

Abstract
Ozone oxidative preconditioning is a prophylactic approach, which favors the antioxidant-prooxidant balance for preservation of cell redox state by the increase of antioxidant endogenous systems in both in vivo and in vitro experimental models. Our aim is to analyze the effect of ozone oxidative preconditioning on serum TNF-α levels and as a modulator of oxidative stress on hepatic tissue in endotoxic shock model (mice treated with lipopolysaccharide (LPS)). Ozone/oxygen gaseous mixture which was administered intraperitoneally (0.2, 0.4, and 1.2 mg/kg) once daily for five days before LPS (0.1 mg/kg, intraperitoneal). TNF-α was measured by cytotoxicity on L-929 cells. Biochemical parameters such as thiobarbituric acid reactive substances (TBARS), enzymatic activity of catalase, glutathione peroxidase, and glutathione-S transferase were measured in hepatic tissue. One hour after LPS injection there was a significant increase in TNF-α levels in mouse serum. Ozone/oxygen gaseous mixture reduced serum TNF-α levels in a dose-dependent manner. Statistically significant decreases in TNF-α levels after LPS injection were observed in mice pretreated with ozone intraperitoneal applications at 0.2 (78%), 0.4 (98%), and 1.2 (99%). Also a significant increase in TBARS content was observed in the hepatic tissue of LPS-treated mice, whereas enzymatic activity of glutathion-S transferase and glutathione peroxidase was decreased. However in ozone-treated animals a significant decrease in TBARS content was appreciated as well as an increase in the activity of antioxidant enzymes. These results indicate that ozone oxidative preconditioning exerts inhibitory effects on TNF-α production and on the other hand it exerts influence on the antioxidant-prooxidant balance for preservation of cell redox state by the increase of endogenous antioxidant systems.
INTRODUCTION
The molecular sequelae of events leading to the adverse and sometimes lethal outcome of sepsis have not been fully elucidated [1]. Cytokines are undoubtedly involved in these processes [2, 3, 4], with the proinflammatory cytokines tumor necrosis factor-α (TNF-α), interleukin-1, macrophage migration inhibitory factor (MIF), and others playing a pivotal role [4, 5]. Cytokines have been considered promising clinical targets in the treatment of shock, but, to date, anticytokine-based therapeutic strategies such as the use of anti-TNF antibodies, soluble TNF receptors, or interleukin-1 receptor antagonists have failed to show a clear clinical benefit [6].
Experimental and clinical studies have demonstrated that exposure to endotoxin (lipopolysaccharide (LPS)) results in the release of various inflammatory mediators. TNF-α is a proinflammatory cytokine greatly involved in the pathophysiological changes associated with acute and chronic inflammatory conditions, including septic shock, autoimmune diseases, rheumatoid arthritis, inflammatory bowel disease, and the respiratory distress syndrome [7, 8].
In addition some antioxidants and reactive oxygen species (ROS) scavengers exert a protective action against endotoxic shock in rodents by inhibiting TNF-α [9].
Ozone/oxygen mixture has a strong microbiocidal activity in vitro [6] comparable to the potent bactericidal activity of NO [10], and might therefore act as a regulator or modulator of many inflammatory processes in vivo. Ozone/oxygen mixture exhibits various effects on the immune system [11], such as the modulation of phagocytic activity of peritoneal [12], and alveolar [13], macrophages, which generates the first line of defense against bacteria and/or its toxins. Therefore, it can be hypothesized that ozone/oxygen mixture enhances the production/release of proinflammatory cytokine in different abdominal organs (eg, spleen, liver) and may be able to influence the outcome of a severe infection.
Repeated rectal administrations of ozone have induced a sort of cross-tolerance to free radicals released after hepatic and renal ischemia-reperfusion [14, 15, 16, 17]. Also it has been demonstrated that low doses of ozone increased antioxidant endogenous systems involving glutathione (GSH), superoxide dismutase (SOD), and catalase (CAT), preparing the host to face physiopathological conditions mediated by ROS [14, 15, 16, 17, 18], and demonstrating that ozone, probably by means of an oxidative preconditioning mechanism, similarly to ischemic preconditioning, protected these organs from the damage produced by ROS, which induced improvement of antioxidant-prooxidant balance and the concomitant preservation of cell redox state [19]. Ozone therapy has also been proved efficient in the treatment of many diseases and beneficial effects have been observed with it [17, 18, 20, 21]. Ozone oxidative preconditioning (OOP) also showed a beneficial effect in lethal polymicrobial sepsis in rats because it increased animal survival and protected them from death [22, 23].
Recently it has also been demonstrated that OOP administered into the abdomen of rats leads to altered sleeping time and to changes in the loss of reflexes [24]. These alterations in the strength of anesthesia are paralleled by a modulation of proinflammatory cytokines levels beyond those seen for a given anesthetic drug alone [25].
Taking into account the former results, we decided to analyze the effect of OOP on serum TNF-α levels and as a modulator of oxidative stress on hepatic tissue in endotoxic shock model mice treated with LPS.
MATERIALS AND METHODS
Animals and treatments
Adult male BALB/c mice obtained from the National Center for Laboratory Animal Production (Havana, Cuba) weighing 18−20 g were used in this study (n = 35). Mice were housed in plexiglass cages, maintained in an air-filtered and temperature-conditioned (20−22°C) room with a relative humidity of 50%–52% and under an artificial light/dark cycle of 12 hours.
Animals were fed with standard laboratory chow and water ad libitum. LPS (Sigma, St. Louis, Mo; Serotype 055:B5 from Escherichia coli) was given intraperitoneally (IP) at 0.1 mg/kg, dissolved in sterile pyrogen-free saline solution.
Ozone (O3) was generated by OZOMED 01 equipment manufactured by the Ozone Research Center (Cuba). Ozone obtained from medicinal grade oxygen was used immediately. The ozone concentration was measured by using a UV spectrophotometer at 254 nm.
Experimental design
Ozone/oxygen mixture was administered IP at doses of 0.2, 0.4, and 1.2 mg/kg. The volume of gaseous mixture administered to each animal was approximately 1 mL. Oxidative preconditioning was performed with five applications (one daily) of the ozone/oxygen mixture. LPS was injected twenty-four hours after the last ozone/oxygen administration.
A control group receiving LPS and two other groups receiving saline or ozone/oxygen mixture alone were also included. Dexamethasone (30 mg/kg) used as a reference drug was administered IP in saline solution 30 minutes before LPS. The mice were bled from the retroorbital plexus under light anesthesia and serum TNF-α was measured 1 hour after administration of LPS.
Effect of OOP on kinetics of TNF-α release in serum after LPS injection
Another experiment was performed in order to study whether OOP has some influence on the normal kinetic TNF-α release after LPS injection.
Ozone/oxygen mixture was administered as described before but just using the highest dose (1.2 mg/kg) IP. LPS was injected twenty-four hours after the last ozone/oxygen mixture administration.
A control group receiving LPS and two other groups receiving saline or ozone/oxygen mixture alone were also included. Dexamethasone (30 mg/kg) used as a reference drug was administered IP in saline solution 30 minutes before LPS. The mice were bled from the retroorbital plexus under light anesthesia and serum TNF-α was measured 0.5, 1, 2, and 3 hours after administration of LPS. Biochemical parameters such as thiobarbituric acid reactive substances (TBARS), enzymatic activity of CAT, glutathione peroxidase (GSH-Px), and glutathione-S transferase (GST) were measured in hepatic tissue.
The experiments were conducted in accordance with the ethical guidelines for investigations in laboratory animals and were approved by the Ethical Committee for Animal Experimentation of the National Center for Scientific Research (CNIC), Havana, Cuba.
TNF-α assay
TNF-α was measured by cytotoxicity on L929 cells [17]. TNF levels were determined using recombinant human TNF-α (BASF/Knoll, Ludwigshafen, Germany; specific activity 107 U/mg) and expressed in pg/mL. Five mice per group were used and results were compared by Student t test. Results are expressed as an inhibition percentage of TNF-α production. All experiments were repeated at least twice.
Biochemical assays
Liver homogenates were obtained using a tissue homogenator Ultraturrax T25 Polytron at 4°C. The homogenates (10% w/v) for GSH-Px and GST were prepared by using KCl 1.15% in aqueous solution. Liver homogenates for CAT enzymatic assay were obtained with a 50 mM phosphate buffer (pH 7) containing 1% Triton X-100 (1 : 9 w/v) (buffer 2).
The homogenates were centrifuged at 600 g for 60 minutes at 4°C and the supernatants were taken for biochemical determinations.
Determination of TBARS
Thiobarbituric acid reactive substances (TBARS) were determined spectrophotometrically by a modified version of the method of Botsoglou et al [26]. The homogenate for TBARS was prepared by transferring 2 g of the sample to a tube, and volumes of 5% aqueous trichloroacetic acid (TCA) (8 mL) and 0.8% butylated hydroxy toluene (BHT) in hexane (5 mL) were immediately added. The content of the tube was ultraturraxed for 30 seconds at high speed and centrifuged for 3 minutes at 3000 g, and the top hexane layer was discarded. The bottom aqueous layer was made to 10 mL volume with 5% TCA, and a 2.5 mL aliquot was pipetted into a screw-capped tube to which a volume (1.5 mL) of 0.8% aqueous TBA, besides 1.5 mL of TCA 5%, 0.2 mL of sodium dodecyl sulphate (SDS) 8.1%, and 0.6 mL of distilled water, was also added. Following incubation for 30 minutes at 70°C, the tube was cooled under tap water and 5 mL of butanol-pyridine (15 : 1) mixture was added. The content of the tube was strongly vortexed and centrifuged for 15 minutes at 3000 g at room temperature. The optical density of the top layer was measured at 532 nm using 1,1,3,3-tetraethoxypropane as a standard.
Determination of GST
The GST activity was determined using GSH and 1-chloro-2,4-dinitrobenzene (CDNB) as the second substrate, according to the method of Habig et al [27]. An aliquot of 0.015 mL of the supernatant was added to the reaction mixture containing 0.2380 mL of 0.1 M potassium phosphate buffer (pH 6.5) and 0.1 mL of CDNB 1 mM in ethanolic solution. Afterward, 0.5 mL of GSH 10 mM was added to the reaction mixture and optical density was measured during 5 minutes in the spectrophotometer. A molar extinction coefficient of 9.6 × 103 M cm−1 was used to determine the activity of GST.
Determination of CAT activity
CAT was determined according to the method of Rice-Evans and Diplock [28]. Liver homogenate was diluted with buffer 2, as described before, to obtain an adequate dilution of the enzyme. Then, 2 mL of the enzyme dilution were added to the cuvette and mixed with 1 mL of 30 mM H2O2, and then the absorbance was measured at 240 nm, for 30 seconds in the spectrophotometer. Initial absorbance of the reaction mixture must be around 0.5. The enzyme activity is expressed as the first order constant that describes the decomposition of H2O2 at room temperature.
Determination of GSH-Px activity
GSH-Px was measured using a modified version of the method of Faraji et al [29]. All reaction mixtures were dissolved in 20 mM sodium phosphate buffer containing 6 mM EDTA (pH 7.0). The reaction mixture consisted of 98.8 μL of phosphate buffer, 700 μL of 2.86 mM GSH, 100 μL of 1 mM sodium azide, 100 μL of 1 mM NADPH, and 4.2 μL of GSH reductase (0.5 units). Then, 10 μL of the tissue homogenate supernatant was added to the reaction mixture and incubated at room temperature for 10–15 minutes. Afterward, 10 μL of 30 mM t-butyl hydroperoxide (dissolved in bidistilled water) was added to the reaction mixture and measured at 340 nm for 7 minutes in the spectrophotometer. A molar extinction coefficient of 6.22 × 103 M cm−1 was used to determine the activity of GSH-Px. The enzyme activity is expressed in international units of enzymatic activity/mg of protein. International units are expressed in μ mol of hydroperoxides transformed /min/mL of enzyme.
Protein assay
Protein concentrations were determined by the method of Lowry et al [30] using bovine serum albumin as a standard.
Statistical analysis
Data were expressed in means ± SEM and analyzed statistically using one-way student t test, whereas Kruskall-Wallis test followed by Mann-Whitney test was applied for the rest of the markers. The 0.05 level of probability was used as statistical significance.
RESULTS
Effect of ozone oxidative preconditioning on TNF-α levels in mouse serum
One hour after LPS injection there was a significant increase in TNF-α levels (Table 1). OOP reduced TNF-α release provoked by LPS in a dose-dependent manner. This reduction was statistically significant in all the administered doses. Ozone/oxygen IP applications at 0.4 and 1.2 mg/kg produced an almost total inhibition of TNF-α release. The effects of ozone/oxygen IP applications at 0.4 and 1.2 mg/kg were quite similar to the inhibition achieved by dexamethasone, a well-known inhibitor of TNF-α release. When ozone/oxygen mixture (1.2 mg/kg) was administered alone, no effect on TNF-α induction was observed (Table 1).
Table 1.
Effects of ozone oxidative preconditioning on serum TNF-α levels in mice treated with LPS.
| Treatment (mg/kg) | TNF-α (pg/mL) | |
| Mean ± SEM | Inhibition (%) | |
| Saline | 0 | 0 |
| Ozone /oxygen (1.2), IP | 0 | 0 |
| LPS (0.1 mg/kg) | 2200 ± 780 | — |
| Ozone /oxygen IP (0.2) + LPS | 485 ± 120∗ | 78 |
| Ozone /oxygen IP (0.4) + LPS | 33 ± 17∗∗ | 98.5 |
| Ozone /oxygen IP (1.2) + LPS | 30.6 ± 5∗∗ | 99 |
| Dexamethasone (30 mg/kg) + LPS | 29 ± 10∗∗ | 98.7 |
∗P < .05.
∗∗P < .01 versus respective control (LPS) by Student t test (n = 5). The animals were pretreated with five IP applications of ozone (one daily) at 0.2, 0.4, and 1.2 mg/kg twenty four hours after the last ozone administration LPS was given (0.1 mg/kg) and 1 hour later blood samples were taken to measure TNF-α levels in serum. Dexamethasone (30 mg/kg IP, 30 minutes before LPS) was used as a control drug.
Effect of OOP on kinetics of TNF-α release in serum after LPS injection
A preliminary kinetics study of TNF-α production in serum 0.5, 1, 2, and 3 hours after LPS injection showed that the maximal level of TNF-α in serum occurred 1 hour after LPS injection for all groups studied while after 2 and 3 hours the levels of TNF-α were lower than 10 pg/mL, and were considered as zero. It has demonstrated that OOP did not affect the normal kinetics of TNF-α release in serum from macrophages in the period after LPS injection in mice (Figure 1).
Figure 1.
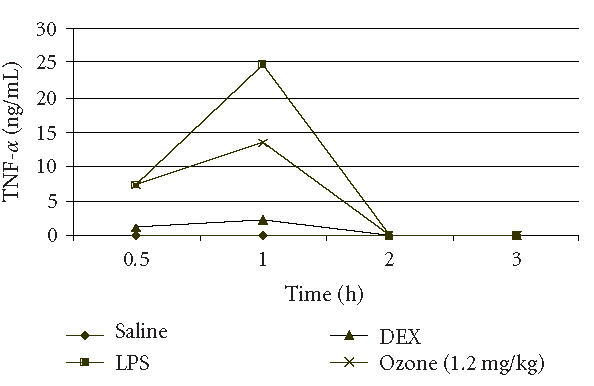
Time curve of serum TNF-α levels in mice after LPS injection. Data less than 10 pg/mL were considered as 0.
Biochemical parameters
In spite of the significant increase in TNF-α level 1 hour after the LPS injection, there was no difference in biochemical markers measured 0.5, 1, 2, and 3 hours after LPS (data not shown). Table 1 shows the changes in TBARS, CAT, GSH-Px, and GST in the hepatic tissue of mice. In LPS-treated mice the enzymatic activity of CAT was slightly increased, whereas a significant decrease was observed in GSH-Px and GST. TBARS were augmented to 238% in comparison with nontreated controls.
Dexamethasone was applied 30 minutes before LPS as a reference anti-inflammatory drug, showing no significant changes in CAT activity in these conditions. On the contrary, activity of GSH-Px was significantly increased compared with nontreated and LPS-treated mice. Dexamethasone showed 40% of enhancement of GSH-Px activity whereas GST activity was unchanged. This drug also reduced TBARS to values even lower compared with nontreated mice, showing a diminution of 89.9% with respect to LPS-treated mice.
Ozone oxidative preconditioning before LPS 0.1 mg/kg has not shown any significant change in CAT activity, whereas GSH-Px activity was increased to 78% compared with nontreated control. Also we have observed a significant increase in GST activity to 56.9% and TBARS levels were diminished to 60% as compared to nontreated control (Table 2).
Table 2.
Biochemical determinations in experimental groups.
| Groups | TBARS (nmol/mg | CAT (k15/g of wet tissue) | GSH-Px (IU/mg of protein) | GST (IU/mg of protein) |
| of protein) | ||||
| Nontreated control | 0.97 ± 0.066a | 30.48 ± 0.316a | 7136.3 ± 786.49a | 1592.6 ± 195.03a |
| LPS (0.1 mg/kg) | 3.28 ± 0.582b | 34.20 ± 2.410b | 5764.6 ± 1012.73b | 1273.4 ± 250.53b |
| DEX (30 mg/kg) + LPS | 0.33 ± 0.041c | 30.33 ± 5.200a | 10 006.2 ± 1457.82c | 1602.8 ± 310.01a |
| O3 (1.2 mg/kg) + LPS | 0.39 ± 0.075c | 30.68 ± 1.140a | 12 745.37 ± 2653.08c | 2498.3 ± 225.77c |
Data are presented as mean ± standard error of the mean for ten animals per group.
Different letters mean significant differences between groups (P < .05). CAT activity is described as the first-order constant of the decomposition of H2O2 at 25° C/g of wet tissue. k15 is the constant kinetic of the first order that describes the decomposition of H2O2 at room tempreture. The GSH-Px activity is expressed in international units of enzymatic activity/mg of protein. International units are expressed in μ mol of transformed hydroperoxides/min/mL of enzyme. The GST activity is expressed in international units of enzymatic activity/mg of protein.
DISCUSSION
Oxidative preconditioning [14] is analogous to other phenomena such as ischemic preconditioning [31], thermal preconditioning [32], and chemical preconditioning [33]. All of these processes have in common that a repeated and controlled stress is able to provide protection against a prolonged and severe stress. Intrarectal ozone has conferred protection against the hepatic ischemia-reperfusion injury by the adenosine accumulation and by blocking the xanthine/xanthine oxidase pathway, decreasing ROS generation after reperfusion [16]. Recently, it has been demonstrated that intrarectal ozone/oxygen mixture reduced ROS by the stimulation and/or preservation of the endogenous antioxidant systems in experimental models of liver and renal ischemia-reperfusion, respectively [14, 15, 16, 17, 33, 34, 35, 36]. Also, it has been shown that there is an increase in the activity of antioxidant enzymes such as SOD and GSH-Px and a decrease of malondialdehyde after ozone preconditioning in cardiopathy patients [18].
We observed a significant inhibitory effect of serum TNF-α release on mice pretreated with ozone/oxygen mixture by IP route before induction of endotoxic shock by LPS.
Intraperitoneal injections of ozone/oxygen mixture might produce a direct effect on peritoneal macrophages, modulating their phagocytic activity [12], hypothesizing that this treatment might modify the production/release of proinflammatory cytokines in different abdominal organs as it occurs in the ozonized blood in vitro which causes a release of different cytokines such as TNF-α, GM-CSF, IL-2, or IFN-α [35, 36, 37].
To our knowledge, this is the first report on the inhibitory effect of OOP on TNF-α release in serum in an experimental model of endotoxic shock in mice.
In our opinion, the inhibitory effects of ozone/oxygen mixture on TNF-α levels in the serum of mice treated with endotoxin are a consequence of the stimulation of the antioxidant defenses induced by ozone therapy. This point of view is scientifically supported by the fact that ROS are strongly involved in the induction and development of the inflammatory process and in the pathogenesis of endotoxic shock [38].
On the other hand, various antioxidants, with ROS scavenging properties, protected mice against endotoxin-mediated organ injury and reduced TNF-α levels in blood serum [38]. It has been ascribed by some authors to the effect of antioxidant agent on the inhibition of the nuclear transcription factor kappa B (NF-κB) activation. NF-κB is activated by ROS, with the subsequent induction of various cytokines and enzymes such as TNF-α, which are involved in the induction and development of endotoxic shock [37].
In the late 1980s and early 1990s, observations led to the conclusion that TNF was a prerequisite for the induction of many other inflammatory cytokines. Injection of LPS into experimental animals [39] or into human volunteers [40] led to the appearance of TNF in the bloodstream before any other cytokine. The maximal plasma levels for TNF are 1.5 hours after injection [41, 42]. These results are in agreement with the obtained by us, in which the maximal point of TNF-α level in serum was found after 1 hour.
Endotoxaemia, sepsis, and septic shock are associated with the generation of ROS. The overproduction of ROS in shock leads to a considerable oxidant stress as indicated by lipid peroxidation, high blood levels of malondialdehyde and conjugated dienes, as well as the consumption of the endogenous antioxidant vitamins C and E. Some authors reported a decline in the expression of copper/zinc SOD in rats with endotoxic shock using LPS dose of 10 mg/kg [43]. Moreover, mice injected with LPS 100 mg/kg showed a significant increase of ROS production, TNF-α, and IL-1β release by peritoneal leukocytes, generating an oxidative stress [44].
In this work we test some markers of oxidative stress in hepatic tissue of mice injected with LPS 0.1 mg/kg. We observed that LPS at 0.1 mg/kg provoked a slight increase in CAT activity, as well as a decrease in GSH-Px and a significant increase in malondialdehyde levels, measured as TBARS. The slight increase in CAT activity in LPS-injected mice might be related with the generation of ROS such as hydrogen peroxide and peroxynitrite associated with endotoxaemia.
Ozone oxidative preconditioning appears to restore the oxidant balance, minimizing tissue injury caused by endotoxaemia. Selenium-dependent GSH-Px and GST were found to be biomarkers of endovenous ozone therapy [45]. Both enzymes are involved in detoxification of lipoperoxides, although GST is a toxicologically multifunctional enzyme that catalyzes the conjugation of xenobiotics with GSH, among others. Both GSH-Px and GST activities were enhanced by ozone IP applications in this experiment.
Considering either the effects of diminution of serum TNF-α release, the enhancement of antioxidant enzymes, or the decrease in malondialdehyde levels in hepatic tissue, ozone/oxygen IP applications exert beneficial effects in this model of shock, avoiding the establishment of a chronic inflammatory response and an oxidative stress. In our opinion, the inhibitory effects of the ozone/oxygen mixture in TNF-α levels in serum of mice injected with endotoxin and the stimulation of the antioxidant activity on hepatic tissue might be ascribed to the inhibition of the activation of the NF-κB which are activated by ROS with the subsequent induction and expression of various cytokines and enzymes such as TNF-α and GSH-Px, respectively, which are involved in the induction and development of endotoxic shock.
Taking into account our results and the former findings of other authors described above it is conceivable that, as occurs with other antioxidants and ROS scavengers, OOP might exert its effects on endotoxic shock by inhibition of NF-κB activation. This may explain its inhibitory effects on TNF-α. However, further studies will be needed to elucidate the mechanisms underlying the beneficial effect of OOP.
ACKNOWLEDGMENT
We would like to thank Dr Carlos Hernández, Dr Cheyla Romay, and Dr Ricardo González for their help in the writing of this paper.
References
- 1.Astiz ME, Rackow EC. Septic shock. (Review) Lancet. 1998;351(9114):1501–1505. doi: 10.1016/S0140-6736(98)01134-9. [DOI] [PubMed] [Google Scholar]
- 2.Gutierrez-Ramos JC, Bluethmann H. Molecules and mechanisms operating in septic shock: lessons from knockout mice. Immunol Today. 1997;18(7):329–334. doi: 10.1016/s0167-5699(97)01085-2. [DOI] [PubMed] [Google Scholar]
- 3.Wilson M, Henderson B, Poole S. Bacteria/Cytokine Interactions in Health and Disease. London, UK: Portland Press; 1998. [Google Scholar]
- 4.Tracey KJ, Abraham E. From mouse to man: or what have we learned about cytokine-based anti-inflammatory therapies? (Review) Shock. 1999;11(3):224–225. [PubMed] [Google Scholar]
- 5.Bernhagen J, Calandra T, Mitchell RA, et al. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature. 1993;365(6448):765–759. doi: 10.1038/365756a0. [DOI] [PubMed] [Google Scholar]
- 6.Vincent JL. New therapies in sepsis. (Review) Chest. 1997;112(suppl 6):330S–338S. doi: 10.1378/chest.112.6_supplement.330s. [DOI] [PubMed] [Google Scholar]
- 7.Beutler B, Cerami A. The biology of cachectin/TNF-a primary mediator of the host response. Annu Rev Immunol. 1989;7:625–655. doi: 10.1146/annurev.iy.07.040189.003205. [DOI] [PubMed] [Google Scholar]
- 8.Vassalli P. The pathophysiology of tumor necrosis factors. Annu Rev Immunol. 1992;10:411–452. doi: 10.1146/annurev.iy.10.040192.002211. [DOI] [PubMed] [Google Scholar]
- 9.Newton RC, Decicco CP. Therapeutic potential and strategies for inhibiting tumor necrosis factor-alpha. J Med Chem. 1999;42(13):2295–2314. doi: 10.1021/jm980541n. [DOI] [PubMed] [Google Scholar]
- 10.Billiar TR. Nitric oxide. Novel biology with clinical relevance. Ann Surg. 1995;221(4):339–349. doi: 10.1097/00000658-199504000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jakab GJ, Spannhake EW, Canning BJ, Kleeberger SR, Gilmour MI. The effects of ozone on immune function. Environ Health Perspect. 1995;103(suppl 2):77–89. doi: 10.1289/ehp.95103s277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Canning BJ, Hmieleski RR, Spannhake EW, Jakab GJ. Ozone reduces murine alveolar and peritoneal macrophage phagocytosis: the role of prostanoids. Am J Physiol. 1991;261(4 pt 1):L277–282. doi: 10.1152/ajplung.1991.261.4.L277. [DOI] [PubMed] [Google Scholar]
- 13.Chatterjee D, Mukherjee SK. Destruction of phagocytosis-suppressing activity of aflatoxin B1 by ozone. Lett Appl Microbiol. 1993;17(2):52–54. doi: 10.1111/j.1472-765x.1993.tb00368.x. [DOI] [PubMed] [Google Scholar]
- 14.Leon OS, Menendez S, Merino N, et al. Ozone oxidative preconditioning: a protection against cellular damage by free radicals. Mediators Inflamm. 1998;7(4):289–294. doi: 10.1080/09629359890983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Candelario-Jalil E, Mohammed-Al-Dalain S, Fernandez OS, et al. Oxidative preconditioning affords protection against carbon tetrachloride-induced glycogen depletion and oxidative stress in rats. J Appl Toxicol. 2001;21(4):297–301. doi: 10.1002/jat.758. [DOI] [PubMed] [Google Scholar]
- 16.Peralta C, Leon OS, Xaus C, et al. Protective effect of ozone treatment on the injury associated with hepatic ischemia-reperfusion: antioxidant-prooxidant balance. Free Radic Res. 1999;31(3):191–196. doi: 10.1080/10715769900300741. [DOI] [PubMed] [Google Scholar]
- 17.Barber E, Menendez S, Leon OS, et al. Prevention of renal injury after induction of ozone tolerance in rats submitted to warm ischaemia. Mediators Inflamm. 1999;8(1):37–41. doi: 10.1080/09629359990702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hernandez F, Menendez S, Wong R. Decrease of blood cholesterol and stimulation of antioxidative response in cardiopathy patients treated with endovenous ozone therapy. Free Radic Biol Med. 1995;19(1):115–119. doi: 10.1016/0891-5849(94)00201-t. [DOI] [PubMed] [Google Scholar]
- 19.Bocci V. Does ozone therapy normalize the cellular redox balance? Implications for therapy of human immunodeficiency virus infection and several other diseases. Med Hypotheses. 1996;46(2):150–154. doi: 10.1016/s0306-9877(96)90016-x. [DOI] [PubMed] [Google Scholar]
- 20.Romero Valdes A, Menendez Cepero S, Gomez Moraleda M, Ley Pozo J. Ozone therapy in the advanced stages of arteriosclerosis obliterans. Angiologia. 1993;45(4):146–148. [PubMed] [Google Scholar]
- 21.Bocci V. Biological and clinical effects of ozone. Has ozone therapy a future in medicine? Br J Biomed Sci. 1999;56(4):270–279. [PubMed] [Google Scholar]
- 22.Schulz S, Rodriguez ZZ, Mutters R, Menendez S, Bette M. Repetitive pneumoperitoneum with ozonized oxygen as a preventive in lethal polymicrobial sepsis in rats. Eur Surg Res. 2003;35(1):26–34. doi: 10.1159/000067032. [DOI] [PubMed] [Google Scholar]
- 23.Zamora Z, Schulz S, Gonzalez Y, Menendez S. Intraperitoneal application of ozone in a postoperative sepsis model in rats. Rev. Mex. Farmacia. 2002;33(2):13–17. [Google Scholar]
- 24.Schulz S, Bette M, Schlimme S, Mutters R, Hoffmann S, Menendez S. Influence of O(3)/O(2)-pneumoperitoneum as an oxidative stressor on duration of anaesthesia, loss of different reflexes and cytokine mRNA expression. Lab Anim. 2004;38(3):261–271. doi: 10.1258/002367704323133646. [DOI] [PubMed] [Google Scholar]
- 25.Bette M, Schlimme S, Mutters R, Menendez S, Hoffmann S, Schulz S. Influence of different anaesthetics on pro-inflammatory cytokine expression in rat spleen. Lab Anim. 2004;38(3):272–279. doi: 10.1258/002367704323133655. [DOI] [PubMed] [Google Scholar]
- 26.Botsoglou NA, Fletouris DJ, Papageorgiou GE, Vassilopoulos VN, Mantis AJ, Trakatellis A. Rapid, sensitive, and specific thiobarbituric acid method formeasuring lipid peroxidation in animal tissue, food, and feedstuff samples. J. Agric. Food Chem. 1994;42:1931–1937. [Google Scholar]
- 27.Habig WH, Pabst MJ, Jakoby WB. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J Biol Chem. 1974;249(22):7130–7139. [PubMed] [Google Scholar]
- 28.Rice-Evans CA, Diplock AT. Laboratory techniques in biochemistry and molecular biology. In: van Knippenber PH, Burdon RH, editors. Techniques in Free Radical Research. Vol. 22. Amsterdam: Elsevier; 1991. pp. 199–201. [Google Scholar]
- 29.Faraji B, Kang HK, Valentine JL. Methods compared for determining glutathione peroxidase activity in blood. Clin Chem. 1987;33(4):539–543. [PubMed] [Google Scholar]
- 30.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193(1):265–275. [PubMed] [Google Scholar]
- 31.Murry CE, Richard VJ, Reimer KA, Jennings RB. Ischemic preconditioning slows energy metabolism and delays ultrastructural damage during a sustained ischemic episode. Circ Res. 1990;66(4):913–931. doi: 10.1161/01.res.66.4.913. [DOI] [PubMed] [Google Scholar]
- 32.Neschis DG, Safford SD, Raghunath PN, et al. Thermal preconditioning before rat arterial balloon injury: limitation of injury and sustained reduction of intimal thickening. Arterioscler Thromb Vasc Biol. 1998;18(1):120–126. doi: 10.1161/01.atv.18.1.120. [DOI] [PubMed] [Google Scholar]
- 33.Riepe MW, Ludolph AC. Chemical preconditioning: a cytoprotective strategy. Mol Cell Biochem. 1997;174(1-2):249–254. [PubMed] [Google Scholar]
- 34.Peralta C, Hotter G, Closa D, Gelpi E, Bulbena O, Rosello-Catafau J. Protective effect of preconditioning on the injury associated to hepatic ischemia-reperfusion in the rat: role of nitric oxide and adenosine. Hepatology. 1997;25(4):934–937. doi: 10.1002/hep.510250424. [DOI] [PubMed] [Google Scholar]
- 35.Peralta C, Closa D, Xaus C, Gelpi E, Rosello-Catafau J, Hotter G. Hepatic preconditioning in rats is defined by a balance of adenosine and xanthine. Hepatology. 1998;28(3):768–737. doi: 10.1002/hep.510280325. [DOI] [PubMed] [Google Scholar]
- 36.Peralta C, Xaus C, Bartrons R, Leon OS, Gelpi E, Rosello-Catafau J. Effect of ozone treatment on reactive oxygen species and adenosine production during hepatic ischemia-reperfusion. Free Radic Res. 2000;33(5):595–605. doi: 10.1080/10715760000301121. [DOI] [PubMed] [Google Scholar]
- 37.Klosterhalfen B, Bhardwaj RS. Septic shock. Gen Pharmacol. 1998;31(1):25–32. doi: 10.1016/s0306-3623(97)00424-2. [DOI] [PubMed] [Google Scholar]
- 38.Novelli GP. Role of free radicals in septic shock. J Physiol Pharmacol. 1997;48(4):517–527. [PubMed] [Google Scholar]
- 39.Redl H, Schlag G, Bahrami S, Schade U, Ceska M, Stutz P. Plasma neutrophil-activating peptide-1/interleukin-8 and neutrophil elastase in a primate bacteremia model. J Infect Dis. 1991;164(2):383–388. doi: 10.1093/infdis/164.2.383. [DOI] [PubMed] [Google Scholar]
- 40.Suffredini AF, Reda D, Banks SM, Tropea M, Agosti JM, Miller R. Effects of recombinant dimeric TNF receptor on human inflammatory responses following intravenous endotoxin administration. J Immunol. 1995;155(10):5038–5045. [PubMed] [Google Scholar]
- 41.Voisin L, Breuille D, Ruot B, et al. Cytokine modulation by PX differently affects specific acute phase proteins during sepsis in rats. Am J Physiol. 1998;275(5 pt 2):R1412–R1419. doi: 10.1152/ajpregu.1998.275.5.R1412. [DOI] [PubMed] [Google Scholar]
- 42.Cavaillon JM, Adib-Conquy M, Fitting C, Adrie C, Payen D. Cytokine cascade in sepsis. Scand J Infect Dis. 2003;35(9):535–544. doi: 10.1080/00365540310015935. [DOI] [PubMed] [Google Scholar]
- 43.Leach M, Frank S, Olbrich A, Pfeilschifter J, Thiemermann C. Decline in the expression of copper/zinc superoxide dismutase in the kidney of rats with endotoxic shock: effects of the superoxide anion radical scavenger, tempol, on organ injury. Br J Pharmacol. 1998;125(4):817–825. doi: 10.1038/sj.bjp.0702123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Victor VM, De la Fuente M. Several functions of immune cells in mice changed by oxidative stress caused by endotoxin. Physiol Res. 2003;52(6):789–796. [PubMed] [Google Scholar]
- 45.Dierickx PJ, Nuffel GV, Alvarez I. Glutathione protection against hydrogen peroxide, tert-butyl hydroperoxide and diamide cytotoxicity in rat hepatoma-derived Fa32 cells. Hum Exp Toxicol. 1999;18(10):627–633. doi: 10.1191/096032799678839482. [DOI] [PubMed] [Google Scholar]