 4c for completeness. The alkyl side chain has density through the fifth carbon when contoured at 0.9 σ (Figure 1c)
Figure 4.
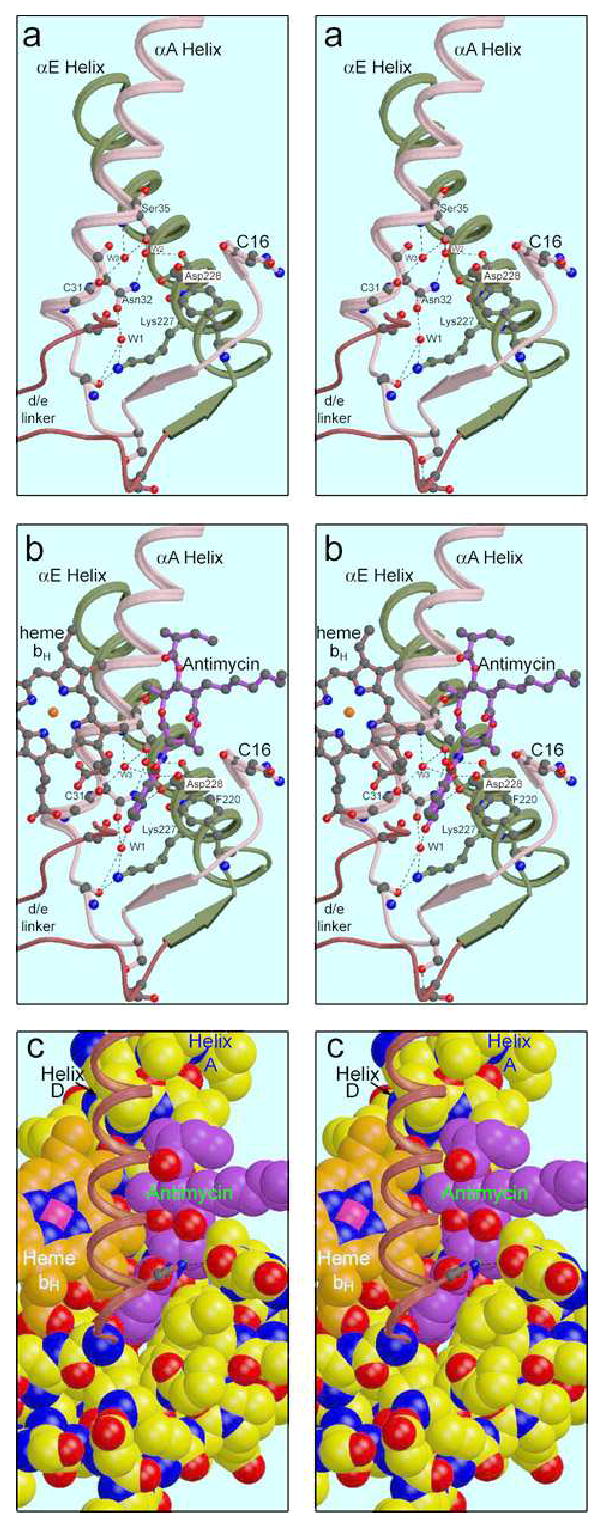
Structure of the Qi site and interaction with bound antimycin. The Qi site lies in a triangular volume formed by helices B and E crossing at an angle (a). The N-side (N-terminal) ends of these helices are held together by β-type H-bonding between residues just preceding the helices (arrows in cartoon) which bounds the third side of the volume, and by Lys228 of helix E which H-bonds with a backbone O of residue 27, and to a water molecule bonded to that atom and to Asn32 Oγ1 of Helix A. These bonds are part of a more extensive H-bonding chain involving also Trp31, Ser35 and two other waters. In (b) antimycin (magenta bonds) and heme bH (orange bonds) are added. The methyl and propionate substituents of the “A” ring of heme protrude from the four-helix bundle between helices A and D (helix D removed for clarity), forming part of the surface of the binding site. The formylaminosalicylate headgroup of antimycin inserts into the triangular volume described above, sandwiched between Phe220 of helix E and the heme propionate, and H-bonding with Asp228 and (via another water) Lys227. In (c) the protein elements shown in (a) are rendered as space-filling model to show the surface of the binding site. Antimycin (magenta carbons) and heme bH (orange carbons) are also space-filled to show the intimate contact between these moieties and the snug fit of the antimycin headgroup in the protein. The binding pocket is completed by the α-a surface helix (shown here starting with residue 15) and the D transmembrane helix, left as a ribbon for clarity. There may be H-bonds involving His201 in helix D with the amide carbonyl oxygen of antimycin and with a backbone oxygen in the α-a helix. At the lower extreme of antimycin is the aromatic ring, viewed edge-on and inserted between the bent propionate of heme bH and Phe220 in helix E. The carbonyl oxygen of the amide linkage is directed toward the viewer, seen beneath His201 of helix D. Higher up, the alkyl side chain extends to the right into the lipid-filled cleft. At the top is the acyl group, with the ester carbonyl oxygen direct towards the viewer. Note the close contact with heme bH, involving not only the aromatic ring of antimycin but also part of the dilactone ring.
The dihedral angles of the formylamino group38 are approximately 0° (Θ1) and 180° (Θ2), in agreement with values found in an energy-minimized structure38. Similar values were found in the small-molecule structure42 and for the bound inhibitor in structure 1NTK. These angles put the formylamino group in the plane of the salicyl ring, directed away from the OH and carboxylate groups and toward Lys227 of cyt b (Figure 6). The observation40 that a methyl group at position 4 but not at position 5 diminishes binding of antimycin analogs is consistent because the methyl at position 4 (compound 17) would prevent the formylamino group from taking on this conformation38.
The dihedral angle Θ3 between the phenyl ring and carbonyl carbon of the salicylate moiety is approximately 180°; that is the amide group is rotated 180° relative to the salicyl ring from the small-molecule structure. This means that the internal H-bond between the phenolic OH and amide carbonyl oxygen, which was proposed to be important for inhibition38, and which was observed in the small-molecule structure42 (indicated in Figure 1a), is actually not present in the enzyme-bound form (Figure 1b and 1c). The observed orientation of the amide moiety with respect to the salicylate ring is contrary to that modeled in structure 1NTK. This and other discrepancies will be considered in the Discussion section.
The 9-membered dilactone ring of antimycin is puckered with alternating members directed up and down except Cβ of the threoninef, which is between members facing up and down. The chirality of the chiral centers meshes with the puckering in such a way that the three bulky substituents as well as one methyl side chain (C5 of the valeric acid moiety) project equatorially, i.e. more or less in the plane of the dilactone ring, while the two carbonyl oxygens project perpendicular to the ring. The other methyl group (Cγ of threonine) projects at an intermediate angle. The planes of the ester and amide substituents and the salicyl ring are nearly perpendicular to the dilactone ring. This differs from the small-molecule structure, in which the plane of the salicyl ring and amide are approximately 45° from that of the dilactone ring (Compare Figures 1a and 1b; in both of which the salicylamide is in the plane of the picture).
The antimycin-binding site
Figure 4 shows the make-up of the antimycin binding site in different levels of dissection, and Table 4 lists contacts between antimycin and the protein. Briefly, the antimycin headgroup is found in a pocket which is bounded by helices αA, αD, αE, and α-a; as well as the edge of heme bH exposed between helices αA and αD of the four-helix bundle. Strong hydrogen bonds are formed directly with Asp228 in helix E and via ordered water molecules to Lys227 in Helix E and Ser35 in Helix A.
Table 4. Residues surrounding Antimycin at the Qi site.
For each contact closer than 4 Å is given the residue type, number, and atom; the atom of antimycin, and the contact distance in each monomer. For distances greater than 3 Å, only the closest contact of each protein residue is given. Except for waters, all the residues contacting antimycin belong to cyt b (chains C and P). The “water” modeled between His201 and antimycin O3 does not account for the density as currently modeled (Figure 6).
| Helix A and Waters (W) | Helix D & E | Heme bH | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| distance(Å) | distance(Å) | distance(Å) | ||||||||||
| protein atom | Anti atom | C | P | protein atom | Anti atom | C | P | Heme atom | Anti atom | C | P | |
| ALA17 O | C10 | 3.6 | 3.7 | HEM502 | CMA | N2 | 3.8 | 3.8 | ||||
| ILE27 CD1 | O2 | 3.5 | 3.5 | MET190 CG | C23 | 4.0 | 3.7 | HEM502 | CMA | C9 | 3.8 | 3.7 |
| TRP31 O | N1 | 3.4 | 3.4 | MET194 CG | O9 | 3.6 | 3.5 | HEM502 | CMA | O4 | 3.2 | 3.2 |
| ASN32 OD1 | C8 | 3.9 | 3.9 | LEU197 CD1 | O4 | 3.2 | 3.1 | HEM502 | CMA | C20 | 3.3 | 3.3 |
| GLY34 O | C27 | 3.9 | 4.0 | SER205 OG | C4 | 3.5 | 3.5 | HEM502 | CMA | O7 | 3.7 | 3.7 |
| SER35 CA | O7 | 3.0 | 3.1 | HEM502 | CMA | C27 | 3.9 | 3.9 | ||||
| GLY38 CA | C27 | 3.8 | 3.8 | PHE220 CE1 | C1 | 3.3 | 3.4 | HEM502 | CAA | C5 | 3.8 | 3.9 |
| LEU41 CD2 | C25 | 3.9 | 3.7 | TYR224 CD1 | O2 | 3.3 | 3.3 | HEM502 | CAA | C6 | 3.8 | 3.9 |
| ASP228 OD1** | N1 | 2.8 | 2.8 | HEM502 | CAA | C7 | 3.9 | 4.0 | ||||
| W3 O ** | O1 | 2.9 | 2.8 | ASP228 OD2** | O1 | 2.6 | 2.6 | HEM502 | CBA | C1 | 3.8 | 3.8 |
| W1 O ** | O2 | 2.6 | 2.6 | HEM502 | CBA | C5 | 3.7 | 3.8 | ||||
| “W” O | O3 | 2.6 | 2.8 | HEM502 | CBA | C6 | 3.6 | 3.6 | ||||
| W1203 O | C5 | --- | 3.7 | |||||||||
Figure 4a shows the underpinnings of the antimycin binding site in helices A and E. These helices cross at an angle, with Van der Waals contacts at the crossing between the side chains of Leu43 and Leu239 (not shown). The N-terminal (N-sideb) ends of these helices are connected by β-bridges between residues in the sequence preceding the helices: residues 21, 23, and 25 in the region before helix A make backbone H-bonds with residues 221, 220, and 218 before helix E. In addition a strong hydrogen bond between the side-chains of Asp216 and Ser25 hold these two residues together. These β-bridges are represented by the antiparallel arrows in Figure 4, and together with helices A and E they bound a triangular volume that encloses the Qi site. Another connection between the A and E helices is made by Lysine 227 in the E helix which H-bonds with the backbone oxygen of residue 27 and a highly ordered water molecule attached to helix A. These bonds are part of a more extensive H-bonding chain that is involved in antimycin binding but is present in both structures 1PPJ (with antimycin) and 1PP9 (without). This chain is shown in stereo in Figure 4a. Lys227 and the first water (W1) are bonded to each other and to the carbonyl oxygen of residue 27. W1 is also bonded to Oγ1 of Asn32. Nγ2 of this residue H-bonds a second water (W2) which in turn bonds to Ser35Oγ and to the carbonyl oxygen of Asp228, further linking the A and E helices. A third water (W3) also bonds with Ser35Oγ and additionally with the backbone O and N of residues 31 and 35, respectively (Figure 4a). As these latter two atoms would normally be involved in the α-helical H-bonding of helix A, W3 can be seen as “intercalated” into the helixg.
In addition to the A and E helices, both the D helix and the α-a surface helix contribute to the Qi site. Helix D is omitted in Figures 4a and 4b for clarity (the view is from the position of helix D), but the linker polypeptide connecting helix D to helix E is shown. Ser205, early in the D/E linker is shown as ball-and-stick. This residue has been implicated in quinone binding at this site 24; 33; 52; 61, and will be discussed further below. The α-a surface helix, starting with residue 15 is shown. This turn borders on the Qi site, and there is some indication that the carbonyl oxygens of residue 16 or 17 may H-bond with the sidechain of His201. Unfortunately this region is poorly ordered in both monomers of these crystals, and its contribution to the Qi site can better be seen in the yeast (e.g. 1KB9) or chicken (3BCC) structures.
In Figure 4b antimycin and heme bH are added to the picture, and Figure 4c shows a spacefilling model of everything represented in 4b and also adds helix D as a thin ribbon. The H-bonding contacts of antimycin can be seen in Figure 4b and, in greater detail, in Figures 5 and 6. The only direct H-bonds with the protein involve conserved Asp228, the carboxylate of which binds to the phenolic OH and the formylamino NH. It seems reasonable to assume that Asp228 is deprotonated at the pH of the crystal, and serves as H-bond acceptor in both these bonds. The phenolic OH also has an H-bond with the intercalated water W3, as well as the intramolecular H-bond mentioned above with the amide NH. Following the reasoning above it must be the acceptor in both of these bonds, as its one proton is being donated to Asp228. The formylamino oxygen H-bonds to water W1 discussed above. Otherwise the contacts appear all to be hydrophobic. Strikingly, no H-bond is made with the dilactone ring or its acyl side chain.
The substituents of the “A” pyrrole ring of heme bH (the bent propionate described above and a methyl group) protrude from the 4-helix bundle between the A and D helices, forming part of the surface of the Qi site. The space-filling model in Figure 4c illustrates the intimate contact between heme (orange) and antimycin (magenta), consistent with the electronic interactions required to explain the quenching of antimycin fluorescence and the spectral shift of cytochrome bH on binding. The aromatic headgroup of antimycin is inserted into a cavity between the bent propionate and Phe220. The aromatic ring of Phe220 is not quite perpendicular to the salicyl ring, the actual angle being 77°. The axial methyl group of the dilactone ring interdigitates loosely with the methyl groups on pyrrole rings A and B of the heme.
Van der Waals contact with Ser205, a possible ubiquinone ligand
On the other side of the Formylaminosalicyl ring from Asp228, potential H-bonding partners are Ser205 and His201 (Figure 4b and c, Figure 6), both believed to be important in ubiquinone binding at the Qi site33; 52; 61; 62. Ser205 makes Van der Waals contact with C5 of the salicylate ring, but there is no H-bonding partner on this area of antimycin.
In antimycin analogs lacking the 3-formylamino group, inhibition can be restored by 3- or 5-NO2 groups, and to some extent by a 5-formylamino group41. This has been attributed to a requirement for an electron-withdrawing substituent to increase the acidity of the phenolic OH16, however, based on a more extensive set of analogs, Tokutake et al decided that electron-withdrawal did not correlate well with activity, and concluded specific interactions of both the formylamino and phenolic OH with the protein were involved. It seems likely that a nitro group in the 5- position would H-bond Ser205, while one in the 3- position would H-bond Asp228. Superposition of 3- or 5-nitrosalicylate on the salicylate moiety of antimycin in 1PPJ (not shown) results in too-close contacts (1.7 and 1.3 Å) of the 3-nitro group oxygen with Asp 228 and the 5-nitro group with Ser205. Small adjustments in the positions of these side chains, or slight repositioning of the ring, could allow a good fit.
Thus H-bonding may be as important as electron-withdrawal in explaining the effects of nitro- substituents. However in light of our assumption that the phenolic OH is the H-bond donor in the bond with Asp228, it has to be questioned whether the more acidic nitro- compounds, which might be deprotonated at neutral pH, could bind in the same way as antimycin. In fact the detailed inhibition pattern of nitrosalicylate compounds has been found to be quite different from antimycin and other 3-formylamino compounds, the nitrosalicylates being ineffective in eliciting either the “double-kill” behavior or oxidant-induced reduction seen with antimycin40; 63.
His201 conformation
The most enigmatic part of the structure around the antimycin binding site is the critical conserved residue His201, which is believed to be a ligand for quinone52; 61; 62 either directly61 or indirectly through a water molecule52. When contoured at a 1.5σ density level (Figure 6a) the imidazole ring seems well localized, but poorly shaped for this resolution. His201 here is modeled in a position close to rotamer 5h, as in all vertebrate cyt bc1 structures currently available, and there is really no possibility of modeling it in with a significantly different value of chi-1.
The distance from His201Nɛ2 to the antimycin O is 4.20 Å, precluding any significant direct H-bonding. An extra bit of density has been modeled as water, with distances 2.81 Å from the water to the amide carbonyl O of antimycin and 2.63 Å to the Ne2 of His201. In fact the density is too close to the histidine to be a water molecule. Non-bonded interaction terms in the refinement process have pushed the modeled water outside of the density peak, and displaced His201 slightly from its best fit. When the water is moved to its density peak, the distances are 1.82 to the His201 and 3.07 to antimycin. When the water is removed and the model is subjected to further positional refinement, His201 moves only slightly closer to antimycin, distance 4.12 Å (not shown).
When contoured at a lower level (1.0σ, Figure 6b), the density around the imidazole spreads out in a triangular shape and merges with the peak assigned to water. This may result from a mixture of two alternative conformations: one in which the water is present at its density peak but His201 rotates back to be farther from it, and another in which the water is absent and His201 rotates forward toward the water position and bonds directly with antimycin.
The two conformations postulated here, and the presence or absence of a water molecule between the histidine and the Qi-site occupant, should not be confused with another interesting difference between available structures. In all the yeast structures presented to date, His202 (corresponding to His201 in the bovine sequence) is positioned close to rotamer 3h. A water molecule bridges between the Nɛ2 nitrogen and the carbonyl oxygen of ubiquinone. In the chicken bc1 structures with ubiquinone, His202 is close to rotamer 5, which allows a direct H-bond to ubiquinone. It seems quite reasonable that both these conformations are correct, depending perhaps on pH or ionic strength, and that the direct involvement of a water may be part of the mechanism for uptake of the protons involved in quinone reduction at the Qi site. However that may be, all of the vertebrate bc1 structures available today have His202 (201) in rotamer 5, and all the yeast structures have rotamer 3. At first glance the bovine structure 1NTZ seems to be an exception, as the H-bond between quinone and His201 is mediated by a water molecule as in the yeast structures. However superposition of the chicken and yeast structures shows that in 1NTZ, His201 is in the chicken position (rotamer 5) and the quinone is deeper in the pocket than in the yeast or chicken structures, making room for the mediating water.
Unknown molecule in hydrophobic site between dilactone and helix A
There is a strong density between the dilactone ring of antimycin and helix A which during most of the refinement of the structure was modeled as a water. Since there are no nearby H-bonding partners to account for stabilizing a water molecule in this position, it was removed before submitting the structure to the PDB. However this leaves a strong peak in difference Fourier maps, indicating that something is there, even if it is not a water. The peak is oblong and about the right size for a diatomic molecule. In consideration of the hydrophobic nature of the environment and the lack of H-bonding partners we think it may be a nonpolar gas such as nitrogen or oxygen. It is modeled as O2 in Figure 6, and labeled “unknown”. The closest contacts in antimycin are O7 and C11 (3.7 and 4.0 Å). The closest contacts in helix A are the carbonyl O of ser35 (4.0 Å) and sidechains of Ile39 and Ile42 (4.0 – 4.2 Å).
Comparison of antimycin and ubiquinone binding positions; local conformational changes induced by antimycin
Disappointingly, the ubiquinone molecule in the crystals without antimycin (1PP9 and Y21) is much less well ordered than antimycin in 1PPJ. There is density in the position indicated for the ubiquinone ring by previous structures (1BCC and 1EZV), however the shape is not well-defined. It seems likely that the occupancy is significantly less than one, due to dissociation during the purification in detergent-containing, quinone-free buffers. Water or other molecules may have entered the unoccupied Qi sites, resulting in the poorly defined density of the crystallographic average. Current experiments are aimed at maintaining a high quinone occupancy by purification in detergent micelles doped with ubiquinone.
There is no strong indication of asymmetric ubiquinone occupancy such as reported64; 65 for the yeast enzyme with one mole of cytochrome c bound per dimer: The peaks of the density attributed to ubiquinone in monomers 1 and 2 were 3.0 and 2.1σ in 1PP9, but 2.6 and 2.8 σ in Y21. The shapes were similar. Ubiquinone has been modeled into the density based on the previous structures, and refinement of the model did not lead to significant discrepancies from those structures. Thus the results obtained superimposing Y21 below are essentially the same as would be obtained superimposing the yeast or chicken structures. Another bovine structure (1NTZ) presents a slightly shifted position for ubiquinone.
Figure 7 shows superposition of the Qi sites of 1PPJ and Y21 based on a rigid core of cytochrome b, including the four-helix bundlei. The quinone ring does not superimpose with the aromatic salicylate ring of antimycin, rather it is centered on the carbonyl carbon of the amide group. Carbonyl oxygen O1 of the quinone is positioned near the phenolic OH oxygen of antimycin (0.63 Å) making the same H-bonds to Asp228 and water W3 as that atom makes. The other carbonyl oxygen, O4, extends toward His201, reaching farther than the carbonyl oxygen of the antimycin amide and making a direct H-bond (or a water-mediated H-bond in the yeast structures) with that residue.
Figure 7.
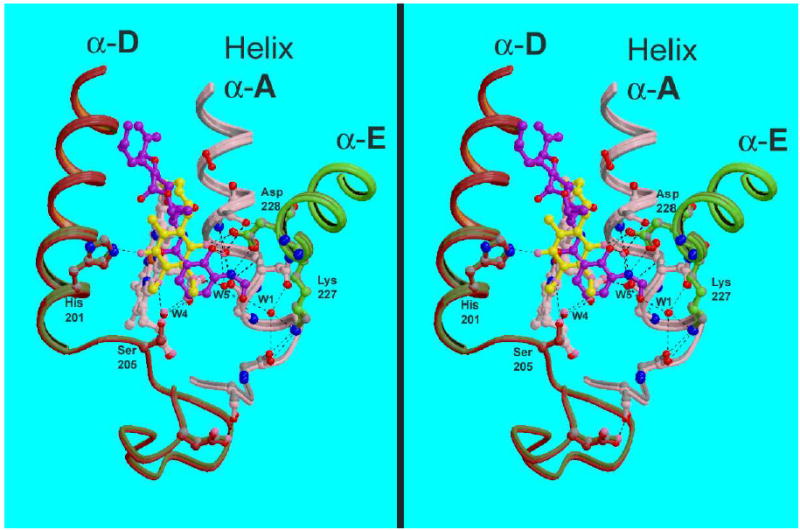
Comparison of Qi-site residues and ligands in structures 1PPJ and Y21. The two structures were superimposed based on cyt. b residues 32–51, 79–99, 113–145, 161–201, and 263–300. The backbone is shown for parts of transmembrane helices A (pink), D (red), and E (green), in color for 1PPJ and gray for Y21; as well as some of the linker region preceding helices A and D. Relevant side-chains are drawn with bonds and carbon atoms the same color as the backbone. Water molecules are shown as red spheres for 1PPJ and pink spheres for Y21. Antimycin from 1PPJ is shown as a purple ball-and stick figure with red oxygen atoms, while ubiquinone from structure Y21 is yellow. Note the relatively invariant positions of the backbone and side-chains, and the positioning of the ubiquinone ring over the amide moiety of antimycin.
There is surprisingly little rearrangement in the protein backbone and even sidechains surrounding antimycin, as compared with for example the rearrangement of protein around the Qo site upon binding inhibitors23; 66. Gao et al.24 also remarked on the structural rigidity of cyt b on antimycin binding, but reported significant conformational changes of cytochrome b residues Ser35, His201, Phe15, and Met194. Comparing structure 1PPJ with 1PP9 or Y21 (Figure 7) showed Ser35 to be identical and His201 not to have changed significantly. We see both conformations for Met194 in the presence or absence of antimycin. The side chain of Phe15 is completely disordered in all three structures.
Long-range Conformational changes induced by antimycin binding and by different crystal packing forces
As described in the introduction, there are several reasons to believe that antimycin binding triggers a long-range conformational change in the bc1 complex. Cursory comparison of the structures described here gives no indication of such a change: the differences between crystals with and without antimycin are smaller than differences between crystals with the same Qo-site occupancy but different cell parameters, attributed to crystal packing distortions. The comprehensive comparison that would be required to put an upper limit on the magnitude of change that might be due to antimycin binding is beyond the scope of this work, however some preliminary quantitative comparisons will be described which indicate that the change must be quite small. The conformational changes observed are also of interest to indicate the modes of flexibility of the protein, whether due to crystal packing forces or inhibitor binding.
In order to look for conformational changes induced by antimycin binding, we compared C-α positions of cytochrome b in structure 1PPJ with structures lacking antimycin (Figure 8 and Table 5). Structure 1PP9 lacks antimycin but has significantly different cell parameters (Table 1a) from 1PPJ, so differences may be due to different packing forces. A third structure of similar resolution (optical resolution 1.70) and lacking antimycin but with cell parameters similar to 1PPJ was compared to control for these changes.
Figure 8.
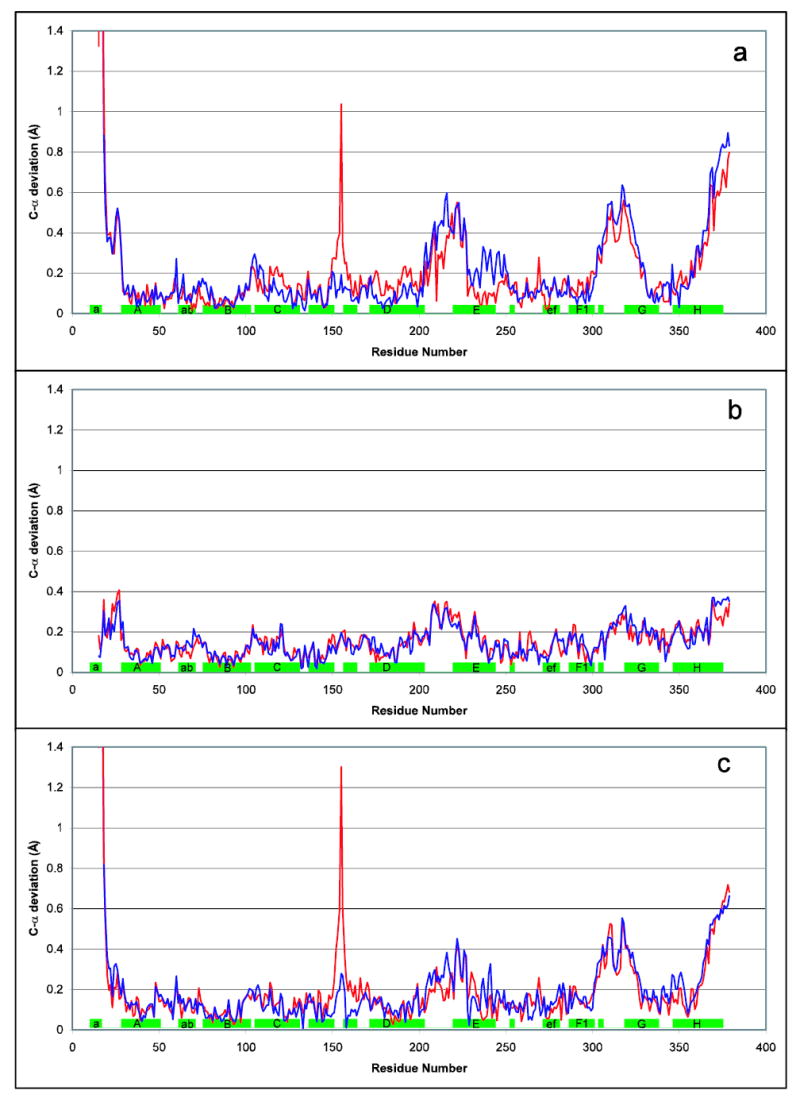
Flexibility in cyt b induced by antimycin and/or crystal packing forces. Various cyt b structures were aligned based on the relatively rigid core consisting of residues 32–51, 79–99, 113–145, 161–201, and 263–300. Deviations of Cα position are plotted vs residue number for selected pairs of structures. For each pair the differences in chain C are shown in red while those for chain P are in blue. The green rectangles along the x axis indicate the position of helices in the sequence. PDB entries 1PP9 (without) and 1PPJ (with antimycin) are the structures featured in this article, while structure Y21 is from a crystal with cell parameters nearly identical to those of 1PPJ. Thus comparison of cyt b from the same monomer between 1PPJ with Y21 (b) gives the best indication of antimycin-induced changes, while comparison of two monomers in the same crystal, or of 1PP9 with Y21 (c), should show only crystal-packing-induced changes. Comparison of 1PP9 with 1PPJ (a) superimposes both sets of changes.
Table 5.
Relative motion of domains of cyt. b between three crystals:
| F2,G,H helices vs cyt. b core | ||||||
|---|---|---|---|---|---|---|
| ------Chain C--------- | -------Chain P------- | |||||
| angle
|
max disp,
|
atom
|
angle
|
max disp,
|
atom
|
|
| 1PP9 vs 1PPJ | 1.392° | 0.8389 | C378 | 1.169° | 0.6904 | P378 |
| 1PPJ vs Y21 | 0.437° | 0.3072 | C378 | 0.310° | 0.2654 | P377 |
| 1PP9 vs Y21 | 0.960° | 0.5969 | C378 | 0.944° | 0.5979 | P378 |
| E helix after 227 vs cyt. b core | ||||||
| ------Chain C--------- | -------Chain P------- | |||||
| angle
|
max disp,
|
atom
|
angle
|
max disp,
|
atom
|
|
| 1PP9 vs 1PPJ | 0.927° | 0.3315 | C245 | 0.503° | 0.1145 | P245 |
| 1PPJ vs Y21 | 0.301° | 0.1486 | C245 | 0.513° | 0.1814 | P230 |
| 1PP9 vs Y21 | 0.859° | 0.2438 | C245 | 0.349° | 0.1600 | P228 |
In the structures compared here (all containing stigmatellin), a core domain of cytochrome b consisting of the four TMH of the four-helix bundle plus much of cd1-cd2, the ef-linker, and the F helix before the kink at 300 (αF1) could be superimposed with rmsd 0.133 Å or below and maximum deviation 0.28Å. The exact residues included in this core are listed in the legend to Figure 8, which shows the by-residue deviation, between 1PPJ (with antimycin) and two structures without antimycin, when cytochrome b is thus superimposed.
The N-terminus and the de linker, both involved in the Qi site, were significantly different between 1PPJ and 1PP9, while the ab and bc linkers showed minor deviations. The mobile region in the de loop actually extends into the N-terminal part of helix E, as far as residue Lys227. Although significant, these movements are very slight, as can be seen in Figure 7.
Helix E starting at 228 could be included in the core domain but gave a slight but perhaps significant increase in rms deviation between the C chains of 1PPJ and 1PP9 (but not between P chains). To test the hypothesis that the E helix transmits a conformational signal to the P side upon antimycin binding at the Qi site, helix E was excluded from the core region used for superposition, and treated as a separate domain.
With the cytochrome b chains thus superimposed, distance between corresponding atoms in pairs of chains were plotted in Figure 8. Any large effect of antimycin should be seen as differences between 1PPJ (with antimycin) and 1PP9 or Y21, and not between 1PP9 and Y21 (both without antimycin).
Such by-residue plots of atom deviation are limited in sensitivity by the inherent noise in the structure, the estimated standard deviation for atom positions being 0.3 – 0.5 Å for these structures (Table 1b). If it is assumed that sections of protein move as rigid bodies, much smaller movements can be detected because positions of all the atoms in the body contribute to determining its position and the “jitter” in individual atomic positions averages out. We tested two domains for such rigid body movement relative to the core domain used for superposition. One was the helix F2+G+H region discussed below, which seems to be moving independently between 1PPJ and 1PP9, and the other was helix E, which shows significant deviations between these structures in the C chain (Figure 8a). To do this the operator best superimposing the domain in question was compared with the operator superimposing the core domain, giving an operator for the additional movement required to superimpose the domain in question after the core domain has been superimposed. This operator was then expressed as a rotation angle and as the largest movement of any atom in the domain upon application of the operator.
As a control to test the significances of the differences observed, the Y21 structure was resolved twice starting with structures 1PPJ in one case and 1PP9 in the other. After positioning the models using the now-well-known intercrystal operators followed by rigid-body refinement and a few rounds of alternating positional minimization and restrained atomic B-factor refinement, the entire cyt b backbone (excluding the disordered region before residue 20j) was superimposable with maximum deviation 0.16 Å and RMSD 0.04. This implies that the small differences observed in this region between 1PPJ and 1PP9 are within the radius of convergence of positional refinement, and thus reflect real differences in the data and not results of accidental differences in the history of model-building.
The results from the comparison of interdomain operators are listed in Table 5. The G and H helices together with Helix F after the kink at residue 299 (αF2) form a separate domain which is rotated significantly with respect to the core domain described above: a rotation of 1.3° with maximum C-α displacement (at C378) of 0.81 Å in the case of C chains of 1PPJ and 1PP9). However the differences correspond more to differences in cell parameters and which of the two monomers in the dimer is being compared than to the presence or absence of antimycin, suggesting they result from different packing forces rather than an antimycin-induced change. The largest changes are seen comparing 1PP9 and 1PPJ, which differ in both presence of antimycin and cell parameters. Comparing the Y21 structure with 1PP9 (difference in cell parameters) and with 1PPJ (presence or absence of antimycin), 1PP9 shows the greatest movement of the F2GH domain in both C and P chains, and the largest movement in helix E for chain C. While movement of the E helix in chain P was largest in 1PPJ, that movement was a barely significant 0.51° rotation with maximal atomic displacement of 0.18 Å. This would appear to limit any antimycin-induced, long-range, static, conformational changes to a very subtle effect, at least in the presence of stigmatellin.
Ramachandran outliers
It is normal for well-refined structures even at high resolution to have 0.1–0.5% of residues in the “Forbidden” zone of the Ramachandran plot. However a Ramachandran outlier can also be an indication of a mis-built residue. Therefore we have examined the outliers in the two structures presented here to see how well the conformation presented is supported by the density.
Tyr155 of cytochrome b is a particularly interesting outlier that is well supported by the density. In bacterial bc1 complexes, there are two conserved glycines in the turn between helices cd1 and cd2, corresponding to positions 155 and 157 in the bovine sequence. Gly157 is also conserved in the mitochondrial complexes, but surprisingly 155 tends to be an aromat- tyrosine in most vertebrates, phenylalanine in yeast. However the backbone phi, psi values for this residue are 66 to 68 and −39 to −42° in the bovine structures and 76.6, −75.0° in the yeast structure. These values lie outside the allowed region on the Ramachandran plot for any residue but glycine. Thus a mitochondrial progenitor has placed an aromat at a position in the fold where only glycine could be accommodated readily, and this strained aromat has been preserved through evolution. We will not speculate about the function, but note that the αcd1-cd2 hairpin helix forms part of the Qo site, and movement of this helix in response to Qo-site occupancy or ISP position has been reported23; 66.
The residue corresponding to Tyr155 is also a Ramachandran outlier in all available structures of the chicken or yeast bc1 complex, and in the bovine complex in tetragonal crystals (e.g. 1L0L). Outlier status is avoided in structures 1BE3 and 1BGY by flipping the plane of the preceding peptide (154–155) relative to all other structures, however this arrangement of the backbone is incompatible with the density in the crystals reported here. The backbone density for this residue is quite strong and unambiguous, however the density on the ring and OH has a peculiar shape. This side chain sticks out into the solvent from the turn of the cd1-cd2 hairpin and makes no contacts with the rest of the protein, so it is not surprising if it is not well ordered. In chain P the tip of the side chain of Tyr155 makes a crystal contact (with chain B of a symmetry-related dimer). This contact varies with cell volume, resulting in the spike at residue 155 in Figure 8, a and c.
Residue Ala171 in chain B (“core II”) also falls in the disallowed region of the Ramachandran plot. This is in a 3–10 helical turn at the end of helix αDd. The electron density leaves little doubt as to the positions of the atoms, so we believe this also is a real outlier. It is an outlier in the yeast (1P84) and tetragonal bovine (1L0L) structures as well, but not in 1BGY, again as a result of flipping the peptide plane (B170–B171) in a way which is inconsistent with the density in our structures.
Residue Met71 in the “tether” region of the iron-sulfur protein differs in the two monomers. The conformation modeled in the first monomer (chain E) is an outlier in the structure deposited for 1PPJ (with antimycin), but not in 1PP9 or the Y21 structure, or in chain R of any structure. This residue appears to have several conformations and is not very well-ordered. It is likely subjected to considerable strain in some positions of the ISP extrinsic domain, which could provide the energy for an unfavorable backbone conformation. This dynamic linker region is worthy of further study to decide if it is really an outlier in some of its conformations, and to better define the different conformations.
The other two Ramachandran outliers are found in poorly defined regions (A223,224 in the interdomain linker of the largest subunit) and probably represent errors in the model.
Discussion
A number of features of the bc1 complex revealed by the presented structures and not discerned in previous structures suggests that this is the most accurate structure of the bc1 complex available. In particular, the binding mode of the inhibitor antimycin is defined to a high level of accuracy. The new structure is consistent with results of structure-activity relationship studies, however it does not support one of the conclusions from those studies: that the intramolecular hydrogen bond between phenolic OH and carbonyl O described for the molecule in solution38 and in the small-molecule crystal42 is important for binding and inhibitory activity. On the contrary, the bound molecule has the carbonyl oxygen facing His201 with a long, weak, or water-mediated hydrogen bond while the amide nitrogen H-bonds to the phenolic hydroxyl oxygen. This is quite understandable because the phenolic OH group is H-bonding to aspartate and is likely to be the donor in that interaction. This would make an H-bond to the carbonyl oxygen impossible, as that atom can only be an H-bond acceptor. The amide nitrogen, on the other hand, has one proton that would be available for H-bond donation. Such a rearrangement of the H-bonding pattern upon binding is not surprising, in fact the possibility was suggested in the small-molecule structure report42. The importance of the intramolecular H-bond was inferred from the fact that an antimycin analog in which the amide is separated from the salicylate benzene ring by an extra carbon (compound B of ref 38) and thus could not form the H-bond, was 104-fold less potent than an analogue with the amide directly connected as in antimycin. However this compound would be equally unable to form the intramolecular H-bond between amide nitrogen and phenolic oxygen that we observe in the bound inhibitor, so these experimental results are consistent with our structure. In fact it could be said that our structure supports the conclusion of that study regarding the importance of an internal H-bond between phenolic oxygen and the amide, but those experiments gave no hint that the amide is flipped; and the amide N, rather than O, is involved in the H-bond.
The structure of bound antimycin and the surrounding protein presented here differs in some significant details from that of the structure 1NTK24. Most importantly, the conformation of antimycin in the binding site is different. While both structures agree that the dilactone and formylaminosalicylate rings are rotated relative to each other as compared to the small-molecule structure, 1NTK keeps the dihedral between the amide moiety and the FSA fixed, preserving the intramolecular H-bond between the phenolic OH and the amide O. In 1PPJ there is 180* rotation about this dihedral relative to the small-molecule structure, breaking the intramolecular bond and forming a new one between the amide NH group and the phenolic O.
In addition two key residues Ser35 and Lys227 have their side chains modeled differently in the two structures, resulting in different roles for these residues in antimycin binding. Gao et al.24 report from structure 1NTK that Ser35 forms H-bonds with the amide carbonyl O and a carbonyl O of the dilactone ring. In 1PPJ Ser35 is in the most stable rotamer, facing away from antimycin (Figure 5a) and H-bonds with two waters (W3, W2) and the carbonyl oxygen of residue 32, but makes no direct H-bond to antimycin. If it were changed to rotamer 2h it would be positioned to H-bond the carbonyl oxygen of the threonine moiety of the dilactone, as in the model of Gao et al. However the electron density (Figure 5a) gives no indication of Ser35 in rotamer 2, even at partial occupancy. On the contrary, as described above W3 mediates an H-bond between Ser35 and the phenolic OH of antimycin. Likewise Lys227Nζ makes a direct bond to the formylamino oxygen of antimycin in 1NTK, but in 1PPJ these atoms are 5.2 Å apart and ordered water W1 binds between them (Figure 5b, 6). Neither structure has Lys227 in one of the five most common rotamers, however in 1PPJ it is in rotamer 26 (3% frequency) of the more extensive rotamer library described in ref 67.
Whenever different results are obtained from two different crystal forms of the same protein under different conditions, it has to be asked whether the different results correctly represent two different states of the protein (possibly corresponding to different steps along a reaction pathway) or whether the feature is actually invariant and one of the structures is in error. In the case of the orientation of the antimycin amide group it seems unlikely that both binding modes are possible. Unfortunately supporting data (structure factors) are not available for the 1NTK structure, which makes it impossible to test whether the data would have been equally consistent with our current model. However the density depicted in the stereodiagram of Figure 2A of ref 24 appears consistent with our structure, having an unaccounted-for protrusion about where we put the carbonyl oxygen, and having the modeled carbonyl oxygen at the edge of contoured density with no surrounding protrusion of the density.
While we want to emphasize that the structure of antimycin in 1PPJ is based on the electron density from x-ray diffraction by a crystal and not on chemical considerations or structure-activity relationships, the flipping of the salicylate amide moiety relative to the small-molecule structure seen here nicely explains why compound D of Reference 38, which is methylated on the amide N, is a poor inhibitor despite still having the internal H-bond (albeit weaker) between phenolic OH and carbonyl oxygen in solution. The amide of a secondary amine cannot be a hydrogen-bond donor, so the intramolecular H-bond that we see between phenolic OH and amide N cannot form, and the methyl group would clash with the phenolic OH preventing the molecule from taking this conformation. The explanation is less obvious with the structure presented in 1NTK, as the amide nitrogen is oriented toward a spacious part of the pocket containing only water molecules that might be expected to be displaceable (although methylation would prevent H-bonding with the water).
As for the discrepancies regarding the roles of Ser35 and Lys128, it seems more possible that the different rotamers observed in the current 1PPJ structure as opposed to 1NTK represent different states depending on pH or ionic strength. However the failure of that structure to correctly orient the amide linkage weakens the argument for different conformations of these residues. Since electron density was not shown supporting the modeled rotamers, and data are not available for independent evaluation, we hesitate to propose alternate conformations for these residues at this time.
Ser35 is not required for antimycin binding, as Rhodobacter and Paracoccus, which have Val or Ile here, are inhibited by antimycin. However Rhodospirillum rubrum has Ser as in mitochondria, and is more sensitive (in whole cells) than Rhodobacter (pers. communication of Fevzi Daldal). Schnaufer et al.68 found that mutation of Ser35 to Ile led to antimycin resistance in L. tarentolae. However the effect of Ser35Ile substitution might be expected due to steric effects, and is not necessarily indicative of a role of this residue in H-bonding to antimycin or stabilizing the waters involved in antimycin binding.
Despite evidence summarized in the introduction for a long-range conformational change induced by antimycin binding, no indication of such a change has been reported from the previous x-ray structures. Our analysis of the present structures also gives no indication of such a change, suggesting it must be a rather subtle change if it exists at all. Much of the evidence for a conformational change is circumstantial, and perhaps amenable to alternative explanations. For example the effect of antimycin on the stability in bile salts may involve strong binding interactions between the inhibitor and the protein serving to hold together the different parts of the sequence contributing to the binding site more strongly than they would be held together in the absence of the inhibitor, perhaps tying down a loose end to prevent some kind of “unraveling” which may initiate the cleavage reaction.
Antimycin may affect conformational dynamics of the protein in solution or embedded in the lipid bilayer, allowing or preventing the visitation of certain conformational states while not affecting the resting state that we see in the crystal, and these transient states may be responsible for the observed effects. It must also be remembered that all structures being compared here have stigmatellin at the Qo site, and it is possible that this tight-binding inhibitor locks the conformation of The Qo region and prevents conformational changes that would otherwise have been induced by antimycin. A similar comparison made with the chicken bc1 crystals in the absence of stigmatellin did not show any clear antimycin-induced change23, but the resolution was lower and refinement not very complete at that time.
Materials And Methods
Bovine hearts were obtained from a slaughterhouse or meat market and either used fresh or stored at −20°C or below before use in the mitochondrial preparation. “Sol-grade” dodecyl β-D-maltopyranoside (DM) and “anagrade” hexyl-β-D-glucopyranoside (HG) were purchased from Anatrace. Stigmatellin and polyethylene glycol (PEG) were from Fluka. Crystallization screen kits mentioned below, as well as cryocrystallography supplies, were from Hampton Research.
Mitochondrial protein was determined by the Lowry method69 with bovine serum albumin as a standard. Cytochrome bc1 concentration was determined from the difference in absorbance of the dithionite-reduced sample at 562 vs 600 nm, for which an extinction coefficient for the bovine enzyme of 70 cm−1mM−1 (E. Berry, unpublished; based on pyridine hemochrome analysis) was used.
Protein purification
Purification was as described70, involving solubilization of mitochondria with 1.0 g DM per gram protein, anion exchange chromatography on DEAE Sepharose CL6B with a gradient from 260–500 mM NaCl (in 50 mM KPi buffer pH 7.5, 0.5 mM EDTA, 0.1 g/l DM) and size-exclusion chromatography on Sepharose CL-6B in “sizing buffer” (20 mM K-MOPS pH 7.2, 100 mM NaCl, 0.5 mM EDTA, 0.1 g/l DM). Pooled fractions from the last column were adjusted to 5 μM cyt bc1 by ultrafiltration or dilution in the same buffer. Stigmatellin and Antimycin A were added to 10 μM (2-fold molar excess) from 10 mM and 15 mM alcoholic stock solutions.
Before setting up crystallization droplets a final step (PEG fractionation) was carried out in which the inhibitor-loaded bc1 complex was mixed with successive portions of a precipitant solution containing 100 mM KMES pH 6.4, 100 g/L PEG 4k, and 0.5 mM EDTA. This procedure clearly separates two populations, a minor fraction (“aggregated material”) which usually precipitates at around 0.3 volumes of precipitant and contains all of the contaminating cytochrome oxidase (present as supercomplex or micelles containing two separate complexes, and incompletely separated by the size-exclusion column) from the major fraction which usually does not begin to precipitate until more than 0.6 volumes have been added. In the case of the antimycin-containing crystal, material precipitating between 0.29 and 0.76 volumes of precipitant was collected by centrifugation and redissolved in several times the original volume of the above-mentioned sizing buffer. To reduce NaCl and residual PEG from precrystallization, the buffer was exchanged by several cycles of dilution in final buffer (20 mM Tris-HCl pH 7.5, 0.5 mM EDTA, 1 g/L UDM) and ultrafiltration on Amicon YM-100 membrane. It is difficult to dissolve the PEG-fractionation pellet directly in a small volume of final buffer- perhaps due to residual PEG it is necessary to have a higher ionic strength, provided by NaCl in the sizing buffer.
Crystallization
Crystallization was by sitting-drop vapor diffusion. Protein in the final buffer described above was mixed with 0.15 volume of 2.5 M HG, and then one volume (usually 10 μl) of this detergent-supplemented protein was mixed with 0.9 volume of major precipitant and 0.1 volume of minor precipitant/additive. The major precipitant consisted of 60 g/L PEG-3350, 100 g/L glycerol, 100 mM Na-cacodylate pH 6.7, 20 mM MgCl2, and 3 mM NaN3, and the minor precipitant/additive was Hampton Research's “Screen II #31”, consisting of 200 g/L Jeffamine M600 in 0.1 M HEPES pH 7.5.
We calculate the final pH to be about 6.86, ignoring buffering by the protein itself. The ionic strength is 72 mM before vapor diffusion. The droplets were allowed to equilibrate by vapor diffusion against a reservoir containing the major precipitant.
Data collection
The crystals were mounted in a nylon loop on a magnetic pin (Hampton Research) and flash-cooled in liquid nitrogen for cryogenic data collection. The diffraction limit and the cell parameters were highly variable, and in some cases warming the crystal to room temperature for 1–2 minutes and refreezing in the cold stream improved the diffraction dramatically. This seems to be related to the extent of dehydration of the crystals, and we are currently working on a way to optimize the diffraction by systematic dehydration.
The crystal from which structure 1PPJ was obtained was mounted in a loop and dipped in a mixture containing equal parts of the mother liquor and cryoprotectant (250 ml/L glycerol, 120 g/L PEG 4k, 10 mM K-MES pH 6.7, 3 mM Azide) before freezing in liquid nitrogen. After a preliminary exposure revealed diffraction to 4 Å and space group P212121 with cell parameters 152 × 178 × 227, the pin was removed from the cold stream and set, base down, at room temperature, so the crystal in the loop was dehydrated by the downdraft produced by the cold copper pin. After 3 minutes the pin was returned to the cold stream for data collection, now with resolution limit 2.0 Å and cell parameters 128, 169, 232.
The crystal for structure 1PP9 (without antimycin) was not intentionally dehydrated, however it was one of only 2 crystals diffracting to around 2 Å from about 40 that were mounted from the same well. It is likely that these two crystals were exposed to air longer than the others before freezing. The cell parameters for 1PP9 are somewhat intermediate between the before and after parameters for 1PPJ, suggesting it is less dehydrated. The other crystal from that well diffracting to 2.0 Å had essentially the same cell parameters as 1PPJ. This is the crystal Y21, mentioned in the discussion of antimycin-induced structural changes.
Diffraction patterns were collected in 0.5° rotations. Even for the best diffracting crystals, the mosaic spread was large (1.0–1.5°). In order to reduce the data to 2.0 Å without excessive overlap, it was necessary to assume a lower mosaic spread (0.6°) during spot integration. This results in sampling spot intensity near the maximum of the rocking curve (“profile peak sampling”) but ignores tails of the measured reflection's rocking curve as well as overlap from tails of neighboring spots in reciprocal space. This together with radiation decay described below contributes to the higher than usual R-merge and R-sym values for these datasets.
Both crystals used in the present work were rod-shaped, with dimensions ~0.2 x 0.2 x 1.5 mm. This allowed collecting several different datasets from each crystal, at different positions along the long axis of the crystal. It was later determined that significant radiation damage occurred during data collection as indicated by increasing B-factor. In the case of the antimycin-containing crystal (1PPJ), the final dataset was constructed by merging early data from each individual dataset, with a cutoff when the B-factor for scaling against a particular reference was more than 15 Å2 greater than that for the first exposures. The measurements from these selected frames from each data collection were individually scaled and merged in scalepack71. The resulting incomplete datasets were merged together using scalepack to make the final dataset. The statistic Rmerge in Table 1 and in the PDB file header refers to the R-merge obtained at this second merging step. For structure 1PP9 data from a single collection was used and Rmerge in Table 1 (Rsym in the PDB entry) refers to the initial merging of frames within the dataset.
To prepare for cross-validated (cv) refinement72 a test set of reflections (“Free-R flags”) was chosen from an ideally generated complete dataset to 1.8 Å, randomly selecting 5% of the reflections. This set of Free-R flags was used with every dataset from this crystal form to avoid biasing the cross-validation.
Structure determination
Phasing
The first (low resolution) dataset from a crystal of this new orthorhombic form was solved by molecular replacement using PDB entry 1BE3 as model. The iron-sulfur proteins were repositioned as in entry 2BCC, and several regions that were observed not to fit the 2Fo-Fc density map were rebuilt. As successively higher resolution datasets were collected, they were phased by molecular replacement using the best available previous structure from the same crystal form. Variation in cell parameters made rigid body refinement of the previous structure against the new data unreliable for positioning the molecule in the cell.
For each crystal, the model was refined by cycles of manual rebuilding using the graphics program O73 alternating with rigid body, multi-rigid-body, positional, and restrained atomic B-factor refinement in CNS74. When significant improvement was achieved in one crystal, the appropriate changes were transferred to the other crystals by refining the improved model to convergence against the other datasets, comparing atomic positions with the previous models for those datasets, and examining the differences in the density to decide which model was appropriate on a crystal-by-crystal and residue-by-residue basis.
Non-crystallographic symmetry
The crystal contains a dimer of the bc1 complex in the asymmetric unit. Initially non-crystallographic symmetry restraints were used for all protein atoms. During rebuilding to fit the electron density it became clear that certain residues did not obey NCS, and the restraints were released for those residues. For a while NCS restraints were eliminated, and the two monomers were refined and rebuilt independently. The resulting structure was examined to locate areas that seemed to violate NCS, and restraints were re-applied everywhere else. The remaining NCS violations were examined to determine whether the electron density supported the violation. If not, the residue was rebuilt in both monomers to be consistent with NCS and the restraint was re-imposed. If the NCS violation appeared real, the surrounding was examined for explanations in the form of crystal contacts. Except in the case of clear NCS violations, application of NCS restraints invariably improved the R-free statistic. It is not known, however to what extent this is due to improvement in the ratio of (data + restraints) to parameters, and to what extent to communication between the test and working sets of reflections (bias) due to the NCS relationship.
In addition to specific violations of NCS, subtle distortions of the protein between the two monomers were present, presumably due to intrinsic flexibility of the protein and the different packing forces. To allow for this flexibility without greatly increasing the number of parameters being fit, the NCS-restrained residues were divided into about 49 NCS groups each of which was allowed its own NCS operator.
Solvent
Water molecules were added with the water_pick program of CNS, and refreshed periodically by removal of waters flagged by whatcheck as too far from protein and picking of new waters. As the density improved, some of the solvent molecules took on distinct oblong or trigonal shapes, and some of these were modeled as oxygen or azide and glycerol, respectively. Phospholipid and detergent molecules appeared in varying states of disorder, and some of the best-defined have been modeled.
Validation
The refined structures were subjected to validation using procheck50, sfcheck48 and whatcheck75. Residues flagged as unusual were examined and in many cases rebuilt, then the refinement was repeated before testing again. ARP/wARP version 6.0 was used to eliminate model bias and confirm the well-determined parts of the structure by automated rebuilding from free atoms refined by the ARP/wARP76 process “automated model building starting from existing model”. ARP/wARP was able to trace the protein in as few as 53 chains containing over 3700 residues, as compared to 20 chains containing ~4020 residues in the final models.
When these steps ceased to yield further improvement, the model was saved and then submitted to a final round of non-cross-validated refinement (positional and B-individual) using all the data, with all parameters the same as during the final cv refinement. No manual adjustment was performed on the final refined structure. Refinement statistics for deposition were obtained by the “xtal_pdbsubmission” routine of CNS using both the final cv-refined structure and the final structure refined against all the data. The coordinates of the latter structure and the data used in refinement were deposited in the PDB.
Protein Data Bank accession number
Structure factors and coordinates have been submitted to the Protein Data Bank under the accession numbers 1PP9 (without) and 1PPJ (with antimycin). The structure referred to as Y21 is being deposited with accesion number 1???.
Acknowledgments
We would like to thank Antony R. Crofts, Patrick Crowley, Fevzi Daldal, Chris Earnshaw, Hideto Miyoshi, Graham Sexton, Bernard L. Trumpower, and Ya-Jun Zheng for helpful discussions during the course of this work and/or reading the manuscript and providing valuable suggestions. This work was supported by NIH research grants DK44842 from NIDDK and by GM62563 from NIGMS. Lawrence Berkeley National Lab (LBNL) is operated by the Department of Energy, contract DE-AC03-76SF00098 to the University of California.
Diffraction data were collected at the Advanced Light Source (ALS) at LBNL and at the Stanford Synchrotron Radiation Laboratory (SSRL), which is operated by the Department of Energy, Office of Basic Energy Sciences. The SSRL Biotechnology Program is supported by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and by the Department of Energy, Office of Biological and Environmental Research. We would like to thank Henry Belamy, Nick Sauter, Aina Cohen, and Dan Harrington at SSRL, and Keith Henderson and Corie Ralston at the ALS, for help with data collection; and Nick Sauter at LBNL for advice on data processing. The PDB entries 1PP9 and 1PPJ were processed by Shri Jain and Rose Oughtred of RCSB. The crystals leading to structures 1PP9 and Y21 were grown by Mr. Yusef Collins, at that time supported by a supplement to NIH grant GM62563.
Footnotes
Abbreviations used are: antimycin, antimycin A; cyt complex; DM, dodecyl bc1, cytochrome bc1 maltoside; UDM, undecyl maltoside; HG, β-hexyl-D-glucopyranoside; FSA, formylaminosalycylate moiety of antimycin; CC correlation coefficient; PDB, Protein Data Bank; 2Fo-Fc map, a map from Fourier coefficients calculated as twice the measured amplitude minus amplitude calculated from the structure, and phases calculated from the structure. “N-side” and “P-side” refer to the normally negative matrix side and positive inter-membrane side of the inner mitochondrial membrane. Structures that have been deposited in the PDB are referred to by their capitalized 4-character accession codes (e.g. “1P84”). Their authors and literature references can be obtained from the entry at the PDB if not given here. heme bH and heme bL refer to the high and low-potential hemes of cytochrome b. Heme B refers to Fe-protoporphyrin 9 irrespective of oxidation state. Heme C refers to a modified Heme B in which both vinyl groups of the porphyrin have been saturated by addition of cysteine Sγ, forming covalent links to the protein.
A collection of supplementary materials for this article, available from the publisher (), includes the following items.
Procheck validation reports for structures 1PP9 and 1PPJ
sfcheck validation reports for structures 1PP9 and 1PPJ
Set of figures laid out for cross-eyed stereo viewing
Scheme and description of the Q-cycle mechanism
Secondary structure diagram for bovine cytochrome b
Table of interaction distances between stigmatellin and the protein
Expanded Figure 5 with 7 views, one per page:
Stereodiagram of space-filling model of cyt b helix A backbone with intercolated waters W3 and W5.
Stereo views of omit map density for antimycin in 1PPJ.
Stereo views of omit map density for critical residues in 1PPJ C:Ser35, C:Lys227, C:His221-Pro222 (cis-peptide), C:His345-Pro346 (cis-peptide), D:Gly73-Pro74 (cis-peptide).
List of standard rotamers referred to, defined by side-chain dihedral angles:
VRML views of Figures1, 5, and 6 with electron density to allow examination from any angle.
The pyrrole rings of heme referred to here as A, B, C, and D correspond to protoporphyrin rings conventionally labeled IV, I, II, and III.
There are different conventions for naming the atoms in antimycin, so we have tried to specify atoms from a chemical standpoint rather than by name. The protein database maintains two versions of antimycin, AMY (from 3BCC) and ANY (introduced with 1PPJ). These have the same atom names, but in ANY two additional carbon atoms have been added to the end of the acyl chain to allow for the possibility that it is heptyl, and the methyl group on the acyl chain has been moved from the 3- to 2- position in accordance with recent results42. In the Cambridge Structure Database of small molecule structures there is (CCDC # 125007) from the work of Deisenhofer's group42, which uses different atom names. The PDB entry 1NTK uses the atom names from the Cambridge Database but the residue name (AMY) from the Protein Bata Bank. The atom names here, where used in the text and in Figure 1 and Table 4, are from ANY of 1PPJ.
In fact the helical bonding is interrupted, with normal α-helical bonding involving the N atom of residues after 35, 3/10 helical bonding involving the O of residues before 31, and no strong helical bonds of either sort involving O of residue 31 or 32 and N of residue 35. The distance from 31O to 35N is 5.1 Å, and to 34N 4.1 Å. The intercolated water W3 is well ordered, with B-factors 25 and 32 Å2 in 1PPJ (but 30 and 46 Å2 in 1PP9), well below average for the structure, and with density in 2Fo-Fc maps 3.9 to 4.7 σ. The heme-propionate-to-axial-ligand-bridging water W5 mentioned in connection with heme bHI can also be seen as intercolated, with a short H-bond to 33 O, but it is equally close to 33 N and 34 N, with neither being as close as 35 N is to W3 (Table 4).
Rotamer numbers used here refer to the lists of rotamers provided with the molecular visualization/modeling program O73, with the most frequently-occurring being in each case rotamer 1. Lysine rotamer 26 is an exception, coming from the more extensive collection of rotamers in reference67. The actual side-chain dihedrals of the rotamers referred to are as follows: Ser rotamer 1: chi1=63°; Ser 2: chi1= −62°; His 3: chi1 = −169°, chi2= 80°; His 5: chi1 = −59°, chi2 = 169°; Lys 26: chi1 = −66°, chi2= 180°, chi3= 67°, chi4=180°.
The structure Y21 was chosen because it's cell is most nearly isomorphous with 1PPJ, whereas 1PP9 has significantly different cell parameters. However superimposing 1PP9, or for that matter the yeast or chicken structures, puts ubiquinone in essentially the same place.
The N-terminus of cytochrome b up to residue 20 is modeled differently in 1PP9 and 1PPJ, to the extent that manual rebuilding would be required for convergence when refined against the same data. Density is not very clear here and it seems likely that multiple conformation exist for all three structures. This region was modeled differently in the bovine cyt bc1 structures from Uppsala (1BE3, 1BGY) as compared to those from Bethesda (e.g. 1L0L). The possibility has been raised77 that the N-terminus including α-a helix serves to transmit a conformational signal between Qi sites of the two monomers.
References
- 1.Mitchell P. Possible molecular mechanisms of the protonmotive function of cytochrome systems. J Theor Biol. 1976;62:327–67. doi: 10.1016/0022-5193(76)90124-7. [DOI] [PubMed] [Google Scholar]
- 2.Crofts AR, Meinhardt SW. A Q-cycle mechanism for the cyclic electron-transfer chain of Rhodopseudomonas sphaeroides. Biochem Soc Trans. 1982;10:201–3. doi: 10.1042/bst0100201. [DOI] [PubMed] [Google Scholar]
- 3.Mitchell P. A chemiosmotic molecular mechanism for proton-translocating adenosine triphosphatases. FEBS Lett. 1974;43:189–94. doi: 10.1016/0014-5793(74)80997-x. [DOI] [PubMed] [Google Scholar]
- 4.Trumpower BL. Evidence for a protonmotive Q cycle mechanism of electron transfer through the cytochrome b-c1 complex. Biochem Biophys Res Commun. 1976;70:73–80. doi: 10.1016/0006-291x(76)91110-4. [DOI] [PubMed] [Google Scholar]
- 5.Slater EC. The mechanism of action of the respiratory inhibitor, antimycin. Biochim Biophys Acta. 1973;301:129–54. doi: 10.1016/0304-4173(73)90002-5. [DOI] [PubMed] [Google Scholar]
- 6.Berden JA, Slater EC. The allosteric binding of antimycin to cytochrome b in the mitochondrial membrane. Biochim Biophys Acta. 1972;256:199–215. doi: 10.1016/0005-2728(72)90053-9. [DOI] [PubMed] [Google Scholar]
- 7.Deul DH, Thorn MB. Effects of 2,3-dimercaptopropanol and antimycin on absorption spectra of heart-muscle preparations. Biochim Biophys Acta. 1962;59:426–36. doi: 10.1016/0006-3002(62)90193-2. [DOI] [PubMed] [Google Scholar]
- 8.Chance B. The kinetics and inhibition of cytochrome components of the succinic oxidase system. III. Cytochrome b. J Biol Chem. 1958;233:1223–9. [PubMed] [Google Scholar]
- 9.Trumpower BL, Katki A. Controlled reduction of cytochrome b in succinate-cytochrome c reductase complex by succinate in the presence of ascorbate and antimycin. Biochem Biophys Res Commun. 1975;65:16–23. doi: 10.1016/s0006-291x(75)80055-6. [DOI] [PubMed] [Google Scholar]
- 10.Wikstrom MK, Berden JA. Oxidoreduction of cytochrome b in the presence of antimycin. Biochim Biophys Acta. 1972;283:403–20. doi: 10.1016/0005-2728(72)90258-7. [DOI] [PubMed] [Google Scholar]
- 11.Siedow JN, Power S, de la Rosa FF, Palmer G. The preparation and characterization of highly purified, enzymically active complex III from baker's yeast. J Biol Chem. 1978;253:2392–9. [PubMed] [Google Scholar]
- 12.Yu CA, Nagaoka S, Yu L, King TE. Evidence for the existence of a ubiquinone protein and its radical in the cytochromes b and c1 region in the mitochondrial electron transport chain. Biochem Biophys Res Commun. 1978;82:1070–8. doi: 10.1016/0006-291x(78)90296-6. [DOI] [PubMed] [Google Scholar]
- 13.Rieske JS, Baum H, Stoner CD, Lipton SH. On the antimycin-sensitive cleavage of complex 3 of the mitochondrial respiratory chain. J Biol Chem. 1967;242:4854–66. [PubMed] [Google Scholar]
- 14.Van Ark G, Berden JA. Binding of HQNO to beef-heart sub-mitochondrial particles. Biochim Biophys Acta. 1977;459:119–27. doi: 10.1016/0005-2728(77)90014-7. [DOI] [PubMed] [Google Scholar]
- 15.Baum H, Rieske JS, Silman HI, Lipton SH. On the Mechanism of Electron Transfer in Complex III of the Electron Transfer Chain. Proceedings of the National Academy of Sciences of the United States of America. 1967;57:798–805. doi: 10.1073/pnas.57.3.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rieske JS. Inhibitors of respiration at energy-coupling site 2 of the respiratory chain. Pharmacol Ther. 1980;11:415–50. doi: 10.1016/0163-7258(80)90036-4. [DOI] [PubMed] [Google Scholar]
- 17.Valkova-Valchanova M, Darrouzet E, Moomaw CR, Slaughter CA, Daldal F. Proteolytic cleavage of the Fe-S subunit hinge region of Rhodobacter capsulatus bc(1) complex: effects of inhibitors and mutations. Biochemistry. 2000;39:15484–92. doi: 10.1021/bi000751d. [DOI] [PubMed] [Google Scholar]
- 18.Covian R, Gutierrez-Cirlos EB, Trumpower BL. Anti-cooperative oxidation of ubiquinol by the yeast cytochrome bc1 complex. J Biol Chem. 2004;279:15040–9. doi: 10.1074/jbc.M400193200. [DOI] [PubMed] [Google Scholar]
- 19.Brandt U, von Jagow G. Analysis of inhibitor binding to the mitochondrial cytochrome c reductase by fluorescence quench titration. Evidence for a 'catalytic switch' at the Qo center. Eur J Biochem. 1991;195:163–70. doi: 10.1111/j.1432-1033.1991.tb15690.x. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Z, Huang L, Shulmeister VM, Chi YI, Kim KK, Hung LW, Crofts AR, Berry EA, Kim SH. Electron transfer by domain movement in cytochrome bc1. Nature. 1998;392:677–84. doi: 10.1038/33612. [DOI] [PubMed] [Google Scholar]
- 21.Brandt U. The chemistry and mechanics of ubihydroquinone oxidation at center P (Qo) of the cytochrome bc1 complex. Biochim Biophys Acta. 1998;1365:261–8. doi: 10.1016/s0005-2728(98)00078-4. [DOI] [PubMed] [Google Scholar]
- 22.Brandt U. Control of ubiquinol oxidation at center P (Qo) of the cytochrome bc1 complex. J Bioenerg Biomembr. 1999;31:243–50. doi: 10.1023/a:1005471829569. [DOI] [PubMed] [Google Scholar]
- 23.Berry EA, Huang LS, Zhang Z, Kim SH. Structure of the avian mitochondrial cytochrome bc1 complex. J Bioenerg Biomembr. 1999;31:177–90. doi: 10.1023/a:1005459426843. [DOI] [PubMed] [Google Scholar]
- 24.Gao X, Wen X, Esser L, Quinn B, Yu L, Yu CA, Xia D. Structural basis for the quinone reduction in the bc(1) complex: a comparative analysis of crystal structures of mitochondrial cytochrome bc(1) with bound substrate and inhibitors at the Q(i) site. Biochemistry. 2003;42:9067–80. doi: 10.1021/bi0341814. [DOI] [PubMed] [Google Scholar]
- 25.de Vries S, Albracht SP, Leeuwerik FJ. The multiplicity and stoichiometry of the prosthetic groups in QH2: cytochrome c oxidoreductase as studied by EPR. Biochim Biophys Acta. 1979;546:316–33. doi: 10.1016/0005-2728(79)90049-5. [DOI] [PubMed] [Google Scholar]
- 26.Glaser EG, Meinhardt SW, Crofts AR. Reduction of cytochrome b-561 through the antimycin-sensitive site of the ubiquinol-cytochrome c2 oxidoreductase complex of Rhodopseudomonas sphaeroides. FEBS Lett. 1984;178:336–42. doi: 10.1016/0014-5793(84)80629-8. [DOI] [PubMed] [Google Scholar]
- 27.Meinhardt, S. & Crofts, A. (1984). In Advances in Photosynthesis Research (Sybesma, C., ed.), Vol. 1, pp. 649–652. Martinus Nijhoff/Dr. W. Junk Publishers,, The Hague.
- 28.Robertson DE, Prince RC, Bowyer JR, Matsuura K, Dutton PL, Ohnishi T. Thermodynamic properties of the semiquinone and its binding site in the ubiquinol-cytochrome c (c2) oxidoreductase of respiratory and photosynthetic systems. J Biol Chem. 1984;259:1758–63. [PubMed] [Google Scholar]
- 29.Salerno JC, Xu Y, Osgood MP, Kim CH, King TE. Thermodynamic and spectroscopic characteristics of the cytochrome bc1 complex. Role of quinone in the behavior of cytochrome b562. J Biol Chem. 1989;264:15398–403. [PubMed] [Google Scholar]
- 30.Rich PR, Jeal AE, Madgwick SA, Moody AJ. Inhibitor effects on redox-linked protonations of the b haems of the mitochondrial bc1 complex. Biochim Biophys Acta. 1990;1018:29–40. doi: 10.1016/0005-2728(90)90106-e. [DOI] [PubMed] [Google Scholar]
- 31.Crofts, A., Barquera, B., Bechmann, G., Guergova, M., Salcedo-Hernandez, R., Hacker, B., Hong, S. & Gennis, R. (1995). In Photosynthesis: from light to biosphere. (Mathis, P., ed.), Vol. Vol. II, pp. 493–500. Kluwer Academic Publ.,, Dordrecht.
- 32.Kunz WS, Konstantinov AA. Effect of b-c1-site inhibitors on the midpoint potentials of mitochondrial cytochromes b. FEBS Lett. 1983;155:237–40. doi: 10.1016/0014-5793(82)80611-x. [DOI] [PubMed] [Google Scholar]
- 33.Dikanov SA, Samoilova RI, Kolling DR, Holland JT, Crofts AR. Hydrogen bonds involved in binding the Qi-site semiquinone in the bc1 complex, identified through deuterium exchange using pulsed EPR. J Biol Chem. 2004;279:15814–23. doi: 10.1074/jbc.M313417200. [DOI] [PubMed] [Google Scholar]
- 34.Mitani S, Araki S, Takii Y, Ohshima T, Matsuo N, Miyoshi H. The Biochemical Mode of Action of the Novel Selective Fungicide Cyazofamid: Specific Inhibition of Mitochondrial Complex III in Phythium spinosum. Pesticide Biochemistry and Physiology. 2001;71(9):107–115. [Google Scholar]
- 35.Ohshima T, Komyoji T. Development of a novel fungicide, cyazofamid, Mitani, S.; Matsuo, N.; Nakajima, T. J Pestic Sci. 2004;29:136–138. [Google Scholar]
- 36.Dickie JP, Loomans ME, Farley TM, Strong FM. The Chemistry of Antimycin A. Xi. N-Substituted 3-Formamidosalicylic Amides. J Med Chem. 1963;122:424–7. doi: 10.1021/jm00340a018. [DOI] [PubMed] [Google Scholar]
- 37.Miyoshi H, Kondo H, Oritani T, Saitoh I, Iwamura H. Inhibition of electron transport of rat liver mitochondria by unnatural (−)-antimycin A3. FEBS Lett. 1991;292:61–3. doi: 10.1016/0014-5793(91)80834-p. [DOI] [PubMed] [Google Scholar]
- 38.Miyoshi H, Tokutake N, Imaeda Y, Akagi T, Iwamura H. A model of antimycin A binding based on structure-activity studies of synthetic antimycin A analogues. Biochim Biophys Acta. 1995;1229:149–54. doi: 10.1016/0005-2728(94)00185-8. [DOI] [PubMed] [Google Scholar]
- 39.Tokutake N, Miyoshi H, Nakazato H, Iwamura H. Inhibition of electron transport of rat-liver mitochondria by synthesized antimycin A analogs. Biochim Biophys Acta. 1993;1142:262–8. doi: 10.1016/0005-2728(93)90154-8. [DOI] [PubMed] [Google Scholar]
- 40.Tokutake N, Miyoshi H, Satoh T, Hatano T, Iwamura H. Structural factors of antimycin A molecule required for inhibitory action. Biochim Biophys Acta. 1994;1185:271–8. doi: 10.1016/0005-2728(94)90241-0. [DOI] [PubMed] [Google Scholar]
- 41.Neft N, Farley TM. Inhibition of electron transport by substituted salicyl-N-(n-octadecyl)amides. J Med Chem. 1971;14:1169–70. doi: 10.1021/jm00294a007. [DOI] [PubMed] [Google Scholar]
- 42.Kim H, Esser L, Hossain MB, Xia D, Yu CA, Rizo J, Helm Dvd, Deisenhofer J. Structure of Antimycin A1, a Specific Electron Transfer Inhibitor of Ubiquinol-Cytochrome c Oxidoreductase. Journal of the American Chemical Society. 1999;121:4902 – 4903. [Google Scholar]
- 43.Birch J Chem Soc. 1961;1961:889. [Google Scholar]
- 44.van Tamelan EE, Dickie JP, Loomans ME, Dewey RS, Strong FM. J Am Chem Soc. 1961;83:1639–1646. [Google Scholar]
- 45.Ha ST, Wilkins CL, Abidi SL. Analysis of antimycin A by reversed-phase liquid chromatography/nuclear magnetic resonance spectrometry. Anal Chem. 1989;61:404–8. doi: 10.1021/ac00180a005. [DOI] [PubMed] [Google Scholar]
- 46.Xia D, Yu CA, Kim H, Xia JZ, Kachurin AM, Zhang L, Yu L, Deisenhofer J. Crystal structure of the cytochrome bc1 complex from bovine heart mitochondria [published erratum appears in Science 1997 Dec 19;278(5346):2037] Science. 1997;277:60–6. doi: 10.1126/science.277.5322.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weiss MS. Global indicators of X-ray data quality. Journal of Applied Crystallography. 2001;34:130–135. [Google Scholar]
- 48.Vaguine AA, Richelle J, Wodak SJ. SFCHECK: a unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr D Biol Crystallogr. 1999;55 ( Pt 1):191–205. doi: 10.1107/S0907444998006684. [DOI] [PubMed] [Google Scholar]
- 49.COLLABORATIVE COMPUTATIONAL PROJECT, N. The CCP4 Suite: Programs for Protein Crystallography''. Acta Cryst D. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 50.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. J App Cryst. 1993;26:283. [Google Scholar]
- 51.Iwata S, Lee JW, Okada K, Lee JK, Iwata M, Rasmussen B, Link TA, Ramaswamy S, Jap BK. Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex [see comments] Science. 1998;281:64–71. doi: 10.1126/science.281.5373.64. [DOI] [PubMed] [Google Scholar]
- 52.Hunte C, Koepke J, Lange C, Rossmanith T, Michel H. Structure at 2.3 A resolution of the cytochrome bc(1) complex from the yeast Saccharomyces cerevisiae co-crystallized with an antibody Fv fragment. Structure Fold Des. 2000;8:669–84. doi: 10.1016/s0969-2126(00)00152-0. [DOI] [PubMed] [Google Scholar]
- 53.Silman HI, Rieske JS, Lipton SH, Baum H. A new protein component of complex 3 of the mitochondrial electron transfer chain. J Biol Chem. 1967;242:4867–75. [PubMed] [Google Scholar]
- 54.Schagger H, Brandt U, Gencic S, von Jagow G. Ubiquinol-cytochrome-c reductase from human and bovine mitochondria. Methods Enzymol. 1995;260:82–96. doi: 10.1016/0076-6879(95)60132-5. [DOI] [PubMed] [Google Scholar]
- 55.Enders D, Geibel G, Osborne S. Diastereo- and enantioselective total synthesis of stigmatellin A. Chemistry. 2000;6:1302–9. doi: 10.1002/(sici)1521-3765(20000417)6:8<1302::aid-chem1302>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 56.Iwata, M. (2001). Structural Studies on cytochrome bc1 complex from Bovine Heart Mitochondria, Uppsala University.
- 57.Berry EA, Huang LS. Observations concerning the quinol oxidation site of the cytochrome bc1 complex. FEBS Lett. 2003;555:13–20. doi: 10.1016/s0014-5793(03)01099-8. [DOI] [PubMed] [Google Scholar]
- 58.Saraste M. Location of haem-binding sites in the mitochondrial cytochrome b. FEBS Lett. 1984;166:367–72. doi: 10.1016/0014-5793(84)80114-3. [DOI] [PubMed] [Google Scholar]
- 59.Widger WR, Cramer WA, Herrmann RG, Trebst A. Sequence homology and structural similarity between cytochrome b of mitochondrial complex III and the chloroplast b6-f complex: position of the cytochrome b hemes in the membrane. Proc Natl Acad Sci U S A. 1984;81:674–8. doi: 10.1073/pnas.81.3.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crofts, A., Robinson, H., Andrews, K., van Doren, S. & Berry, E. (1988). Catalytic Sites for Reduction and Oxidation of Quinones. In Cytochrome Systems, (Papa, S., ed.), pp. 617–624. Plenum Press, N.Y.
- 61.Berry EA, Zhang Z, Huang LS, Kim SH. Structures of Quinone binding sites in bc complexes: Functional implications. Biochem. Soc. Transactions. 1999;27:565–572. doi: 10.1042/bst0270565. [DOI] [PubMed] [Google Scholar]
- 62.Gray KA, Dutton PL, Daldal F. Requirement of histidine 217 for ubiquinone reductase activity (Qi site) in the cytochrome bc1 complex. Biochemistry. 1994;33:723–33. doi: 10.1021/bi00169a014. [DOI] [PubMed] [Google Scholar]
- 63.Xu JX, Xiao Y, Wang YH, Li X, Gu LQ. Comparison between the properties of 3-nitrosalicyl-N-alkylamide and antimycin A acting on QH2:cytochrome c reductase. Biochim Biophys Acta. 1993;1142:83–7. doi: 10.1016/0005-2728(93)90087-v. [DOI] [PubMed] [Google Scholar]
- 64.Lange C, Hunte C. Crystal structure of the yeast cytochrome bc1 complex with its bound substrate cytochrome c. Proc Natl Acad Sci U S A. 2002;99:2800–5. doi: 10.1073/pnas.052704699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hunte C, Solmaz S, Lange C. Electron transfer between yeast cytochrome bc(1) complex and cytochrome c: a structural analysis. Biochim Biophys Acta. 2002;1555:21–8. doi: 10.1016/s0005-2728(02)00249-9. [DOI] [PubMed] [Google Scholar]
- 66.Gao X, Wen X, Yu C, Esser L, Tsao S, Quinn B, Zhang L, Yu L, Xia D. The crystal structure of mitochondrial cytochrome bc1 in complex with famoxadone: the role of aromatic-aromatic interaction in inhibition. Biochemistry. 2002;41:11692–702. doi: 10.1021/bi026252p. [DOI] [PubMed] [Google Scholar]
- 67.Lovell SC, Word JM, Richardson JS, Richardson DC. The penultimate rotamer library. Proteins. 2000;40:389–408. [PubMed] [Google Scholar]
- 68.Schnaufer A, Sbicego S, Blum B. Antimycin A resistance in a mutant Leishmania tarentolae strain is correlated to a point mutation in the mitochondrial apocytochrome b gene. Curr Genet. 2000;37:234–41. doi: 10.1007/s002940050525. [DOI] [PubMed] [Google Scholar]
- 69.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–75. [PubMed] [Google Scholar]
- 70.Berry EA, Huang LS, DeRose VJ. Ubiquinol-cytochrome c oxidoreductase of higher plants. Isolation and characterization of the bc1 complex from potato tuber mitochondria. J Biol Chem. 1991;266:9064–77. [PubMed] [Google Scholar]
- 71.Otwinowski, Z. & Minor, W. (1997). Processing of X-ray Diffraction Data Collected in Oscillation Mode “, , Volume :, p., ,, Eds., . In Macromolecular Crystallography, part A (C.W. Carter, J. & Sweet, R. M., eds.), Vol. 276, pp. 307–326. Academic Press, New York. [DOI] [PubMed]
- 72.Brunger AT. The Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–474. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 73.Jones TA, Zhou J-Y, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors on these models. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 74.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 75.Hooft RW, Vriend G, Sander C, Abola EE. Errors in protein structures. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- 76.Perrakis A, Harkiolaki M, Wilson KS, Lamzin VS. ARP/wARP and molecular replacement. Acta Crystallogr D Biol Crystallogr. 2001;57:1445–50. doi: 10.1107/s0907444901014007. [DOI] [PubMed] [Google Scholar]
- 77.Berry EA, Huang LS, Saechao LK, Pon NG, Valkova-Valchanova M, Daldal F. X-Ray Structure of Rhodobacter Capsulatus Cytochrome bc1: Comparison with its Mitochondrial and Chloroplast Counterparts. Photosynthesis Research. 2004;81:251–275. doi: 10.1023/B:PRES.0000036888.18223.0e. [DOI] [PubMed] [Google Scholar]