

Abstract
We report three new structures of mitochondrial respiratory Complex II (succinate ubiquinone oxidoreductase, E.C. 1.3.5.1) at up to 2.1 Å resolution, with various inhibitors. The structures define the conformation of the bound inhibitors and suggest the residues involved in substrate binding and catalysis at the dicarboxylate site. In particular they support the role of Arg297 as a general base catalyst accepting a proton in the dehydrogenation of succinate. The dicarboxylate ligand in oxaloacetate-containing crystals appears to be the same as that reported for Shewanella flavocytochrome c treated with fumarate. The plant and fungal toxin 3-nitropropionic acid, an irreversible inactivator of succinate dehydrogenase, forms a covalent adduct with the side chain of Arg297. The modification eliminates a trypsin cleavage site in the flavoprotein, and tandem mass spectroscopic analysis of the new fragment shows the mass of Arg 297 to be increased by 83 Da and to have potential of losing 44 Da, consistent with decarboxylation, during fragmentation.
The toxin 3-nitropropionic acid (3-NP1) is produced by certain plants and fungi. It is a specific inhibitor of mitochondrial respiratory complex II. Fatalities after eating moldy sugarcane have been linked to 3-NP toxicity(1,2). Ruminants grazing in regions with 3-NP-producing plants acquire resistance due to reduction of the nitro group to an amine by ruminal bacteria(3).
The effectiveness of 3-NP in vivo after injection or oral administration has made it useful in studies involving tissues or whole animals. Ingestion of 3-NP results in neurodegeneration with symptoms resembling those of Huntington's disease(4), and conversely Huntington's disease results in loss of complex II activity(5), thus 3-NP has been used to produce an animal model for study of Huntington's(6,7). Symptoms also include convulsions, and 3-NP is being looked at for inducing a model of epilepsy(8). Prior subacute 3-NP poisoning seems to provide resistance to ischemic damage to nervous tissue by a preconditioning effect(9) similar to that resulting from mild ischemia.
The target of 3-NP is Complex II, which is both a member of the Krebs tricarboxylic acid cycle (oxidizing succinate to fumarate) and an entry-point for electrons into the respiratory chain at the level of ubiquinol. It consists of a large flavoprotein subunit (FP) containing covalently bound FAD, an iron-sulfur protein (IP) with three different iron-sulfur clusters, and two small membrane anchor subunits (chains C and D) ligating a single low-spin heme of type B. Human genetic defects in the IP subunits or chains C or D lead to development of paragangliomas(10,11). A mutation in chain C leads to premature aging in nematodes, presumably through excessive production of free radicals(12). Bacterial homologs succinate:quinone oxidoreductase (SQR) and menaquinol:fumarate oxidoreductase (FRD) in E. coli have been studied as genetically manipulable models for the mitochondrial protein. Recent reviews cover this family of enzymes (13–18). X-ray crystallographic structures are available for a number of members of the family. Available mitochondrial structures and representative bacterial examples are listed in Table 1.
Table 1.
Tabulation of some x-ray structures available for members of the SQR/FRD family.
| enzyme | source | ref | PDB Code | resolution | relevance |
|---|---|---|---|---|---|
| SQR | chicken | 1YQ3 | 2.2 | crystallized with OAA | |
| 1YQ4 | 2.4 | crystallized with 3-NPA | |||
| 9ZZZ | 2.1 | crystal soaked with carboxin | |||
| pig | (24) | 1ZOY | 2.4 | Arg(A298) out of active site | |
| 1ZP0 | 3.5 | with 3-NPA and TTFA | |||
| E. coli | (23) | 1NEK | 2.6 | Arg(A286) out of active site | |
| FCc | Shewanella | (38) | 1QJD | 1.8 | dicarboxylate site like SQR |
| FRD | Wolinella | (49) | 1QLA/B | 2.2 | "open" CAP domain |
| "" | (46) | 1QO8 | 3.0 | Arg(A301) disordered | |
| FRD | E. coli | (22) | 1KY6 | 2.7 | CAP domain slightly open |
The toxin 3-NP, structurally similar to and isoelectronic with the substrate succinate, is believed to be a suicide inactivator of Complex II. Alston and co-workers(19) proposed, based on previous observations and on their own experience with another flavo-protein, that the normal reaction pathway involves a temporary adduct with the N5 nitrogen of flavin, which in the case of 3-NP collapses to a stable adduct resulting in permanent inactivation. Irreversible covalent modification of the flavin was ruled out by later work(20) examining the spectral change induced and showing that unmodified flavin-peptide could be isolated from the inactivated complex by mild proteolysis. It was proposed that 3-NP is oxidized to 3-nitroacrylate, an unstable molecule which then reacts with some residue in the active site. A cysteine which was believed to be in the active site and essential for activity and for tight binding of another inhibitor, oxaloacetate (OAA), was suggested to be the residue involved.
Later studies showed this cysteine is not essential for activity or OAA binding. Recent elucidation of the structures of the E. coli FRD (21,22) and SQR (23) proteins showed that in fact the cysteine in question (residue 247 in FRD and 257 in SQR) is some 7–8 Å from the active site. A recent report of the structure of porcine complex II reveals for the first time the overall architecture of the mitochondrial enzyme(24) at 2.4 Å resolution. The location of difference density in the substrate-binding site after 3-NP treatment was also reported, however considering the lower resolution of that structure (3.5 Å), the specific model proposed for bound 3-NP has to be regarded as tentative. In any case the non-covalent binding described provides no explanation for the completely irreversible inactivation that is found with 3-NP.
We recently developed a method for reproducible crystallization of mitochondrial Complex II from chicken (25). We report here three structures of avian complex II: one treated with OAA, one treated with 3-NP, and one with no dicarboxylate-site inhibitors but with the quinone-site inhibitor carboxin. In the structure with added OAA, or in that with no added dicarboxylate ligand, the carboxylate site contains a malate-like ligand. The ligand and its surroundings are well-ordered, allowing assignment of the residues involved in substrate binding and putative catalytic roles at this site. In particular, the structure confirms that Arg297 is positioned for the role of general acid-base catalyst abstracting a proton during conversion of succinate to fumarate, which has not been clearly seen in any of the membrane bound SQR or FRD structures to date.
In the structure of 3-NP – treated protein, the density for the ligand is quite different, and can be modeled as a cyclic adduct of 3-NP with the catalytic Arg297. While the chemistry involved has not yet been elucidated, we suppose that 3-nitroacrylate or some intermediate derived from it reacts with Arg297 in the active site to form a cyclic adduct such as obtained by treating arginine with 1,2 or 1,3 dicarbonyls (26–30).
Materials and Methods
Purification, crystallization and phasing of the avian complex II protein were described in a preliminary report(25). As described, either one of two different crystal forms were obtained depending on conditions we have not yet determined. Type 1 crystals are orthorhombic with a monomer in the asymmetric unit (The same crystal form was reported(24) for the porcine enzyme), while type 2 crystals are monoclinic and contain a dimer. OAA and 3-NP were added to separate batches of the final purified protein in 2-fold molar excess before adding precipitant and additives, yielding type 1 crystals from both. Carboxin was soaked into a type 2 crystal by adding 0.5 μl of a 25 mM solution to a drop (initially set up with 15 μl each of protein solution and precipitant, and supplemented with 1 μl 15 mM MnCl2, 47.5% PEG as described(25)) after crystal growth was complete. Data was collected at the Advanced Light Source (Berkeley, California) and the Stanford Synchrotron Radiation Laboratory (Stanford, California). Data was processed using denzo and scalepack (31), Other crystallographic calculations utilized the CCP4 suite (32) including molecular replacement by amore (33).
The initial model was built in a type 1 crystal by the ARP/wARP program(34) using phases from the molecular replacement model (after considerable manual rebuilding), and was refined for each crystal by many cycles of automated refinement with CNS 1.1 (35) alternating with manual inspection and rebuilding using the molecular graphics program O(36). The monoclinic Type 2 crystals were solved by molecular replacement using the structure from the type 1 crystals. The two molecules in the asymmetric unit are related by a 2-fold axis perpendicular to the crystallographic screw axis, resulting in pseudo-orthorhombic symmetry broken only by a slight screw component (0 to 6 Å) along the noncrystallographic 2-fold. The pdb entry codes and final refinement statistics for the three structures presented here are shown in Table 2, with more detailed statistics in the online supplemental materials part S1. Figures 1, 2, 4, and 6 were made with molscript and raster3d. The electron density maps in Figures 2, 4, and 6 were made using CCP4 programs, calculating structure factors from the model with sfall, scaling Fobs to Fcalc with rstats, and calculating the 2Fo−Fc maps with fft.
Table 2.
Key refinement statistics for three Complex II structures used in this work. Additional statistics from the data processing and refinement work are available in supplemental materials.
| PDB ID code | 1YQ3 | 1YQ4 | 9ZZZ |
|---|---|---|---|
| Added Ligand | OAA | 3-NP | Carboxin |
| Space Group | P212121 | P212121 | P21 |
| Cell parameters | 70.0 × 84.4 × 289.5 | 69.6 × 83.5 × 288.6 | 122.0 × 203.4 × 69.0, 90.0 90.0 90.0 |
| Resolution range (last shell) | 8.66−2.20 2.24−2.19) | 56.38 − 2.33 (2.33 – 2.38) | 34.87 − 2.10 (2.1–2.15) |
| Completeness | 89.2% (47.1%) | 93.9% (80.2%) | 86.0 (42.8) |
| # Reflections | 78719 (2623) | 68501 (3672) | 166872 (5572) |
| Cryst. R Value | 0.176 (0.267) | 0.205 (0.32) | 0.195 (0.30) |
| Free R Value | 0.225 (0.313) | 0.259 (0.38) | 0.235 (0.36) |
| B Values | |||
| From Wilson Plot | 34.4 Å2 | 30.9 Å2 | 11.9 Å2 |
| Mean atomic B Val | 47.8 Å2 | 47.1 Å2 | 31.3 Å2 |
| RMS Deviations from Ideality: | |||
| Bond Lengths | 0.022 Å | 0.019 Å | 0.032 |
| Bond Angles | 1.8° | 1.9° | 2.0° |
| Dihedral Angles | 22.4° | 22.3° | 22.8 |
| Improper Angles | 1.02° | 1.08° | 1.15 |
Figure 1. Overall structure of mitochondrial complex II.
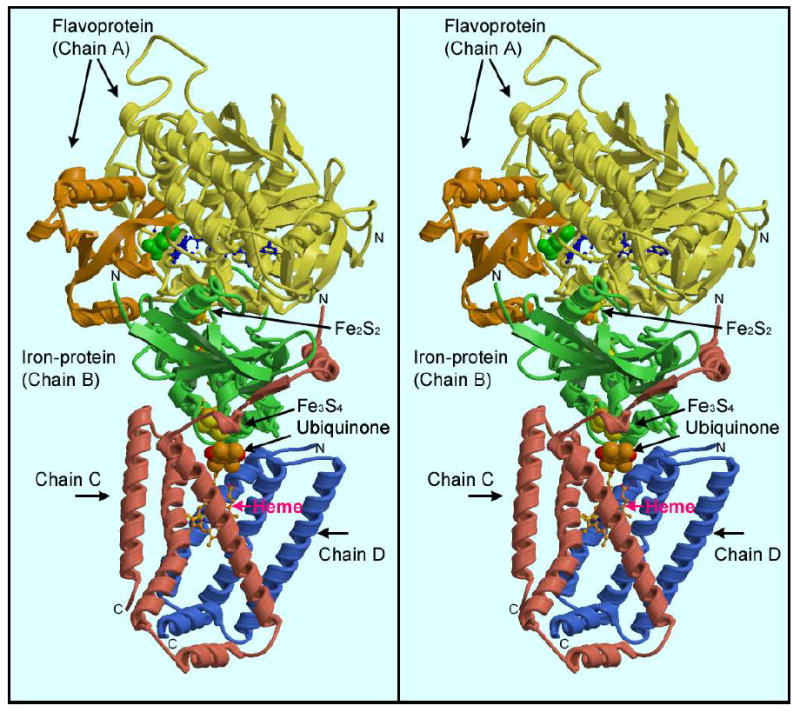
The stereo ribbon diagram is colored yellow & brown (flavoprotein), green (iron protein), pink (large anchor polypeptide, chain C), and blue (small anchor peptide, chain D). The CAP domain of the FP is colored brown. The green spacefilling model at the intersection between the CAP domain and the rest of the FP is the malate-like ligand, and the extended blue ball-and-stick model starting just to the right of that is the FAD cofactor. Note the first helix of anchor peptide (Chain C) packs against a helix of the IP.
Figure 2. The dicarboxylate site and its ligand in the untreated enzyme.
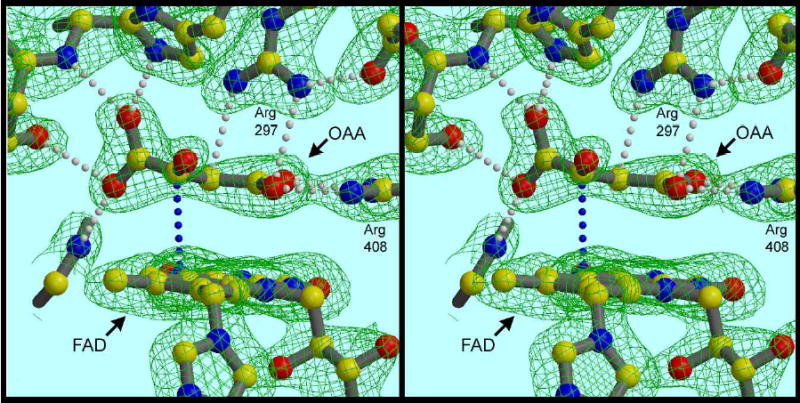
The occupant is nonplanar, suggesting that if oxaloacetate, it is highly strained. Note the H-bond between C2 of the ligand and catalytic base Arg297, and the close approach of C3 to the flavin N5 atom (blue dotted line). Essentially the same arrangement was seen when crystallization was carried out in the presence of excess OAA. The density is a 2Fo−Fc map contoured at 1.7 σ.
Figure 4. The dicarboxylate site in the 3-NP-treated enzyme.
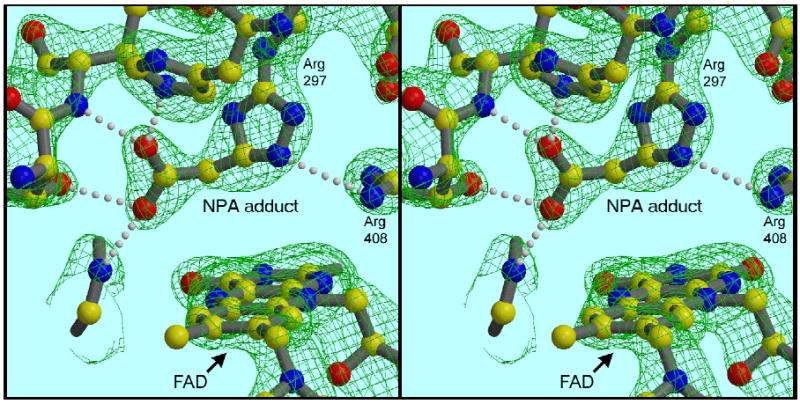
The density attributed to the ligand has shrunk and moved away from the flavin toward Arg297 and His253. It can be modeled by assuming two atoms from the backbone of 3-NP (C3 and N) fuse with the guanidino group to form a 5-membered ring, with loss of the two nitro oxygens. The carboxylate at the other end fits into the clearly forked density branching off the ring. 2Fo−Fc map contoured at 1.7 σ
Figure 6. Binding mode of carboxin, and a H-bonded network involving heme propionate and residues from three subunits.
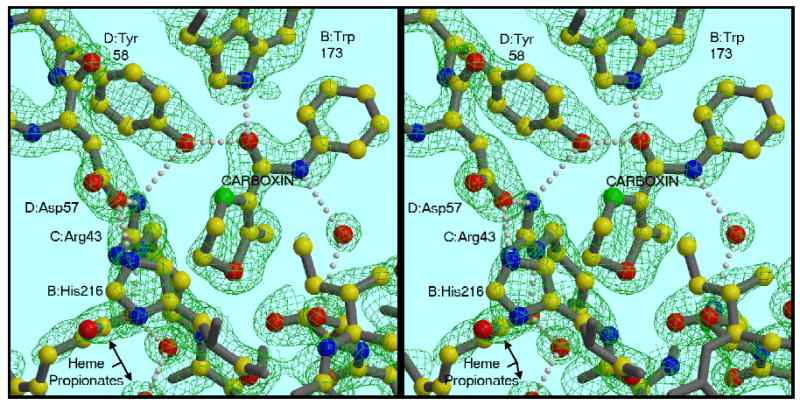
The carbonyl oxygen of the inhibitor is H-bonded to Tyr58 of chain D and Trp173 of chain B. D:Tyr58 is also H-bonded to C:Arg43 which makes H-bonds with a heme propionate and with D:Asp57. The latter H-bonds B:His216. The density is a 2Fo−Fc map contoured at 1.8 σ.
For mass spectral analysis, a sample of purified complex II (in 20 mM TrisHCl pH 7.5, 0.5 mM EDTA, 0.1 g/l dodecyl maltoside) was concentrated to 0.9 mM, treated with 3-NPA at 3 mM final concentration (from a 0.1 M stock solution in ethanol) and incubated four days at 4°. Another sample was analyzed without treatment. The samples were electrophoresed on parallel lanes of a Tricine-SDS-PAGE gel as described previously (37). Following electrophoresis and Coomassie staining, the bands containing control and 3-NP treated complex II subunits were sliced from the gel and tryptic in-gel digestions performed after reducing and alkylating cysteine (37). The tryptic peptides were extracted with 50% acetonitrile/5% formic acid and analyzed by LC/MS/MS using a Finnigan LTQ linear ion trap MS system, coupled with a nano-flow capillary HPLC column (100×0.18mm, Biobasic-18, Thermo-electron) and a 10-μm i.d. PicoTip nanospray emitter (New Objective, Woburn, MA). After dilution with 0.1% formic acid, the tryptic peptides were chromatographed with a gradient of 0%–80% CH3CN-0.1% formic acid at 500 nL/min with the ion source operated at 1.9 kV. The digest was analyzed by data-dependent acquisition of full scan mass spectra and MS/MS scans for the most abundant ion. The data obtained were processed by searching for modified residues in the tandem mass spectra against the chicken complex II sequence using TurboSequest. Further interpretation of the MS/MS spectrum of the modified peptide DLASR*DVVSR was performed manually with the aid of the web-based program MS-Product (www.prospector.ucsf.edu/ucsfhtml4.0/msprod.htm).
RESULTS
Overall structure
The overall structure of the mitochondrial protein has been described for the porcine complex(24), and is generally confirmed by the present higher resolution structure of the avian complex (Figure 1). Particularly noticeable is the packing of the N-terminal helix of chain C with the IP, which is not seen in the bacterial structures.
The overall folds of the porcine and chicken enzymes are essentially identical, as expected from the phylogenetic proximity of these organisms. Comparing the porcine structure 1ZOY1 with a chicken structure in the same crystal form, the 1089 residues that were modeled in both structures can be superimposed with rmsd 0.71 Å. Major differences are at N- and C- termini; a loop around A259; the distal part (around A568) of a free-floating loop of the FP between the helical domain and the C-terminal domain; and the transverse helix of chain C (comprising residues 66 to 79 in the chicken sequence). Excluding 17 residues with greatest differences gave rmsd 0.57 Å, decreasing to 0.45, 0.33, 0.57, and 0.42 Å when the individual subunits were superimposed separately. Comparing the same 1072 residues in the porcine structure with the 3-NP treated structure 1YQ4 and the two monomers of the carboxin-loaded structure 9ZZZ gave rmsd's of 0.60, 0.90 and 0.83 Å.
Architecture of the succinate-binding site and identification of key residues involved in binding and catalysis
In contrast to the backbone, a large number of side chains are clearly different than modeled in the porcine structure. This includes key residues of the dicarboxylate-binding site. Figure 2 is a stereo view of this site in a crystal (PDB entry 9ZZZ) to which no inhibitor of this site had been added. The same result was obtained when stoichiometrically excess OAA was present during crystallization (1YQ3). The 4-carbon backbone of the ligand, and the oxygens of the C4 carboxylate and C2 keto group, lie in a plane which is nearly parallel to the flavin isoalloxazine ring and in van-der-Waals contact with it. The C1 carboxylate moiety (adjacent to the keto group), which extends beyond the edge of the ring, is rotated about 60° out of the plane of the rest of the molecule. This conformation would interrupt the resonance stabilization due to conjugation between the vicinal keto and carboxylate groups of OAA. The significance of this conformation and the specific identity of the ligand in the dicarboxylate site will be considered below. In any case superimposing our structure with fumarate-treated flavocytochrome c (FCc) from Shewanella (1QJD) shows the ligand to be identical in conformation and orientation to the "malate-like intermediate" found in that structure.
Furthermore all of the side chains in the active site superimpose well with those of 1QJD, and even though Met236 and Met375 are not conserved, the side chains of Phe130 and Leu263 which replace them occupy as nearly as possible the same space. These residues were proposed to provide steric constraints which together with the H-bonding pattern result in twisting the carboxylate of the substrate out of plane(38). That appears to be the case in Complex II as well. Each of the four carboxylate oxygens of the ligand makes two H-bonds with the protein, as illustrated schematically in Figure 3. The ligand is correspondingly well ordered, with average B-factors 38, 24, and 24 Å2 in 1YQ3 and the two monomers of 9ZZZ, well below the average for all atoms. This strong similarity with the relatively distantly related Shewanella FCc was unexpected, as previous SQR and FRD structures from pig, E.coli, and Wollinella are rather more different.
Figure 3. Schematic of interactions of the ligand with the dicarboxylate site residues.
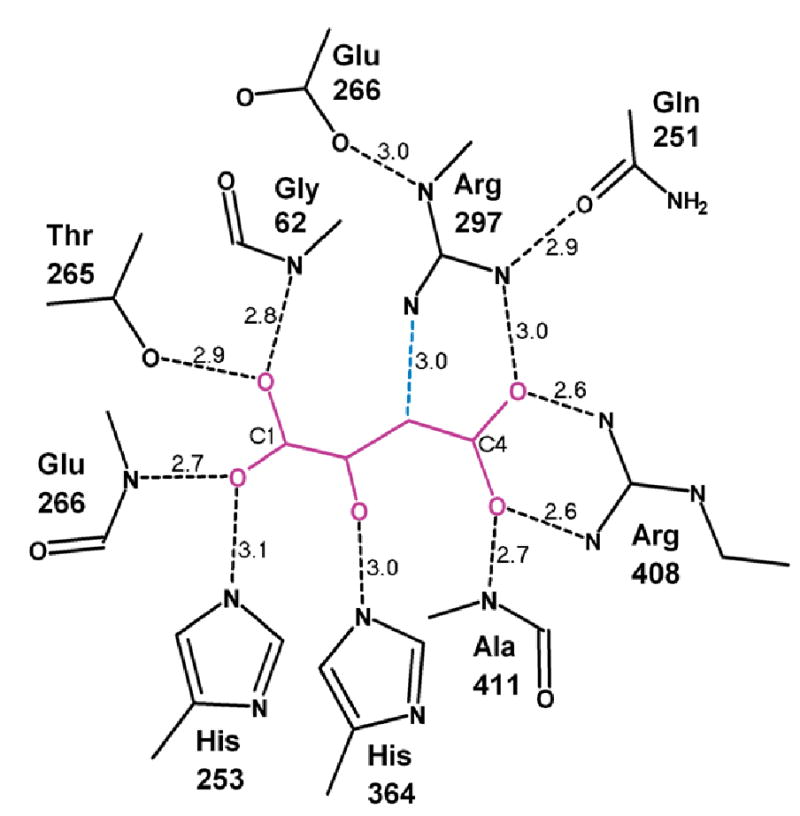
Eight strong hydrogen bonds to the four carboxylate oxygens locate the ligand in the binding site and maintain a nonplanar conformation. In addition, His364 is positioned to interact with carboxylate oxygen O1 or an oxygen on C3 in the inhibitor or a reaction intermediate. The green dotted line shows the interaction of catalytic base Arg297 with C3 of the ligand, from which a proton is extracted during oxidation of succinate.
The residue equivalent to Arg297 is believed from structural studies and site-directed mutagenesis to serve as a catalytic acid in the soluble FCc FRD (38,39), donating a proton to one end of the double bond while a hydride is transferred from flavin to the other end. Assuming that succinate oxidation in SQR occurs by the reverse of this mechanism, Arg297 should act as a general base catalyst to abstract a proton from one of the central carbons, while a hydride is transferred from the other to the flavin. In fact Arg297 is well positioned to abstract a proton, if it is assumed that succinate and fumarate bind as does the ligand in the 1YQ3 structure, in which a terminal nitrogen atom of Arg297 is approximately 3.0 Å from C3 of OAA. Furthermore the other two nitrogens of the guanidino group H-bond to carboxylates, which would tend to make Arg297 a stronger base: NH1 binds to the substrate carboxylate oxygen O1, while Nɛ binds the carboxylate of conserved Glu266. A third H-bond to Gln251, together with that to Glu266, serves to position the guanidino group. At the same time, C2 of OAA is about 3.1 Å from N5 of the flavin moiety (blue dotted line in Figures 2 and 3). This presumably represents the path of the hydride transfer.
If in fact the same intermediate is obtained starting with fumarate (as in structure 1QJD) or with OAA (as in the present studies), it suggests that SQR can carry out the Kreb's cycle reactions normally catalyzed by fumarase and malate dehydrogenase, although perhaps at very low rate. It is known that malate can be oxidized to OAA by FRD or SQR(40,41), and there is evidence that OAA is bound as the enol tautomer(42), with the double bond between C2 and C3, rather than the keto form which is more stable in aqueous solution. The geometry about C2 (the oxygen-bearing central carbon) is nearly planar rather than tetrahedral, which suggests sp2 hybridization. This would be consistent with either tautomer of OAA, but would imply the C1 carboxylate, which is about 60° out of plane of the C2 center, is in a strained conformation: resonance stabilization would favor an in-plane conformation. The unsaturated "malate-like intermediate" proposed for the ligand in 1QJD (Figure 3b of 38) would allow the out-of-plane C1 carboxylate, as the double bond between C3 and C4 is not conjugated with the carbonyl of C1. This would however result in sp3 hybridization for C2. C2 in the ligand of 1QJD in fact has (R) chirality as reported(38), however the improper angle is only 11°, which is more nearly planar than tetrahedral.
Regardless of the nature of the dicarboxylate in the active site of the OAA-inhibited enzyme, acid denaturation(43) or reductive activation(44) results in the release of a species that can be recovered as OAA in the medium. Since it seems unlikely that reductive activation would result in oxidation of malate to OAA, it seems more likely that the ligand in the inhibited enzyme is an OAA molecule in one of its tautomers.
Caution is warranted in interpreting the x-ray structures because x-ray irradiation generates reducing equivalents in materials. It is possible that the flavin is reduced early in the course of data collection, and we are looking at the structure of the reductively activated complex. The dissociation constant for OAA from the reduced enzyme is around 0.16 μM (45), so with the protein concentration in the crystal of 3.5 – 4 mM, only a small fraction would dissociate even after reduction. It is also possible that the ligand was originally OAA but has been reduced to malate by radiation-generated reducing equivalents, perhaps funneled through flavin and the normal catalytic mechanism.
In any case, it is clear that the observed structure contains a four-carbon dicarboxyllic acid. It seems reasonable to assume that succinate and fumarate bind in a similar fashion, and that Arg297 plays the role of catalytic base as suggested by the present structure. This is entirely consistent with the mechanism proposed for Shewanella FCc fumarate reductase (38,39), but was not supported by structures of the membrane proteins SQR (23) or FRD (21,22) from E. coli or porcine Complex II (24). A low-resolution structure of Wolinella FRD in a third crystal form (46) was interpreted as supporting this mechanism, however the arginine was disordered and not apparent in 2Fo−Fc electron density, so the inference was somewhat indirect.
Nitropropionic acid treatment results in covalent modification of Arg297 with formation of a substituted ring structure
In an attempt to explain the irreversible inactivation of Complex II by 3-NP, we treated a sample of Complex II with an excess of 3-NP before crystallization. The pattern of density in the dicarboxylate site in crystals from 3-NP – treated protein was dramatically different from that seen in the untreated or OAA-treated enzyme, with the density attributed to the dicarboxylate shrinking and shifting up towards the guanidino group of Arg297. Initial attempts to model the density with a free dicarboxylate resulted in strong positive difference density between Arg297 and the ligand unless the H-bonds connecting them were shortened to the dimensions of covalent bonds. The density could be fit well by assuming 3-NP fuses with Arg297 to make a ring structure (Figure 4), with loss of two oxygens from that end of 3-NP. We want to emphasize that this is only a model and that the resolution of the x-ray structure is not high enough to reliably determine the atomic structure. Furthermore we are not able to propose a detailed chemical mechanism by which this product would be obtained by reaction of nitropropionate or nitroacrylate with arginine. The chemical identity of the adduct and the chemistry leading to its formation is still under investigation.
In order to confirm the modification of Arg297 and potentially obtain information about the nature of the adduct, trypsin digestion and LC/MS/MS analyses (37) were carried out on the native and modified protein. A tryptic peptide present in the treated, but not in the untreated sample, had the mass/charge ratio consistent with a mass of 1200.9 Da, which could be accounted for by the predicted tryptic peptide plus an additional 83 Da. Tandem mass spectroscopy (Figure 5) confirmed this sequence, except that in place of R297 was a residue the mass of arginine plus 83 Da. This mass is consistent with the adduct depicted in Figure 4, but would also be consistent with one in which the orientation of the 3-NP backbone is reversed or one in which 3-NP undergoes cycloaddition of the guanidino group across the C=C double bond. However the latter possibility is not supported by the electron density seen crystallographically, and the second is made unlikely by identification of additional peaks in the tandem mass spectrum of the peptide in question, corresponding to predicted fragments, all containing Arg297, from which a mass of 44 had been lost. These are consistent with decarboxylation of the depicted adduct, but can not be readily explained if the ring substituent is a nitromethyl group as in the second scenario.
Figure 5. LTQ linear ion trap tandem mass spectrum obtained from fragmenting 3-NP modified peptide DLASR*DVVSR.
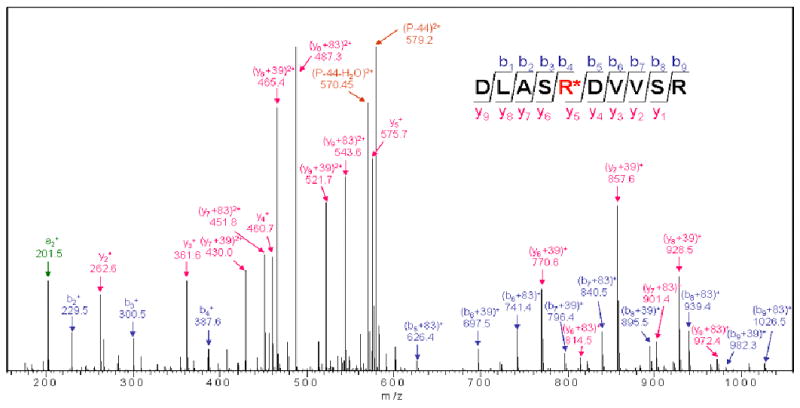
The doubly charged precursor ion of m/z 600.9 corresponds to a mass increment of 83 Da. This peptide was present in the digest of the 3-NP-treated sample but not the control. The peaks can be assigned as "b" and "y" fragments of the indicated sequence (corresponding to D293 – R302 of the mature flavoprotein sequence, and including the modified catalytic base R297) if it is assumed that R297 has an additional mass of 83 Da or 39 Da, consistent with the proposed adduct (NCCCO2) or its decarboxylation product (NCC). Peaks corresponding to the decarboxylation (P-44) and dehydrated decarboxylation (P-44-H2O) products of the parent peptide (P) are also indicated.
Carboxin and TTFA binding at the presumed quinone binding site
The residues making up the quinone binding site defined in the E. coli SQR structure are well conserved in the mitochondrial enzymes, and similarly positioned except that the side-chains of Asp57 in chain D and His218 in chain B are directly H-bonded, whereas they were farther apart and bridged by a water molecule in the E. coli structure 1NEK. There is density that would accommodate a quinone as built in 1NEK, however it is poorly ordered and cannot be unequivocally modeled. We have tentatively placed a ubiquinone headgroup in the density, but cannot be sure it is oriented correctly or even that ubiquinone is present.
On the other hand the crystals from protein treated with 2-methyl-1,4-oxathiin-3-carboxanilide, (carboxin) or thenoyl trifluoro-acetone (TTFA) had density of a different and better defined shape. In particular the density for the carboxin-treated enzyme (Figure 6) showed a puckered ring with a projection consistent with the methyloxathiin ring, and good density for the carbonyl group. The oxygen of the latter is in the position of quinone carbonyl oxygen of structure 1NEK, H-binding to Trp173 of chain B and Tyr58 of chain D. Density for the phenyl ring is less interpretable, perhaps this ring is rotationally disordered.
TTFA binds in a similar fashion (not shown), with the thenoyl group in the position of the oxathiin ring of carboxin and the carbonyl group of the thenoyl linkage H-bonding to B:Trp173 and D:Tyr58. This is essentially the same arrangement as reported for TTFA1 of the porcine enzyme(24). We see no evidence for carboxin or TTFA binding at the position of TTFA2 in that structure.
Other features of the structures
Possibly long heme-ligand distances
Sun et al. reported for the porcine enzyme that the histidine-Fe bonds by which the heme is ligated are 2.2 Å in length, which is rather longer than is seen in other bis-histidyl cytochromes b such as those of the cyt. bc1 complex (~2.00 Å) or model compounds(47). If we release the constraint on this bond length it does in fact increase to about 2.15 Å with refinement, but we do not feel this is highly significant in a structure of this resolution. Based on the wealth of information from related model compounds, including sterically hindered ligands that would tend to stretch the bond (reviewed in ref. 47), we restrained this length to 2.0 Å in the final refinement.
As usual, heme ligation is by histidine Nɛ2 atoms, the ligands being C:His98 and D:His46 in this case. The Nδ1 atom of C:His98 makes long, weak H-bonds to Oγ1 atom of Thr45 and O and Nɛ2 atoms of His42 in the same chain. The Nδ1 atom of D:His46 has a short H-bond to a water and a long bond to Oγ atom of D:Ser17.
Ordered metal atoms
Two of the density peaks initially picked as "waters" had unusually high density. A Bivoet difference map calculated from a dataset collected at 1.74154 Å revealed a significant anomalous signal associated with these peaks. The anomalous peaks of the two atoms were stronger than those of sulfurs (cysteine and methionine) but weaker than those of irons. Coordination for both was roughly octahedral, with ligand distances in the range 2.6 – 3.3 Å. They have been assigned as K+ ions at unit occupancy, and refined to B factors of 28–45 Å2 for K1, and 59–66 Å2 for K2 in various type 1 crystals. Both were also present in the type 2 crystal form, with B-values of 20 and 27 Å2 for K1, 40 and 41 Å2 for K2 in the two monomers of 9ZZZ.
K1 is ligated by flavoprotein residues: the phenolic OH of Tyr365 and carbonyl oxygens of residues 366, 367, 368 (in the segment between strand E2 and F1), and residues 397 and 399 (in short strand F2). In this site in flavocytochrome c (1QJD) a sodium ion has been modeled, E. coli FRD (1KF6) has a potassium, and the E. coli SQR (1NEK) has a calcium ion. It is not known whether the metal at this site is readily exchangeable and thus reflects the crystallization medium, but the fact that this metal binding site has been preserved throughout evolution of this protein family suggests it plays an important role, perhaps a structural role as suggested for FCc(38).
K2 is on the surface of the IP, ligated by carbonyl oxygens of residues 191, 193, 196; OG1 of Thr199, and two water molecules. A water has been modeled here in 1KF6. Nothing is in the same place in 1NEK, but a Ca++ ion is modeled in the position of one of the H-bonded waters. This site is on the surface and would be expected to be exchangeable.
In the present structures the coordination distances2 make Ca an unlikely assignment for either K1 or K2, and the anomalous signal is too strong for Na, for which f" is much less than that of S at 1.742 Å.
Cis-serine peptide
The peptide linkage between Ala401 and Ser402 in the flavoprotein subunit has cis geometry. This is unusual, especially when the residue (i+1) is not proline, but not unprecedented. A study(48) of 571 protein sequences with less than 25% sequence identity and structures of better than 3.5 Å resolution found 43 out of 145796 Xaa-nonPro peptides, or 0.029%, to be in cis conformation. The fraction increased to 27 out of 69160 or 0.039% when only structures of resolution 2.0 Å or better were considered, attesting to the frequency with which these bonds are misbuilt as trans in low-resolution structures.
The region is quite well ordered, and the density leaves no doubt that it is modeled correctly in the current structures. A simulated-annealing omit map showing the density for these residues is included in the supplemental materials. The cis-peptide occurs in a compound turn between short strand F2 and helix 14, comprising residues 401–411. Also in this turn is Arg408, involved in binding OAA at the carboxylate site. The cis-peptide is well outside the active site, but the entire turn and start of helix α12 is conserved in SQR flavoproteins (401[AV]SVHGANRL GANSL[LI]DLVVFGRA423), and conservatively modified in FRD's. Ser402 becomes Gly or Asp in FRD from E. coli or Wolinella, and there is a single-residue gap at or adjacent to the dipeptide in question in Shewanella. Comparing the chicken structure to that of Shewanella (1QJD), residues 400–402 replace residues 537 and 538 of the latter structure, with the cis-peptide generating a bulge which accommodates the extra residue in the mitochondrial structure. It is interesting to note the proximity of this site to the highly conserved metal-binding site K1, for which A:397 and 399 are ligands. While the cis-peptide does not interact with the metal, it may be that both features contribute to a unique conformation of this stretch of the backbone that holds Arg408 in position for binding the dicarboxylate ligand.
DISCUSSION
The three new structures of avian mitochondrial respiratory Complex II reported here provide strong evidence for a mechanism for the succinate/fumarate site similar to that of the distantly-related Shewanella FCc fumarate reductase. The structures with endogenous or added OAA bound have the ligand and surrounding side chains in the same orientation as the structures of that enzyme treated with fumarate. As in that structure, Arg 297 is positioned to abstract or donate a proton to C2 or C3 of a dicarboxylate
While Arg297 is conserved in all the SQR/FRD family, previous structures have shown the corresponding residue to be in different conformations or positions relative to the ligand, and thus raise the question whether a different mechanism is operative in the membrane-bound, quinone-coupled succinate/fumarate oxidoreductases.
The high-resolution structures of the Wolinella SQR enzyme (1QLA, 1QLB) have the CAP domain, to which Arg297 is attached, in an "open" position which puts this residue too far from the ligand to serve a catalytic role. This led to the proposal(49) that a water molecule donates the proton to the substrate during fumarate reduction. A later low-resolution structure in a third crystal form (46) had the CAP domain in the closed position. This structure was interpreted as supporting the Shewanella mechanism, however the arginine was disordered and not apparent in 2Fo−Fc electron density, so the inference was somewhat indirect.
The structures of E. coli FRD (e.g. 1KF6) also have the CAP domain slightly displaced which prevents interaction between the residue corresponding to Arg297 and the ligand. The structures of SQR from E. coli or pig have the CAP domain in essentially the same closed position observed in the chicken structures (or the structures from Shewanella FCc or Wolinella SQR in crystal form C), however the residue corresponding to Arg297 has been built in a different conformation which does not reach the ligand-binding site. We remodeled this residue in 1ZOY and refined against the data submitted by the authors in support of the structure (See supplemental materials). From this experiment we conclude that the data are consistent with this residue being in the same conformation we modeled in the chicken structure. No data have been deposited in support of the E.coli QFR structures.
The toxin 3-NP irreversibly inactivates Complex II, and various invetigations of the mechanism(19,20) have all proposed covalent modification of the catalytic machinery. The noncovalent binding mode presented by Sun et al.(24) in structure 1ZP0 must have come as a surprise to many students of this enzyme. Superimposing this structure on our 1YQ4 shows good correlation between the density, if not the structure. Comparing with that structure, it appears the authors have placed the inhibitor in density which belongs mainly to the guanidino group of Arg297 (298 in the pig sequence), which they built in an unusual rotamer (a Chi-1 outlier by the PROCHECK analysis). This sort of error is not unexpected in low-resolution structures. Again, rebuilding this residue as in the chicken structure and refining against the authors' deposited data led to a better fit to the density (supplemental materials), although the interpretation is not as clear as with the higher-resolution 1ZOY structure.
Supplementary Material
Footnotes
The crystallographic parts of this work were supported by NIH R01 grants GM62563) and DK44842 and were carried out at Lawrence Berkeley National Lab, which is operated by the Department of Energy, contract DE-AC03-76SF00098 to the University of California. Diffraction data were collected at the Advanced Light Source (ALS) at LBNL and at the Stanford Synchrotron Radiation Laboratory, which is operated by the Department of Energy, Office of Basic Energy Sciences. We thank staff at the various beamlines for help and advice in data collection. The mass spectroscopic portions were supported by NIA PO1 AG15885 and were carried out at the Case Center for Proteomics.
Abbreviations used are: 3-NP, 3-nitropropionic acid; OAA, oxaloacetatic acid; PDB, Protein Data Bank; carboxin, 2-methyl-1,4-oxathiin-3-carboxanilide; TTFA, thenoyl trifluoroacetone; FAD, Flavin adenine dinucleotide; SQR, Complex II (succinate:quinone reductase); FRD, fumarate reductase; FCc, flavocytochrome c FRD; FP, flavoprotein of SQR/FRD; IP, iron-sulfur protein of SQR/FRD. Structures deposited at the Protein Databank are refered to by their PDB accession codes, summarized in Table 1.
Predicted coordination distances were calculated as the sum of ionic radii, taking 1.36 Å for O, 1.0 Å for octahedral Ca, and 1.52 for octahedral K, predicting oxygen ligand distances around 2.36 for Ca and 2.88 for K.
Supplemental Data:
S1. Statistics from the structure determination.
S2. Table listing ions detected by tandem mass spectroscopy 3-NP modified peptide, with calculated and observed molecular mass
S3. Selected ion chromatograms of m/z 600.9 obtained by reverse phase HPLC of the tryptic peptides from control and 3-NP-inactivated Complex II.
S4–S6. Simulated-annealing omit maps for residue A:Arg297 and the dicarboxylate ligand, Arg297 adduct after NPA treatment, and cis-peptide A:Ala401, A:Ser402
S7. Evaluation of apparent differences between the avian and porcine structures.
S8. Figure depicting the enol form of OAA as the "malate-like intermediate" in the dicarboxylate site.
References
- 1.Ming L. J Toxicol Clin Toxicol. 1995;33:363–367. doi: 10.3109/15563659509028924. [DOI] [PubMed] [Google Scholar]
- 2.Liu X, Luo X, Hu W. Biomed Environ Sci. 1992;5:161–177. [PubMed] [Google Scholar]
- 3.Anderson RC, Rasmussen MA, Allison MJ. Appl Environ Microbiol. 1993;59:3056–3061. doi: 10.1128/aem.59.9.3056-3061.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT. J Neurosci. 1993;13:4181–4192. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Browne SE, Bowling AC, MacGarvey U, Baik MJ, Berger SC, Muqit MM, Bird ED, Beal MF. Ann Neurol. 1997;41:646–653. doi: 10.1002/ana.410410514. [DOI] [PubMed] [Google Scholar]
- 6.Borlongan CV, Koutouzis TK, Sanberg PR. Neurosci Biobehav Rev. 1997;21:289–293. doi: 10.1016/s0149-7634(96)00027-9. [DOI] [PubMed] [Google Scholar]
- 7.Borlongan CV, Koutouzis TK, Freeman TB, Hauser RA, Cahill DW, Sanberg PR. Brain Res Brain Res Protoc. 1997;1:253–257. doi: 10.1016/s1385-299x(96)00037-2. [DOI] [PubMed] [Google Scholar]
- 8.Urbanska EM. Pol J Pharmacol. 2000;52:55–57. [PubMed] [Google Scholar]
- 9.Brambrink AM, Noga H, Astheimer A, Heimann A, Kempski O. Acta Neurochir Suppl. 2004;89:63–66. doi: 10.1007/978-3-7091-0603-7_8. [DOI] [PubMed] [Google Scholar]
- 10.Niemann S, Muller U. Nat Genet. 2000;26:268–270. doi: 10.1038/81551. [DOI] [PubMed] [Google Scholar]
- 11.Baysal BE, Willett-Brozick JE, Lawrence EC, Drovdlic CM, Savul SA, McLeod DR, Yee HA, Brackmann DE, Slattery WH, 3rd, Myers EN, Ferrell RE, Rubinstein WS. J Med Genet. 2002;39:178–183. doi: 10.1136/jmg.39.3.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Senoo-Matsuda N, Hartman PS, Akatsuka A, Yoshimura S, Ishii N. J Biol Chem. 2003;278:22031–22036. doi: 10.1074/jbc.M211377200. [DOI] [PubMed] [Google Scholar]
- 13.Ackrell BA. FEBS Lett. 2000;466:1–5. doi: 10.1016/s0014-5793(99)01749-4. [DOI] [PubMed] [Google Scholar]
- 14.Ackrell BA. Mol Aspects Med. 2002;23:369–384. doi: 10.1016/s0098-2997(02)00012-2. [DOI] [PubMed] [Google Scholar]
- 15.Cecchini G. Annu Rev Biochem. 2003;72:77–109. doi: 10.1146/annurev.biochem.72.121801.161700. [DOI] [PubMed] [Google Scholar]
- 16.Hagerhall C. Biochim Biophys Acta. 1997;1320:107–141. doi: 10.1016/s0005-2728(97)00019-4. [DOI] [PubMed] [Google Scholar]
- 17.Lancaster CR. FEBS Lett. 2003;555:21–28. doi: 10.1016/s0014-5793(03)01100-1. [DOI] [PubMed] [Google Scholar]
- 18.Ohnishi T, Moser CC, Page CC, Dutton PL, Yano T. Structure Fold Des. 2000;8:R23–32. doi: 10.1016/s0969-2126(00)00098-8. [DOI] [PubMed] [Google Scholar]
- 19.Alston TA, Mela L, Bright HJ. Proc Natl Acad Sci U S A. 1977;74:3767–3771. doi: 10.1073/pnas.74.9.3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coles CJ, Edmondson DE, Singer TP. J Biol Chem. 1979;254:5161–5167. [PubMed] [Google Scholar]
- 21.Iverson TM, Luna-Chavez C, Cecchini G, Rees DC. Science. 1999;284:1961–1966. doi: 10.1126/science.284.5422.1961. [DOI] [PubMed] [Google Scholar]
- 22.Iverson TM, Luna-Chavez C, Croal LR, Cecchini G, Rees DC. J Biol Chem. 2002;277:16124–16130. doi: 10.1074/jbc.M200815200. [DOI] [PubMed] [Google Scholar]
- 23.Yankovskaya V, Horsefield R, Tornroth S, Luna-Chavez C, Miyoshi H, Leger C, Byrne B, Cecchini G, Iwata S. Science. 2003;299:700–704. doi: 10.1126/science.1079605. [DOI] [PubMed] [Google Scholar]
- 24.Sun F, Huo X, Zhai Y, Wang A, Xu J, Su D, Bartlam M, Rao Z. Cell. 2005;121:1043–1057. doi: 10.1016/j.cell.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 25.Huang LS, Borders TM, Shen JT, Wang CJ, Berry EA. Acta Crystallogr D Biol Crystallogr. 2005;61:380–387. doi: 10.1107/S0907444905000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Westwood ME, Thornalley PJ. J Protein Chem. 1995;14:359–372. doi: 10.1007/BF01886793. [DOI] [PubMed] [Google Scholar]
- 27.Toi K, Bynum E, Norris E, Itano HA. J Biol Chem. 1965;240:PC3455–3457. [PubMed] [Google Scholar]
- 28.Toi K, Bynum E, Norris E, Itano HA. J Biol Chem. 1967;242:1036–1043. [PubMed] [Google Scholar]
- 29.Gilbert HF, 3rd, O'Leary MH. Biochemistry. 1975;14:5194–5199. doi: 10.1021/bi00694a027. [DOI] [PubMed] [Google Scholar]
- 30.Oya T, Hattori N, Mizuno Y, Miyata S, Maeda S, Osawa T, Uchida K. J Biol Chem. 1999;274:18492–18502. doi: 10.1074/jbc.274.26.18492. [DOI] [PubMed] [Google Scholar]
- 31.Otwinowski, Z., and Minor, W. (1997) in Macromolecular Crystallography, part A (C.W. Carter, J., and Sweet, R. M., eds) Vol. 276, pp. 307–326, Academic Press, New York
- 32.COLLABORATIVE COMPUTATIONAL PROJECT, N. Acta Cryst D. 1994;D50:760–763. [Google Scholar]
- 33.Navaza J. Acta Cryst A. 1994;A50:157–163. [Google Scholar]
- 34.Lamzin, V. S., Perrakis, A., and Wilson, K. S. (2001) in International Tables for Crystallography.Volume F: Crystallography of biological macromolecules (Rossmann, M. G., and Arnold, E., eds), pp. 720–722, Kluwer Academic Publishers, Dordrecht
- 35.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 36.Jones TA, Zhou J-Y, Cowan SW, Kjeldgaard M. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 37.Sun G, Kinter MT, Anderson VE. J Mass Spectrom. 2003;38:531–539. doi: 10.1002/jms.467. [DOI] [PubMed] [Google Scholar]
- 38.Taylor P, Pealing SL, Reid GA, Chapman SK, Walkinshaw MD. Nat Struct Biol. 1999;6:1108–1112. doi: 10.1038/70045. [DOI] [PubMed] [Google Scholar]
- 39.Doherty MK, Pealing SL, Miles CS, Moysey R, Taylor P, Walkinshaw MD, Reid GA, Chapman SK. Biochemistry. 2000;39:10695–10701. doi: 10.1021/bi000871l. [DOI] [PubMed] [Google Scholar]
- 40.Dervartanian DV, Veeger C. Biochim Biophys Acta. 1965;105:424–436. doi: 10.1016/s0926-6593(65)80228-4. [DOI] [PubMed] [Google Scholar]
- 41.Belikova YO, Kotlyar AB, Vinogradov AD. Biochim Biophys Acta. 1988;936:1–9. doi: 10.1016/0005-2728(88)90245-9. [DOI] [PubMed] [Google Scholar]
- 42.Panchenko MV, Vinogradov AD. FEBS Lett. 1991;286:76–78. doi: 10.1016/0014-5793(91)80944-x. [DOI] [PubMed] [Google Scholar]
- 43.Schroder I, Gunsalus RP, Ackrell BA, Cochran B, Cecchini G. J Biol Chem. 1991;266:13572–13579. [PubMed] [Google Scholar]
- 44.Ackrell BA, Kearney EB, Edmondson D. J Biol Chem. 1975;250:7114–7119. [PubMed] [Google Scholar]
- 45.Kotlyar AB, Vinogradov AD. Biochim Biophys Acta. 1984;784:24–34. doi: 10.1016/0167-4838(84)90168-7. [DOI] [PubMed] [Google Scholar]
- 46.Lancaster CR, Gross R, Simon J. Eur J Biochem. 2001;268:1820–1827. [PubMed] [Google Scholar]
- 47.Walker FA. Chem Rev. 2004;104:589–615. doi: 10.1021/cr020634j. [DOI] [PubMed] [Google Scholar]
- 48.Stewart DE, Sarkar A, Wampler JE. J Mol Biol. 1990;214:253–260. doi: 10.1016/0022-2836(90)90159-J. [DOI] [PubMed] [Google Scholar]
- 49.Lancaster CR, Kroger A, Auer M, Michel H. Nature. 1999;402:377–385. doi: 10.1038/46483. [DOI] [PubMed] [Google Scholar]
- 50.Vaguine AA, Richelle J, Wodak SJ. Acta Crystallogr D Biol Crystallogr. 1999;55 (Pt 1):191–205. doi: 10.1107/S0907444998006684. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.