

Abstract
The authors report a case of a 64-year-old male with Alzheimer’s disease (AD) and dementia with Lewy bodies (DLB) pathology at autopsy who did not manifest the core symptoms of DLB until very late in his clinical course. His initial presentation of early executive and language dysfunction suggested a cortical dementia similar to frontotemporal lobar degeneration (FTLD). Core symptoms of DLB including dementia, hallucination, and parkinsonian symptoms were not apparent until late in the course of his illness. Autopsy revealed both brainstem and cortical Lewy bodies and AD pathology. Family history revealed 7 relatives with a history of dementia including 4 with possible or probable DLB. This case is unique because of the FTLD-like presentation, positive family history of dementia, and autopsy confirmation of DLB.
Keywords: Lewy bodies, familial dementia, Alzheimer’s disease
Dementia with Lewy bodies (DLB) may be the second leading cause of dementia after Alzheimer’s disease (AD).1 Autopsy studies have demonstrated Lewy bodies (LBs) in the brains of at least 15% to 25% of patients with dementia.2–4 Approximately 50% to 70% of DLB cases also have sufficient Alzheimer’s pathology to meet criteria for AD.5 Clinically, DLB is frequently characterized by dementia, fluctuation in cognition and attention, visual hallucinations, and parkinsonian symptoms.1 “Cortical” symptoms, such as executive dysfunction and aphasia, occur in more advanced stages of DLB but have not been reported as presenting symptoms.6–9
In this report, we describe a case of neuropathologically confirmed DLB, with early executive dysfunction and aphasia with a positive family history of dementia.
CASE PRESENTATION
The proband, a 64-year-old right-handed male aerospace engineer, was referred to a local neurologist for evaluation of a 3-year history of mild depressive symptoms, memory and speech disturbance, and deterioration in handwriting after cardiac bypass surgery. He reported new onset of difficulty recalling names of casual professional and social acquaintances. Although these symptoms were continuous and troublesome to the patient, they were not initially apparent to others. He continued to function at work and at home without substantial difficulties. More recently, his cognitive difficulties had become noticeable to his wife with loss of language and decision-making skills. His wife noted relatively intact recall for recent events, although he reported using written notes to assist his memory. He had been able to maintain full-time employment as an aerospace engineer. Past medical history was significant for coronary artery disease and successfully treated pernicious anemia. The neurologist found no signs of parkinsonism on examination.
Initial neuropsychological evaluation (see Table 1) revealed constructional dyspraxia and mild difficulties with executive functioning. He was noted to be fluent with superior intellectual functioning and memory that was “high for age” with no recall deficits.
Table 1.
Summary of Neuropsychological Evaluations
| Date of Neuropsychological Evaluation | September 1983a | January 1990 |
|---|---|---|
| Age at time of testing (years) | 64 | 70 |
| Duration of symptoms at time of testing | 2 years | 8 years |
| Cognitive domain/test | ||
| Intellectual functioning | ||
| WAIS (WAIS-R) full-scale IQ | 124 (95 percentile) | 109 (73 percentile) |
| Verbal IQ | 120 (91 percentile) | 104 (61 percentile) |
| Performance IQ | 124 (95 percentile) | 115 (84 percentile) |
| Highest subtest | Arithmetic | Picture completion |
| Mini-Mental Status Examination | NA | 24/30 |
| Attention | ||
| Trail-making test, part A | WNL | WNL |
| Memory | ||
| Verbal | ||
| Story recall WMS/WMS-R logical memory | ||
| Immediate recall | WNL | Impaired (8 percentile) 2/46 |
| Delayed recall | WNL | Impaired (8 percentile) 0/46 |
| Visual | ||
| WMS/WMS-R visual reproduction | ||
| Immediate recall | WNL | WNL (99 percentile) 11/14 |
| Delayed recall | b | WNL (99 percentile) 10/14 |
| Construction | ||
| Aphasia screening test drawings | Mild difficulties | Mild difficulties |
| WAIS-R block design subtest | b | WNL (84 percentile) SS = 13 |
| Executive functions | ||
| Trail-making test, part B | Impaired (3 errors) | Impaired (6 to 10 percentile) (3 errors) |
| Halstead-Reitan Category Test | WNL (with confusion during subtest 5) | NA |
WAIS =Wechsler Adult Intelligence Scale; NA = not administered; WNL = within normal limits; WMS = Wechsler Memory Scale.
. Details of neuropsychological evaluation were obtained from narrative report. Therefore, individual test scores and percentiles were available only as noted in the report.
. Test administered but actual score unavailable.
His prior evaluation by his primary care physician had been negative and included serum chemistry, sedimentation rate, VDRL and serum B12. Head computed tomography (CT) at that time demonstrated slight prominence of the sylvian fissure area at the left temporal tip with normal-sized ventricles. The neurologist attributed his cognitive problems to age, whereas the neuropsychologist attributed them to mixed anxiety and depression.
He was subsequently referred to a psychiatrist and underwent a series of treatment trials with antidepressants including selective serotonin reuptake inhibitors and tri-cyclic antidepressants. However, his cognitive difficulties continued to progress, leading to retirement at age 65.
At age 70, the patient was evaluated by his primary care physician secondary to subjective reports of cognitive decline. The neurological evaluation revealed no parkinsonian signs and symptoms. His primary care physician felt that his cognitive functioning was normal. However, the patient was referred for neuropsychological reassessment in a dementia specialty clinic. During this evaluation, the patient reiterated his complaints of a progressive decline in memory, speech, and cognition with depressed mood. Evaluating neuropsychologists noted he had significant depression with dysphoric mood, early morning awaking, and thoughts of death and euthanasia. On examination (see Table 1), he was noted to be aphasic with a Folstein Mini-Mental Status Exam (MMSE)10 score of 24/30, reflecting difficulties with serial 7s, delayed recall, and inability to correctly copy intersecting pentagons. Examiners noted impairments in executive functioning, language, and verbal memory with significant word-finding difficulties.
Periodic neurological examinations from age 71 to 77 documented continued cognitive decline. Aphasic symptoms included increasingly severe difficulties with word finding, naming, and word comprehension. He also exhibited evidence of apraxia. He developed behavioral disturbances including auditory and visual hallucinations, verbal outbursts, agitation, pacing, and frequent falls. He was treated with multiple psychotropic medications including antipsychotics, antidepressants, anxiolytics, and anticonvulsants, with poor clinical response. The proband maintained some insight into his disorder stating, “My brain is going away and I cannot convert thoughts to words.”
Parkinsonian symptoms including bradykinesia appeared at approximately age 73 and were noted to worsen after treatment with haloperidol with an increase in tremor, rigidity, abnormal gait, and posture.
General neurological examination at age 76, 15 years after the onset of dementia symptoms, noted continued evidence of clinical parkinsonism with cogwheel rigidity; slow, stooped shuffling gait; and positive snout reflex. Resting tremor, extraocular movement abnormalities, and other parkinsonian signs were not appreciated. Magnetic resonance imaging and computerized axial tomography revealed mild to moderate cerebral atrophy. Electroencephalogram (EEG) demonstrated diffuse slowing.
His functional status worsened, and his behavioral disturbances including severe agitation and assaultive behavior increased, eventually leading to nursing home placement. Neurological examination at age 78 revealed a severe global aphasia and worsening parkinsonism, although without tremor.
He died at age 79 from acute bronchopneumonia and multiple pulmonary emboli.
FAMILY HISTORY
Family history obtained from family members revealed a clinical history of dementia in 7 relatives including his mother and sister (Figure 1).
Figure 1.
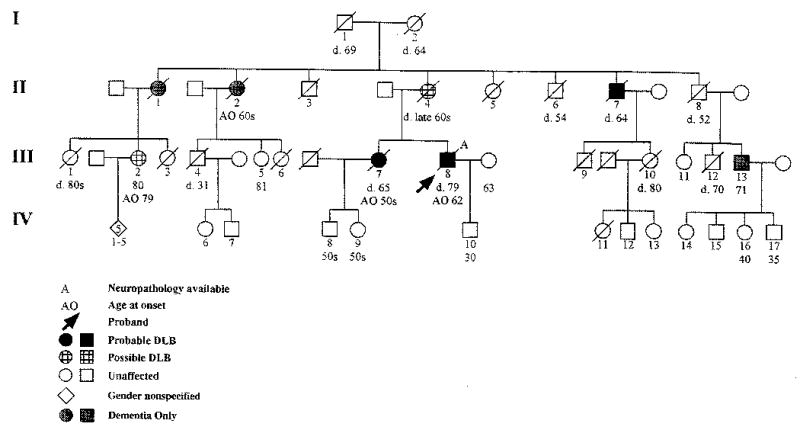
Pedigree of family with dementia.
First-Degree Relatives
The proband’s mother (II 4) was approximately 60 years old when she demonstrated difficulties with memory, organization, planning, and visual spatial deficits. She would wander and become lost in her neighborhood on multiple occasions. Approximately 2 to 3 years into the course of her illness, she demonstrated a marked decline in her ability to perform activities of daily living as well as slowing of her movements, difficulty maintaining balance, and urinary incontinence. There was no history of fluctuating mental status. She died at the age of 65.
The proband’s sister (III 7) first manifested symptoms of dementia when she was in her late 50s. Her family reported difficulties with executive functioning and memory with decline in ability to perform activities of daily living. Neurological evaluation at age 61 found severe and continued deterioration in functioning with normal laboratory parameters and head CT demonstrating generalized atrophy. Reexamination at age 62 documented inappropriate affect, with excessive laughing and joking during the examination; conversation was noted to be “rich in puns.” Cognitive evaluation at that time revealed delayed recall of 2 out of 3 items after 5 minutes. On neurological examination, she demonstrated parkinsonian symptoms with rigidity, cogwheeling, and shuffling gait. She was also noted to have a positive glabellar, bilateral grasp, slight Babinski, and poor upward gaze. Head CT demonstrated severe cortical atrophy, normal lumbar puncture, and EEG slowing of background activity.
Her dementia continued to worsen with cognitive and functional decline, auditory and visual hallucinations, and worsening parkinsonian symptoms. She subsequently developed language impairments, and her mental status fluctuated from episodes of coherence to periods of bizarre behavior. Terminally, she was mute and bedridden, dying at age 65.
Second-Degree Relatives
The proband’s maternal aunt (II 1) had memory problems that began in her 70s. She died in her late 70s/early 80s. She did not demonstrate parkinsonism, hallucinations, or bizarre behaviors.
The proband’s maternal aunt (II 2) initially manifested symptoms of dementia during her 60s. Her family reported difficulties with memory, visuospatial and executive functioning. She would frequently wander the neighborhood, becoming lost. As her illness progressed, she became paranoid and developed aphasia and apraxia. Eventually, she was institutionalized at a state mental hospital secondary to extreme combativeness and resistiveness to care. At the time of death, she was virtually mute.
The proband’s maternal uncle (II 7) had a history of excessive alcohol intake. His dementia symptoms began in his 60s with his family reporting difficulties with memory, apraxia, and visuospatial and executive functioning. Toward the end of his life, he developed parkinsonian symptoms with slow, stooped gait; bradykinesia; and resting tremor. He also manifested symptoms of agitation, paranoia, and visual and auditory hallucinations. Although he continued to use alcohol until his death, his dementia and behavioral symptoms were generally not associated with intoxication or withdrawal.
Third-Degree Relatives
The proband’s cousin (III 2) is 80 years old and reports a 1-year history of increasing difficulty with short-term memory that interferes with daily activities, visuospatial dysfunction with geographic disorientation, and slight impairment of executive functioning. She had a recent Clinical Dementia Rating (CDR) Scale11 score of 1.0, indicating mild dementia, and Neuropsychiatric Inventory Score (NPI)12 of 0, both obtained via a telephone interview with a relative. The CDR and NPI are assessments of cognitive and neuropsychiatric symptomatology, respectively. Both are obtained by a semistructured interview with the caregiver and are easily and reliably performed. Neurological examination revealed an MMSE10 of 27/30, reflecting incorrect date and 1/3 delayed recall. Administration of the Unified Parkinson’s Disease Rating Scale (UPDRS)–Motor Examination13 revealed a score of 8, reflecting mild hypomimia, mild bilateral slowing and/or reduction in amplitude of both hand movements and rapid alternating movements, mild bilateral slowing and/or reduction in leg agility, and minimal bradykinesia and hypokinesia.
The proband’s cousin (III 5) is 81 years old. She reports “occasional forgetfulness.” A telephone interview with an informant showed no substantial behavioral nor functional disturbance (scores of 0 on both the CDR11 and the NPI12). Neurological examination revealed a MMSE10 of 29/30 (she missed 1 out of 3 items on delayed recall). The UPDRS–Motor Examination13 score was 2, reflecting mild slowing and/or reduction in amplitude of left hand movements and left hand rapid alternating movements.
The proband’s cousin (III 13) is 71 years old. He and his family deny memory difficulties. Via telephone interview with a relative, he has scores of 0 on both the CDR11 and NPI12. Neurological examination revealed an MMSE10 of 27/30, reflecting a 0/3 score for delayed recall. Administration of the UPDRS–Motor Examination13 revealed a score of 2, reflecting mild bilateral slowing and/or reduction in leg agility.
NEUROPATHOLOGY
At autopsy, the proband’s brain weighed 1225 grams with gross evidence of moderate to severe fronto-temporal atrophy and mild parietal atrophy. There was mild patchy large vessel atherosclerosis. On gross sectioning, there was severe ventriculomegaly and depigmentation of the substantia nigra. Microscopically, Bielschowsky silver staining demonstrated moderate to severe changes of AD. There was moderate to severe senile plaque formation in the cortex and hippocampal formation. Neurofibrillary tangles were moderate to severe in the medial temporal lobe and hippocampus and mild to moderate in association cortices (Braak Stage V).14 Hematoxylin and Eosin revealed neuronal loss in the substantia nigra with occasional LB inclusions in the remaining neurons (Figure 2a). Immunohistochemistry for alpha-synuclein (LB 509, Zymed) demonstrated widespread inclusions in the substantia nigra, cingulate gyrus, parahippocampal gyrus, and amygdala (Figure 2b). There were immunopositive neurites in these regions and in the CA-2 subfield of the hippocampus. The case fulfilled neuropathologic diagnostic criteria for DLB (neocortical subtype)1 and high likelihood AD.15 There was no evidence grossly or microscopically of any vascular lesions.
Figure 2.

(a) Lewy body inclusion in pigmented substantia nigra neuron (large arrow) and microglial cell cluster, containing pigment, suggestive of neuronal degeneration (small arrow) (125X, hematoxylin and eosin). (b) Alpha-synuclein immunopositive inclusions in neurons of the deep layers of the parahippocampal gyrus (31X, alpha-synuclein immunohistochemistry). Images digitized with adjustment of brightness and contrast.
MOLECULAR GENETICS
Genomic DNA from the proband and 3 family members was screened for alpha-synuclein mutations. The complete genomic sequences encompassing the alpha-synuclein gene is available through Genbank (http://www.ncbi.nlm.nih.gov/Genbank). Polymerase chain reaction amplification of the 5 coding regions (exons) that encoded for the amino acid sequences was conducted. There were no genetic variants in the gene, specifically, the previously reported A53T mutation.16 In addition, apolipoprotein E genotyping was conducted using standard methods17 in the University of Washington Alzheimer’s Disease Research Center Genotyping Core (G. Schellenberg, core principal investigator). The proband’s APOE genotype was ɛ3/ɛ4.
DISCUSSION
Our index case fulfilled both clinical and pathological criteria for DLB, although his early clinical presentation made the diagnosis difficult. Ultimately, he exhibited 2 of the 3 core DLB symptoms1 including hallucinations and motor symptoms of parkinsonism. Fluctuations in mental status were not well documented, although his wife reported nighttime confusion. In addition, his history of repeated falls and exquisite neuroleptic sensitivity support a diagnosis of DLB. However, he did not exhibit any of these DLB symptoms during the first 10 years of his 18-year course. Pathologically, he exhibited both brainstem and cortical LBs consistent with the neocortical category of DLB.1 As with many DLB patients, he also exhibited concomitant AD pathology with moderate to severe senile plaque and neurofibrillary tangle changes.1
The initial clinical presentation of the proband included clinical and neuropsychological evidence of visuospatial, language, and executive dysfunction. Despite early subjective memory complaints, initial neuropsychological testing found superior memory function and argued against a diagnosis of AD. His early language and executive dysfunction, with subsequent development of a severe global aphasia, suggested a cortical disorder similar to fronto-temporal lobar degeneration (FTLD).18 In fact, his clinical course included several core (insidious onset, gradual progression, nonfluent spontaneous speech) and supportive features (age of onset less than 65 years of age, early preservation of word meaning, late mutism, early preservation of social skills, late behavioral changes, late akinesia, and rigidity) of progressive nonfluent aphasia, a proposed clinical subtype of FTLD.18 One exclusionary item for FTLD present during the initial presentation was clinical and neuropsychological evidence of visuospatial dysfunction.
DLB patients generally exhibit neuropsychological deficits quite similar to that observed in AD except for relatively more severe visuospatial deficits and less severe memory dysfunction.1 In several studies, language dysfunction has been found to be equivalent in AD and DLB.7–9 In our proband, the only early symptom that suggested a diagnosis of DLB, versus FTLD, was the visuospatial dysfunction. It was only in the second decade of his clinical course that the core symptoms of DLB became evident. To the best of our knowledge, this is the first case report of an FTLD-like clinical presentation in a neuropathologically verified DLB case.
The family history of our case suggests an autosomal dominant inheritance pattern with similar symptoms in affected relatives. Although autopsy was not available for other affected family members, they also demonstrated dementia characterized by symptoms of executive dysfunction, with the sister demonstrating additional language impairment. Seven of the proband’s relatives also demonstrated dementia, some characterized by symptoms suggestive of DLB including clinical parkinsonism and hallucinations.
There have been multiple reports of familial parkinsonism with dementia.19–26 In the majority of these families, clinical parkinsonism is the predominant presenting symptom with later development of a dementing disorder. Only a few families include individuals that presented with dementia.20,22 However, there are no neuropathological examinations available in these affected individuals. Most recently, Tsuang et al27 and Galvin et al28 described 2 families with 2 or more individuals with autopsy-proven DLB.
Mutations in α-synuclein have also been found in families with clinical parkinsonism and dementia.29 Pathologically, most of these familial cases have brainstem, limbic, and cortical LB pathology.30,31 In this proband, there was no mutation in the α-synuclein gene. Other disorders associated with a familial frontal dementia with parkinsonism include fronto-temporal dementia with parkinsonism associated with chromosome 17 (FTDP-17) and hereditary dysphasic dementia.32,33 The former is characterized by τ-associated pathology and LBs are not characteristic, while the latter is associated with inconsistent parkinsonism and LB pathology.
This family is unique in that all 7 potentially affected cases presented with dementia and the autopsy of the index case demonstrated cortical and brainstem LB pathology. Our index case demonstrates the degree of clinical heterogeneity in DLB. DLB should be considered in the differential diagnosis of demented patients presenting with FTLD-like symptoms, especially if there is early evidence of visuospatial dysfunction. Other disorders characterized by parkinsonian signs and dementia must also be considered in these patients including Parkinson’s disease with dementia,34 progressive supranuclear palsy,34 FTDP-17,32 and AD.34 Further evaluation of this family and a search for additional families with autopsy-proven DLB will help to further elucidate the genetics and pathophysiology of this important dementing disorder.
Acknowledgments
This work was supported by NIA RO1-AG18644, NIA P50 AG05136, and the Department of Veterans Affairs. The authors wish to thank the family members for their valuable time and willingness to participate in research and Lynne Greenup for her technical assistance.
References
- 1.McKeith LG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47:1113–1124. doi: 10.1212/wnl.47.5.1113. [DOI] [PubMed] [Google Scholar]
- 2.Leverenz J, Sumi M. Parkinson’s disease in patients with Alzheimer’s disease. Arch Neurol. 1986;43:662–664. doi: 10.1001/archneur.1986.00520070020010. [DOI] [PubMed] [Google Scholar]
- 3.Lim A, Tsuang D, Kukull W, et al. Clinico-neuropathological correlation of Alzheimer’s disease in a community-based cases series. J Am Geriatr Soc. 1999;47:564–569. doi: 10.1111/j.1532-5415.1999.tb02571.x. [DOI] [PubMed] [Google Scholar]
- 4.Hulette C, Mirra S, Wilkinson W, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part IX. A prospective cliniconeuropathologic study of Parkinson’s features in Alzheimer’s disease. Neurology. 1995;45:1991–1995. doi: 10.1212/wnl.45.11.1991. [DOI] [PubMed] [Google Scholar]
- 5.Dickson D, Crystal H, Davies P, Hardy J. Cytoskeletal and Alzheimer-type pathology in Lewy body disease. In: Perry R, McKeith I, Perry E, eds. Dementia with Lewy bodies: clinical, pathological, and treatment issues. New York: Cambridge University Press, 1996:224–237.
- 6.Gibb WR, Luthert PJ, Janota I, Lantos PL. Cortical Lewy body dementia: clinical features and classification. J Neurol Neurosurg Psychiatry. 1989;52:185–192. doi: 10.1136/jnnp.52.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salmon D, Galasko D. Neuropsychological aspects of Lewy body dementia. In: Perry R, McKeith I, Perry E, eds. Dementia with Lewy bodies. Cambridge, UK: Cambridge University Press, 1996:99–133.
- 8.Gnanalingham K, Byrne E, Thornton A, et al. Motor and cognitive function in Lewy body dementia: comparison with Alzheimer’s and Parkinson’s diseases. J Neurol Neurosurg Psychiatry. 1997;62:243–252. doi: 10.1136/jnnp.62.3.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hansen L, Salmon D, Galasko D, et al. The Lewy body variant of Alzheimer’s disease: a clinical and pathologic entity. Neurology. 1990;40:1–8. doi: 10.1212/wnl.40.1.1. [DOI] [PubMed] [Google Scholar]
- 10.Folstein MF, Folstein SE, McHugh PR. Mini-Mental State: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 11.Hughes CP, Berg L, Danziger WL, et al. A new clinical scale for the staging of dementia. Br J Psychiatry. 1982;140:566–572. doi: 10.1192/bjp.140.6.566. [DOI] [PubMed] [Google Scholar]
- 12.Cummings JL, Mega M, Gray K, et al. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology. 1994;44:2308–2314. doi: 10.1212/wnl.44.12.2308. [DOI] [PubMed] [Google Scholar]
- 13.Fahn S, Elton R, and members of the UPDRS Development Committee. Unified Parkinson’s disease rating scale. In: Fahn S, Marsden C, Calne D, Goldstein M, eds. Recent development in Parkinson’s disease. Florham Park, NJ: Macmillan Health Care Information, 1987:153–163.
- 14.Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropatholo (Berl) 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 15.National Institute on Aging and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Neurobiol Aging. 1997;18:S1–S2. [PubMed] [Google Scholar]
- 16.Polymeropoulos M, Lavedan C, Leroy E, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 17.Hixson J, Vernier D. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res. 1990;31:545–548. [PubMed] [Google Scholar]
- 18.Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 19.Waters CH, Miller CA. Autosomal dominant Lewy body parkinsonism in a four-generation family. Ann Neurol. 1994;35:59–64. doi: 10.1002/ana.410350110. [DOI] [PubMed] [Google Scholar]
- 20.Ohara K, Takauchi S, Kokai M, et al. Familial dementia with Lewy bodies (DLB) Clin Neuropathol. 1999;18:232–239. [PubMed] [Google Scholar]
- 21.Golbe LI, Miller DC, Duvoisin RC. Autosomal dominant Lewy-body Parkinson’s disease. Adv Neurol. 1990;53:287–292. [PubMed] [Google Scholar]
- 22.Wakabayashi K, Hayashi S, Ishikawa A, et al. Autosomal dominant diffuse Lewy body disease. Acta Neuropathol (Berl) 1998;96:207–210. doi: 10.1007/s004010050883. [DOI] [PubMed] [Google Scholar]
- 23.Ishikawa A, Takahashi H, Tanaka H, et al. Clinical features of familial diffuse Lewy body disease. Eur Neurol. 1997;38:34–38. doi: 10.1159/000113459. [DOI] [PubMed] [Google Scholar]
- 24.Denson MA, Wszolek ZK, Pfeiffer RF, et al. Familial parkinsonism, dementia, and Lewy body disease: study of family G. Ann Neurol. 1997;42:638–643. doi: 10.1002/ana.410420415. [DOI] [PubMed] [Google Scholar]
- 25.Muenter MD, Forno LS, Hornykiewicz O, et al. Hereditary form of parkinsonism—dementia. Ann Neurol. 1998;43:768–781. doi: 10.1002/ana.410430612. [DOI] [PubMed] [Google Scholar]
- 26.Golbe LI, Lazzarini AM, Schwarz KO, et al. Autosomal dominant parkinsonism with benign course and typical Lewy-body pathology. Neurology. 1993;43:2222–2227. doi: 10.1212/wnl.43.11.2222. [DOI] [PubMed] [Google Scholar]
- 27.Tsuang D, Dalan A, Eugenio C, et al. Familial dementia with Lewy bodies: a clinico-neuropathological study of two families. Arch Neurol. In press. [DOI] [PubMed]
- 28.Galvin J, Lee S, Perry A, et al. Familial dementia with Lewy bodies: clinicopathologic analysis of two kindreds. Neurology. 2002;58:A251. doi: 10.1212/wnl.59.7.1079. [DOI] [PubMed] [Google Scholar]
- 29.Kruger R, Kuhn W, Leenders KL, et al. Familial parkinsonism with synuclein pathology: clinical and PET studies of A30P mutation carriers. Neurology. 2001;56:1355–1362. doi: 10.1212/wnl.56.10.1355. [DOI] [PubMed] [Google Scholar]
- 30.Golbe L, Di Iorio G, Bonavita V, et al. A large kindred with autosomal dominant Parkinson’s disease. Ann Neurol. 1990;27:276–282. doi: 10.1002/ana.410270309. [DOI] [PubMed] [Google Scholar]
- 31.Spira PJ, Sharpe DM, Halliday G, et al. Clinical and pathological features of a parkinsonian syndrome in a family with an Ala53Thr alpha-synuclein mutation. Ann Neurol. 2001;49:313–319. [PubMed] [Google Scholar]
- 32.Reed LA, Wszolek ZK, Hutton M. Phenotypic-correlations in FTDP-17. Neurobiol Aging. 2001;22:89–107. doi: 10.1016/s0197-4580(00)00202-5. [DOI] [PubMed] [Google Scholar]
- 33.Morris JC, Cole M, Banker BQ, Wright D. Hereditary dysphasic dementia and the Pick-Alzheimer spectrum. Ann Neurol. 1984;16:455–466. doi: 10.1002/ana.410160407. [DOI] [PubMed] [Google Scholar]
- 34.Cummings JL, Benson DF. Dementia: a clinical approach. Boston: Butterworth-Heinemann, 1992.