

Abstract
BphK is a glutathione S-transferase of unclear physiological function that occurs in some bacterial biphenyl catabolic (bph) pathways. We demonstrated that BphK of Burkholderia xenovorans strain LB400 catalyzes the dehalogenation of 3-chloro 2-hydroxy-6-oxo-6-phenyl-2,4-dienoates (HOPDAs), compounds that are produced by the cometabolism of polychlorinated biphenyls (PCBs) by the bph pathway and that inhibit the pathway's hydrolase. A one-column protocol was developed to purify heterologously produced BphK. The purified enzyme had the greatest specificity for 3-Cl HOPDA (kcat/Km, ∼104 M−1 s−1), which it dechlorinated approximately 3 orders of magnitude more efficiently than 4-chlorobenzoate, a previously proposed substrate of BphK. The enzyme also catalyzed the dechlorination of 5-Cl HOPDA and 3,9,11-triCl HOPDA. By contrast, BphK did not detectably transform HOPDA, 4-Cl HOPDA, or chlorinated 2,3-dihydroxybiphenyls. The BphK-catalyzed dehalogenation proceeded via a ternary-complex mechanism and consumed 2 equivalents of glutathione (GSH) (Km for GSH in the presence of 3-Cl HOPDA, ∼0.1 mM). A reaction mechanism consistent with the enzyme's specificity is proposed. The ability of BphK to dehalogenate inhibitory PCB metabolites supports the hypothesis that this enzyme was recruited to facilitate PCB degradation by the bph pathway.
Polychlorinated biphenyls (PCBs) are one of the most widely distributed chlorinated pollutants in the environment (39). The partial transformation of these pollutants by bacteria has spurred research to utilize microbial catabolic activities to bioremediate PCB-contaminated sites. Many PCB congeners, of which there are ∼50 in typical commercial formulations, are aerobically cometabolized by the bph pathway (Fig. 1). The bph pathway catabolizes biphenyl using a strategy that is typical of that of the aerobic catabolism of aromatic compounds (1, 30). Biphenyl dioxygenase catalyzes the NADH-dependent dihydroxylation of biphenyl, yielding a 2,3-dihydrodiol. The latter is transformed to 2,3-dihydroxybiphenyl (DHB). This catechol is oxygenolytically cleaved in an extradiol fashion by the bphC-encoded 2,3-dihydroxy-1,2-dioxygenase (DHBD), yielding 2-hydroxy-6-oxo-6-phenyl-2,4-dienoate (HOPDA). In the final step of the upper bph pathway, catalyzed by BphD, HOPDA is hydrolyzed to benzoate and 2-hydroxypenta-2,4-dienoate.
FIG. 1.

The bph pathway responsible for the degradation of biphenyl. (A) Organization of the bph operon in B. xenovorans LB400. Open reading frames and genes of unknown function are represented by white arrows. (B) bph metabolic pathway. (Adapted from reference 17a.)
The PCB-transforming capabilities of the bph pathway are strain dependent. Nevertheless, even the “best” strains, such as Burkholderia xenovorans LB400 (previously Pseudomonas sp. strain LB400) (13), poorly transform congeners containing more than four chlorine substituents. While most efforts have focused on characterizing and engineering the PCB-transforming abilities of biphenyl droxygenase (7, 8, 36), it is clear that other Bph enzymes are inhibited by specific chlorinated metabolites. For example, 2′,6′-diCl DHB, produced by the transformation of congeners dichlorinated in the ortho position, is a potent competitive inhibitor of DHBD (17) and can inactivate the enzyme through oxidation of the enzyme's catalytically essential ferrous ion (42). As a second example, BphD is competitively inhibited by 3-Cl and 4-Cl HOPDAs (33). Such metabolites can originate from para- and meta-chlorinated congeners, respectively.
BphK is a glutathione S-transferase (GST) of unclear function that occurs in some bph pathways (9). The bphK gene appears to occur in only two of the different types of bph gene clusters reported to date (30): (i) those that are organized as in B. xenovorans LB400 and Pseudomonas pseudoalcaligenes KF707 (38) and (ii) those that are organized as in Sphingobium yanoikuyae B1 (47) and Novosphingobium aromaticivorans F199 (31). The enzyme does not appear to be essential for growth on biphenyl (9). Based on its ability to catalyze the glutathione (GSH)-dependent dehalogenation of 4-chlorobenzoate (4-CBA) (19), it has been hypothesized that BphK was recruited by some strains to facilitate the degradation of PCBs. However, the specific activity of BphK for 4-CBA is very low (<10−4 U/mg) (22), and B. xenovorans LB400 neither grows on nor detectably transforms 4-Cl benzoate (25). Therefore, the physiological substrate of BphK and the enzyme's role in the bph pathway remain unclear.
GSTs are one of nature's most versatile enzymes, catalyzing a wide range of reactions involving the conjugation of GSH to electrophilic compounds (4, 35). Prokaryotic GSTs are generally not as well characterized as their eukaryotic counterparts (43), although genome sequencing efforts indicate that some bacterial strains possess over 10 GST-encoding genes (44). Many bacterial GSTs occur in pathways responsible for the aerobic catabolism of aromatic compounds. Such GSTs may transform catabolites or protect cell components against reactive oxygen species (38). Catabolite transformations include dehalogenation and carbon-carbon double bond isomerization (45). One of the better-studied bacterial GSTs is tetrachlorohydroquinone (TCHQ) dehalogenase from Sphingobium chlorophenolicum which catalyzes the reductive dehalogenation of TCHQ to dichlorohydroquinone via trichlorohydroquinone (23, 24, 46) and appears to have been recently recruited for the degradation of pentachlorophenol (PCP) (15).
Herein we report the highly efficient heterologous production and purification of BphK of B. xenovorans LB400. The ability of BphK to transform a variety of PCB metabolites was investigated. The transformation products and reaction stoichiometries were determined. Steady-state kinetic parameters for the BphK-catalyzed dehalogenation of several chlorinated HOPDAs were evaluated. The results are discussed with respect to potential reaction mechanisms and the ability of bacterial catabolic pathways to degrade xenobiotics that have recently been released into the biosphere.
MATERIALS AND METHODS
Chemicals.
DHB, 4-Cl DHB, 5-Cl DHB, 6-Cl DHB, and 4,3′,5′-triCl DHB were synthesized according to standard procedures (28). HOPDA, 3-Cl HOPDA, 4-Cl HOPDA, 5-Cl HOPDA, and 3,9,11-triCl HOPDA were produced enzymatically from DHB, 4-Cl DHB, 5-Cl DHB, 6-Cl DHB, and 4,3′,5′-triCl DHB, respectively, as described previously (33). The HOPDAs were purified from a methanol-water (0.5% H3PO4) mixture (80:20) by high-performance liquid chromatography (HPLC) using a Waters 2695 separation module fitted with a Prodigy 10-μm-particle-size ODS-Prep column (21.2 by 250 mm; Phenomenex, Torrance, CA). The HOPDAs were eluted in the same methanol/water (0.5% H3PO4) mixture at a flow rate of 7 ml/min. The HOPDA-containing fractions were pooled, acidified to a pH of ∼2 with 2 N HCl, and extracted three times with 0.5 volume of ethyl acetate. The organic extract was dried over anhydrous MgSO4 and evaporated to dryness, yielding the purified HOPDA as a powder. All other chemicals were of analytical grade and used without further purification.
Strains, media, and growth.
Escherichia coli strains DH5α and Nova Blue DE3 (EMD Biosciences, Inc.) were used for DNA propagation and were cultured at 37°C and 200 rpm in Luria-Bertani (LB) broth with the appropriate antibiotics. E. coli GJ1158 (11) transformed with pT7-7 derivatives was used for overexpression and was cultured at 37°C and 250 rpm in low-salt LB with 100 μg/ml ampicillin.
Construction of plasmids and overexpression.
DNA was manipulated using standard protocols (5). The gene bphK was amplified by PCR from a plasmid containing the full bph pathway of B. xenovorans LB400, pAH92, using the following oligonucleotides: 5K (5′-AAGCGCTAGCATGAAACTTTACTACAGCCC-3′ [underlining represents an introduced NheI site]) and 3K (5′-GCGCAAGCTTCGGGAAGGCATCTTC-3′ [underlining represents an introduced HindIII site]). PCR was performed using the Expand High Fidelity DNA polymerase system (Roche Applied Sciences, Laval, Canada) according to the manufacturer's instructions and an annealing temperature of 45°C. The resulting amplicon was digested with NheI and HindIII and cloned into pT7HP20 yielding pT7BK20. A single NdeI restriction site present within bphK was removed by site-directed mutagenesis using the Quick Change Multi mutagenesis kit (Stratagene, La Jolla, CA) and the oligonucleotide KSDM (5′-AAAGATCAA-TGTTGACATAAGCGCTCCAACCGAGCAC-3′), yielding pT7BK21. The bphK gene was amplified from pT7BK21 using the primers 5Knde and 3K. The resulting amplicon was digested with NdeI and HindIII and cloned into pT7-7 (37), yielding pT7BK1. The nucleotide sequences of cloned amplicons were confirmed using an ABI 373 Stretch DNA sequencer (Applied Biosystems, Foster City, CA) and a BigDye v3.1 Terminator kit. BphK was produced using E. coli GJ1158 transformed with pT7BK1. Cells were grown in low-salt LB (10 g tryptone extract and 5 g yeast extract per liter) supplemented with 100 μg/ml ampicillin. When the culture reached an optical density at 600 nm of 0.5, NaCl was added to a final concentration of 0.3 M, and the culture was incubated for an additional 12 h before harvesting.
Purification of BphK.
BphK was affinity purified using a glutathione resin (Amersham Biosciences, Baie d'Urfé, Canada) according to the manufacturer's instructions. Briefly, cells from 1 liter of culture were harvested by centrifugation. The cell pellet (approximately 6 g) was resuspended in 10 ml phosphate-buffered saline (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.3). The cells were disrupted by three successive passages through a continuous-flow French press (Spectronic Instruments Inc., Rochester, NY) operated at a pressure of 10,000 lb/in2. The cell debris was removed by ultracentrifugation at 37,000 rpm for 30 min in a T1250 rotor (DuPont Instruments, Wilmington, DE). The clear supernatant was carefully decanted and filtered using a 0.45-μm-pore-size filter (Sartorius AG, Göttingen, Germany). This fluid, referred to as raw extract, was applied to a 20- by 32-mm column of glutathione Sepharose 4B resin. The resin was washed three times with 100 ml of phosphate-buffered saline and then eluted with 20 ml of 50 mM Tris-Cl (pH 8.0) and 10 mM GSH. The collected fractions were concentrated to >50 mg of protein/ml using a stirred-cell concentrator equipped with a YM10 membrane (Amicon, Oakville, Canada) and frozen as beads in liquid N2. Purified BphK was stored at −80°C for up to 6 months without any significant loss of activity.
Handling and characterization of BphK samples.
Aliquots of BphK were thawed immediately prior to use and were diluted in potassium phosphate buffer, pH 6.5 (I = 0.05 M), supplemented with 0.1 mg/ml bovine serum albumin. Total protein concentrations were determined using the Bradford method (14). BphK concentrations were determined using the absorbance of the protein solution at 280 nm and an extinction coefficient obtained by amino acid analysis (ɛ = 33.3 mM−1 cm−1). The molecular weight of native BphK was determined using a HiLoad 26/60 Superdex 75 gel filtration column (Amersham Pharmacia Biotech, Uppsala, Sweden) operated at a flow rate of 2 ml/min. The elution buffer was 50 mM potassium phosphate containing 0.15 M NaCl, pH 6.5. The column was calibrated using RNase A (13.7 kDa), chymotrypsinogen A (25.0 kDa), ovalbumin (45.0 kDa), and aldolase (158 kDa).
Transformation of PCB metabolites.
Solutions of BphK containing potassium phosphate buffer (pH 6.5; I = 0.05 M) and 1 mM GSH were incubated at 25.0 ± 0.1°C with, separately, 4-chlorobenzoic acid, 4-Cl DHB, 5-Cl DHB, 6-Cl DHB, HOPDA, 3-Cl HOPDA, 4-Cl HOPDA, 5-Cl HOPDA and 3,9,11-triCl HOPDA. Reaction mixtures contained 0.1 to 2.5 mM PCB metabolite and 0.02 to 2 mM enzyme. Aliquots of the reaction mixture were withdrawn periodically and analyzed by HPLC using a Waters 2695 separation module and a C18 Symmetry 3.5-μm-particle-size column (4.6 by 75 mm) (Waters, Mississauga, Canada). The reaction products were eluted using a 60:40 mixture of methanol and water at a flow rate of 1 ml/min. The concentration of chloride ion was measured using a published procedure (10). The concentration of reduced GSH was determined using 5,5′-dinitrothiobenzene (18).
To characterize the transformation product of 3-Cl HOPDA, 20 μmol of 3-Cl HOPDA, 200 μmol of GSH, and 100 nmol of BphK were incubated in 200 ml of 50 mM potassium phosphate buffer (pH 6.5) at 25°C. The reaction was allowed to proceed to >80% completion as determined spectrophotometrically. BphK was removed by ultrafiltration using a stirred cell equipped with a YM10 membrane (Amicon). The protein-free ultrafiltrate containing >80 μM of the reaction product was acidified to a pH of 3 by adding 200 ml of an aqueous solution of 0.5% orthophosphoric acid. The resulting solution was extracted twice with 0.5 volume of ethyl acetate. The ethyl acetate fraction was dried using anhydrous magnesium sulfate and removed using a rotary evaporator, and the product was dissolved in a 1:1 mixture of methanol and water and purified by HPLC using a Waters 2695 separation module and a Prodigy 10-μm-particle-size ODS-Prep column (21.2 by 250 mm; Phenomenex, Torrance, CA). The column was eluted with the same methanol-water mixture at a flow rate of 7 ml/min. The retention time of the product was ∼40 min. The fractions containing the product were pooled, added to 4 volumes of aqueous 0.5% orthophosphoric acid, and then extracted as described above. The product was further characterized by visible-UV spectrophotometry and analytical HPLC.
Physical properties of HOPDAs.
The extinction coefficients of chlorinated HOPDAs were determined essentially as previously described (33), by weighing the corresponding DHBs and following their quantitative cleavage by DHBD using a Clark-type oxygen electrode. Solutions of the freshly prepared HOPDAs had the characteristic yellow color of the enolate anion, absorbing maximally at wavelengths (λmax) between 400 and 438 nm. The extinction coefficient and λmax of each HOPDA in 50 mM phosphate (pH 6.5) at 25°C were as follows: HOPDA, 4.9 mM−1 cm−1 at 430 nm; 3-Cl HOPDA, 29.9 mM−1 cm−1 at 430 nm; 5-Cl HOPDA, 23.5 mM−1 cm−1 at 400 nm; 9,11-diCl HOPDA, 17.9 mM−1 cm−1 at 438 nm; and 3,9,11-triCl HOPDA, 35.6 mM−1 cm−1 at 438 nm.
Steady-state kinetic studies.
The steady-state transformation of chlorinated HOPDAs by BphK was routinely measured spectrophotometrically using a Varian Cary 1E spectrometer equipped with a thermojacketed cuvette holder. The standard assay conditions used potassium phosphate buffer (pH 6.5; I = 0.05 M) at 25.0 ± 0.1°C, 1 mM of GSH, and 50 μM 3-Cl HOPDA. The reaction was monitored at 438 nm. Assays performed using 1 mM 1-chloro-2,4-dinitrobenzene (CDNB) were monitored at 340 nm as previously described (20). The amount of enzyme used in each assay was adjusted so that the progress curve was linear for at least 1 min. Initial velocities were determined from a least-squares analysis of the linear portion of the progress curves using the kinetics module of the Cary software. One unit of enzymatic activity was defined as the quantity of enzyme required to consume 1 μmol of substrate per minute.
The steady-state kinetic parameters for chlorinated HOPDAs were determined using substrate concentrations ranging from 4 to 100 μM and an enzyme concentration of ∼40 nM. The apparent Km for GSH was determined using a concentration of 50 μM 3-Cl HOPDA and 75 μM of either 5-Cl HOPDA or 3,9,11-triCl HOPDA and 0.02 to 10 mM GSH. Steady-state rate equations were fit to data sets using the least-squares and dynamic-weighting options of LEONORA (16).
RESULTS AND DISCUSSION
Expression and purification of BphK.
A highly efficient expression system was developed to facilitate the characterization of BphK. When E. coli GJ1158 and pT7BK1 were used, BphK constituted ∼60% of the soluble cellular protein. By using a single chromatographic step, BphK was purified to greater than 99% homogeneity as judged by the presence of a single 23-kDa band on sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels (not shown). This procedure yielded 169 mg of BphK from 1 liter of cell culture, representing 87% of the activity in the raw extract. The specific activity of the purified enzyme towards the reporter substrate CDNB, 7.8 U/mg, was 1.7-fold higher than that of the raw extract. The molecular mass of native BphK was estimated to be 33.9 kDa by calibrated gel filtration chromatography. Crystallographic data demonstrate that the enzyme is dimeric (41).
Transformation of PCB metabolites.
In investigating the ability of BphK to transform PCB metabolites, we first replicated previous reports that BphK catalyzes the dechlorination of 4-CBA (19). When a solution containing 2.5 mM 4-CBA and 5 mM GSH was incubated in the presence of 2 mM BphK, half of the 4-CBA was dechlorinated after 2.5 h, indicating that 4-CBA is not a particularly good substrate for BphK. Moreover, B. xenovorans LB400 does not grow on 4-CBA (25). We therefore hypothesized that the physiological substrate of BphK might be an inhibitory PCB metabolite such as a DHB halogenated on the catechol ring or a HOPDA halogenated on the dienoate moiety. Accordingly, we tested the ability of BphK to catalyze the transformation of a variety of such PCB metabolites by following the depletion of these compounds using analytical HPLC. In reaction mixtures containing up to 5 mM GSH and 2 mM BphK, the enzyme did not detectably transform any of the following DHBs after 5 h when present at concentrations up to 2.5 mM: 4-Cl DHB, 5-Cl DHB, and 6-Cl DHB. In contrast, BphK transformed 3-Cl, 5-Cl, and 3,9,11-triCl HOPDAs, as described in more detail below. The triCl HOPDA was chosen as an example of a physiologically relevant substrate: 3-Cl and 5-Cl HOPDA are unlikely to occur during the degradation of PCBs by B. xenovorans LB400 as the corresponding monochlorobiphenyls are likely to be attacked on the unchlorinated ring (21, 34). BphK did not detectably catalyze the transformation of either unchlorinated HOPDA or 4-Cl HOPDA.
Dechlorination of 3-Cl HOPDAs.
To better characterize the BphK-catalyzed transformation of 3-Cl HOPDA, we analyzed the products of this reaction by use of a chloride ion assay and HPLC. The former demonstrated that chloride was stoichiometrically released during GSH-dependent transformation of 3-Cl HOPDA (results not shown). Analysis of the reaction mixture by HPLC revealed a product with a retention time (16.1 min) and absorption spectrum (Fig. 2) that corresponded to those of unchlorinated HOPDA (retention time of 3-Cl HOPDA = 15.3 min). Finally, when incubated in the presence of the HOPDA hydrolase BphDLB400, the product of the BphK-catalyzed reaction was transformed to 2-hydroxypentadienoate and benzoate, the two expected products of the BphD-catalyzed reaction. The dechlorination of 12.0 ± 0.8 nmol of 3-Cl HOPDA was coupled to the oxidation of 25.0 ± 0.4 nmol of GSH, indicating that BphK catalyzes the dehalogenation of this PCB metabolite in a reaction that consumes 2 equivalents of GSH.
FIG. 2.

Electronic absorption spectra of 3-Cl HOPDA (solid line) and its BphK-catalyzed transformation product (dotted line). The product was generated by incubating 0.1 mM 3-Cl HOPDA with 1 mM GSH and an excess of BphK under standard reaction conditions for 2 min. 3-Cl HOPDA and the reaction product were injected into the HPLC. Spectra were collected in a 50:50 mixture of methanol and water (0.5% H3PO4) using a Waters 2996 photodiode array detector during HPLC analysis. The retention time and spectrum of the reaction product are identical to those of HOPDA.
Steady-state kinetic analyses.
To investigate the substrate specificity of BphK, we developed a continuous, spectrophotometric assay. This assay was based on the differential absorption of each chlorinated HOPDA and its corresponding dechlorinated product. The wavelength of the assay and extinction coefficients thus depended on the identity of the chlorinated HOPDA. The assay was performed at a pH of 6.5 to maximize differences in the absorption of the reactants and products. The dehalogenation of 3-Cl, 5-Cl, and 3,9,11-triCl HOPDAs were monitored at 430 nm (Δɛ3CH = 21.5 mM−1 cm−1), 400 nm (Δɛ5CH = 18.5 mM−1 cm−1), and 438 nm (ΔɛtriCH = 17.7 mM−1 cm−1), respectively (I = 0.05 M potassium phosphate; pH 6.5).
In the presence of 1 mM GSH, the BphK-catalyzed dehalogenation of 3-Cl-HOPDA obeyed Michaelis-Menten kinetics with respect to 3-Cl HOPDA concentration (Fig. 3). Similar behavior was observed as a function of GSH concentration in the presence of 50 μM 3-Cl HOPDA (Fig. 3, inset) and was also confirmed using either 5-Cl HOPDA or 3,9,11-triCl HOPDA (results not shown). These analyses revealed that at 1 mM GSH, BphK had the greatest apparent specificity for 3-Cl HOPDA [kcat/Km = (5.6 ± 0.9) × 103 M−1 s−1] followed by 5-Cl HOPDA kcat/Km = (2.0 ± 0.5) × 103 M−1 s−1] and 3,9,11-triCl HOPDA [kcat/Km = (0.35 ± 0.03) × 103 M−1 s−1]. However, these analyses also indicated that BphK was not saturated with GSH under the described reaction conditions: using 50 μM 3-Cl HOPDA, the apparent Km for GSH (Km GSH of BphK was 124 μM (Fig. 3); using 75 μM 5-Cl HOPDA, the apparent Km GSH was ∼0.3 mM; and using 75 μM 3,9,11-triCl HOPDA, the apparent Km GSH was 2.0 mM.
FIG. 3.

Steady-state transformation of 3-Cl HOPDA by BphK. The main panel depicts the dependence of initial velocity on the concentration of 3-Cl HOPDA in the presence of 1 mM GSH. The curve represents a best fit of the Michaelis-Menten equation to the data using the least-squares and dynamic-weighting options of LEONORA. The fitted parameters are a value for 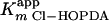 of 22 ± 2 μM and a velocity of 41 ± 2 μM/min. The insert depicts the dependence of initial velocity on the concentration of GSH using 50 μM 3-Cl HOPDA. The fitted parameters are a value for
of 22 ± 2 μM and a velocity of 41 ± 2 μM/min. The insert depicts the dependence of initial velocity on the concentration of GSH using 50 μM 3-Cl HOPDA. The fitted parameters are a value for 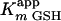 of 120 ± 20 μM and a velocity of 32 ± 3 μM/min. Experiments were performed using potassium phosphate, pH 6.5 (I = 0.05 M), at 25°C.
of 120 ± 20 μM and a velocity of 32 ± 3 μM/min. Experiments were performed using potassium phosphate, pH 6.5 (I = 0.05 M), at 25°C.
To further investigate the steady-state mechanism of the BphK-catalyzed dehalogenation of chlorinated HOPDAs and to better evaluate the steady-state rate parameters, initial reaction rates were measured over a range of GSH and Cl HOPDA concentrations. Three sets of experiments were performed, using (i) 5 to 50 μM 3-Cl HOPDA and 50 to 1,000 μM GSH, (ii) 10 to 100 μM 5-Cl HOPDA and 100 to 2,000 μM GSH, and (iii) 10 to 50 μM 3,9,11-triCl HOPDA and 500 to 3,500 μM GSH. Fitting an equation describing a substituted-enzyme (or “ping-pong”) mechanism to any of these data sets yielded residual plots possessing nonrandom trends. In contrast, an equation describing a ternary-complex mechanism (16) fit all data sets with random trends in the residual plots as well as lower standard errors in the estimated parameters. It was not possible to distinguish between a rapid-equilibrium random-order ternary-complex mechanism and a compulsory-order ternary-complex mechanism using the present data, as the two are kinetically equivalent (16).
The kinetic parameters obtained from the more complete analyses revealed that the specificity of BphK for 3-Cl HOPDA was approximately 5 times higher than for 5-Cl HOPDA and 10 times higher than for 3,9,11-triCl HOPDA (Table 1). Moreover, the identity of the HOPDA influenced the reactivity of BphK with GSH. Thus, the specificity of BphK for GSH was 50 times higher in the presence of 3-Cl HOPDA than in the presence of 3,9,11-triCl HOPDA and 2.5 times higher than in the presence of 5-Cl HOPDA. In these experiments, the errors associated with the kinetic parameters determined for 5-Cl HOPDA were higher as the enzyme could not be saturated with each of the two substrates.
TABLE 1.
Steady-state kinetic parameters of BphK for chlorinated HOPDAs and GSHa
| HOPDA | Km GSH (μM) | Km ClH (μM) | kcat (s−1) | kcat/Km ClH (103 M−1 s−1) | kcat/Km GSH (103 M−1 s−1) |
|---|---|---|---|---|---|
| 3-Cl | 110 (20) | 17 (5) | 0.16 (0.008) | 10 (2) | 1.5 (0.3) |
| 5-Cl | 370 (190) | 110 (40) | 0.24 (0.06) | 2 (1) | 0.6 (0.3) |
| 3,9,11-triCl | 390 (120) | 10 (3) | 0.010 (0.001) | 1.0 (0.2) | 0.03 (0.01) |
Values in parentheses represent standard errors. The kinetic parameters for 5-Cl HOPDA have higher standard errors as the enzyme could not be saturated with its substrates. Experiments were performed using potassium phosphate, pH 6.5 (I = 0.05 M), at 25°C.
Overall, these kinetic studies indicate that the physiological substrate of BphK is made up of 3-Cl and 5-Cl HOPDAs. Thus, the specificity of the enzyme for 3-Cl HOPDAs was over 3 orders of magnitude greater than for 4-CBA, a compound that has been suggested to be the physiological substrate of BphK (19). Previous studies from our laboratory have demonstrated that 3-Cl and 4-Cl HOPDAs inhibit BphD, the hydrolase of the bph pathway (32, 33), blocking the catabolism of biphenyl and its derivatives at this step. In contrast to 4-Cl HOPDAs, which readily undergo nonenzymatic hydrolysis, 3-Cl HOPDAs are quite stable. Thus, the ability of BphK to dehalogenate 3-Cl HOPDAs enables B. xenovorans LB400 to better transform PCBs.
The specificity of BphK for chlorinated HOPDAs is nevertheless relatively low. For example, the specificity constant (kA) of BphK for the physiologically relevant 3,9,11-triCl HOPDA, predicted to be formed by the transformation of 4,3′,5′-trichlorobiphenyl, was ∼103 M−1 s−1, which is approximately 3 orders of magnitude lower than that of human maleylacetoacetate isomerase (MAAI) for maleylacetoacetate (kA of 1.3 × 106 M−1 s−1) (12). However, BphK's kA for 3-Cl HOPDAs is similar to that of TCHQ dehalogenase for TCHQ. TCHQ dehalogenase, a GST that is thought to have been recently recruited for the degradation of PCP, has an apparent kA of ∼7 × 104 M−1 s−1 for THCQ (23). The relatively low specificity of BphK for a critical inhibitory PCB metabolite could indicate a relatively recent recruitment of bphK for PCB degradation. Alternatively, the low specificity of BphK for 3-Cl HOPDAs may reflect the enzyme's recruitment for the transformation of 5-Cl HOPDAs as well. Preserving the ability to attack both types of compounds may require sacrificing efficacy to act on one, as the two reactions are expected to necessitate attack by GSH at different positions as discussed below. However, selective pressure to transform 5-Cl HOPDAs is not expected to be high as these compounds are reasonably good substrates for BphD (33). Finally, it is possible that BphK has higher specificity for chlorinated HPDs, which would be chemically similar to the dienoate portion of HOPDAs on which BphK acts. However, chlorinated HPDs could not arise in vivo if BphD is inhibited.
Mechanism of dehalogenation.
The ternary-complex mechanism of BphK is typical of GSTs (3). Moreover, the oxidation of 2 equivalents of GSH during the dechlorination reaction is typical of GSH-dependent reductive dehalogenases. For example, the dechlorination of 2,5-dichlorohydroquinone, catalyzed by LinD from Sphingomonas paucimobilis UT26 during the degradation of γ-hexachlorocyclohexane (lindane), involves the oxidation of 2 GSH equivalents to glutathione disulfide (27). By contrast, most members of the GST superfamily, including MAAI and maleylpyruvate isomerase (MPI), utilize one molecule of GSH during catalytic turnover (3). TCHQ dehalogenase is of particular note as it catalyzes two different reactions: the reductive dechlorination of THCQ and the isomerization of maleylacetone to fumarylacetone (2). The former reaction requires two GSHs whereas the latter uses, but does not consume, GSH.
Based on the specificity of BphK and studies of other GSTs, we propose that the BphK-catalyzed dehalogenation of 3-Cl HOPDA proceeds via either a simple addition of GS− followed by elimination of chloride or an SN2-mediated displacement of chloride at C-3 by GS− (Fig. 4). A similar mechanism can be outlined for the dechlorination of 5-Cl HOPDA, with attack of GS− at C-5 instead of C-3. Consistent with experimental observations, the proposed mechanism does not allow for the enzyme-catalyzed dechlorination of 4-Cl HOPDA. The release of HOPDA and regeneration of the free enzyme using a second equivalent of GSH would proceed in a manner analogous to that proposed for TCHQ dehalogenase (23). Ongoing biochemical studies of BphK combined with structural data (41) should provide further insight into the mechanism of BphK and GST-catalyzed reactions in general.
FIG. 4.

Proposed mechanisms for the BphK-catalyzed dehalogenation of 3-Cl HOPDA. The upper mechanism proceeds through addition/elimination steps. The lower mechanism is similar to the SN2 reaction that was ruled out for the TCHQ dehalogenase reaction. Enz, enzyme.
Distribution and origins of bphK.
The bphK gene is found only in certain bph gene clusters. Thus, of at least five different types of bph gene clusters reported to date (30), bphK occurs in only those exemplified by B. xenovorans LB400 and Sphingobium yanoikuyae B1 clusters. More specifically, biphenyl degraders such as Rhodococcus globerulus P6 and Pseudomonas putida KF715 do not contain BphK homologs (9). The BphK homologs from strains LB400 and B1 share approximately 55% amino acid sequence identity (6), but it is not clear whether BphK from B1 transforms chlorinated HOPDAs. Indeed, bphK enabled S. yanoikuyae B1 to grow faster on m-toluate. Nevertheless, the BphK homologues of LB400-type clusters from five different strains were >95% identical at the amino acid level, yet the nucleotide sequence was significantly divergent in the regions flanking the bphK genes, suggesting independent recruitment events (9).
The recruitment of BphK by the bph pathway to degrade PCBs is reminiscent of the PCP degradation pathway of S. chlorophenolicum. Several lines of evidence indicate that the latter has recently evolved through the patchwork assembly of two other catabolic pathways (15, 40). Notably, the flavin oxygenase that catalyzes the initial hydroxylation of PCP possesses a remarkably broad and low specificity compared to other flavin oxygenases (26). Also, the reductive dehalogenase catalyzing the second and third steps of the pathway, TCHQ dehalogenase, may have originated from MAAI, normally involved in degrading tyrosine. Indeed, TCHQ dehalogenase still catalyzes the isomerization of MAA (15). Moreover, the dehalogenase is strongly inhibited by its substrates, TCHQ and trichlorohydroquinone, further suggesting that the enzyme is not highly evolved (15). Finally, the genes of the PCP pathway are dispersed throughout the genome of S. chlorophenolicum, and their transcription is poorly regulated (15).
BphK shares relatively high amino acid sequence identity with GSTs from a range of bacteria, including 62% with the GST domain of a predicted transmembrane protein from Rhodoferax ferrireducens DSM 15236 (ZP_00695363), 60% with a GST from Polaromonas naphthalenivorans (ZP_01019796), and 54% with a GST from Sphingomonas paucimobilis (AAC46031.1), whose crystal structure has been reported (29). Unfortunately, neither the substrates nor the physiological roles of these enzymes are known. Nevertheless, the existence of such homologues suggests that BphK could have been readily recruited from one of these pathways to facilitate the degradation of PCBs. Ongoing genomic analyses and biochemical studies should continue to provide insights into the physiological role of the BphK homologues and the similarity of their respective reactions with those catalyzed by BphK.
Concluding remarks.
The characterization of BphK reported herein constitutes the first report of a dehalogenase activity of this enzyme against inhibitory PCB metabolites. In light of the 3-Cl HOPDA-dechlorinating activity, it appears that the latter are not significant bottlenecks in PCB degradation in all strains. Transient-state kinetic analyses, mutational studies, and ongoing collaborative crystallographic studies should provide considerable insight into the mechanism of the dehalogenase reaction. Unfortunately, the reported data are insufficient to evaluate whether BphK-containing strains degrade PCBs more completely or confer an improved ability to survive in the presence of these compounds. Disruption of bphK may help establish the role of the enzyme in PCB degradation. Moreover, a directed evolution strategy could be used to improve the activity of BphK against 3-Cl HOPDAs and related inhibitory PCB metabolites. Such an approach could help identify the structural determinants of specificity and contribute to our understanding of the chemistry underlying the dehalogenation reaction.
Acknowledgments
This work was supported by Operating and Strategic grants from the Natural Sciences and Engineering Research Council of Canada (NSERC). P.D.F. was the recipient of FCAR and NSERC postgraduate scholarships.
We thank Victor Snieckus (Queen's University, Canada) and Walter Reineke (Wüppertal, Germany) for the synthesis of chlorinated DHBs.
REFERENCES
- 1.Abramowicz, D. A. 1990. Aerobic and anaerobic biodegradation of PCBs: a review. Crit. Rev. Biotechnol. 10:241-251. [Google Scholar]
- 2.Anandarajah, K., P. M. Kiefer, Jr., B. S. Donohoe, and S. D. Copley. 2000. Recruitment of a double bond isomerase to serve as a reductive dehalogenase during biodegradation of pentachlorophenol. Biochemistry 39:5303-5311. [DOI] [PubMed] [Google Scholar]
- 3.Armstrong, R. N. 1991. Glutathione S-transferases: reaction mechanism, structure, and function. Chem. Res. Toxicol. 4:131-140. [DOI] [PubMed] [Google Scholar]
- 4.Armstrong, R. N. 1997. Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem. Res. Toxicol. 10:2-18. [DOI] [PubMed] [Google Scholar]
- 5.Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl. 2000. Current protocols in molecular biology. J. Wiley & Sons Inc., New York, N.Y.
- 6.Bae, M., W. J. Sul, S.-C. Koh, J. H. Lee, G. J. Zylstra, Y. M. Kim, and E. Kim. 2003. Implication of two glutathione S-transferases in the optimal metabolism of m-toluate by Sphingomonas yanoikuyae B1. Antonie Leeuwenhoek 84:25-30. [DOI] [PubMed] [Google Scholar]
- 7.Barriault, D., M.-M. Plante, and M. Sylvestre. 2002. Family shuffling of a targeted bphA region to engineer biphenyl dioxygenase. J. Bacteriol. 184:3794-3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barriault, D., and M. Sylvestre. 2004. Evolution of the biphenyl dioxygenase BphA from Burkholderia xenovorans LB400 by random mutagenesis of multiple sites in region III. J. Biol. Chem. 279:47480-47488. [DOI] [PubMed] [Google Scholar]
- 9.Bartels, F., S. Backhaus, E. R. B. Moore, K. N. Timmis, and B. Hofer. 1999. Occurrence and expression of glutathione-S-transferase-encoding bphK genes in Burkholderia sp. strain LB400 and other biphenyl-utilizing bacteria. Microbiology 145:2821-2834. [DOI] [PubMed] [Google Scholar]
- 10.Bergmann, J. G., and J. Sanik, Jr. 1957. Determination of trace amounts of chlorine in naphtha. Anal. Chem. 29:241-243. [Google Scholar]
- 11.Bhandari, P., and J. Gowrishankar. 1997. An Escherichia coli host strain useful for efficient overproduction of cloned gene products with NaCl as the inducer. J. Bacteriol. 179:4403-4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Board, P. G., M. C. Taylor, M. Coggan, M. W. Parker, H. B. Lantum, and M. W. Anders. 2003. Clarification of the role of key active site residues of glutathione transferase zeta/maleylacetoacetate isomerase by a new spectrophotometric technique. Biochem. J. 374:731-737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bopp, L. H. 1986. Degradation of highly chlorinated PCBs by Pseudomonas strain LB400. J. Ind. Microbiol. 1:23-29. [Google Scholar]
- 14.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 15.Copley, S. D. 2000. Evolution of a metabolic pathway for degradation of a toxic xenobiotic: the patchwork approach. Trends Biochem. Sci. 25:261-265. [DOI] [PubMed] [Google Scholar]
- 16.Cornish-Bowden, A. 1995. Analysis of enzyme kinetic data. Oxford University Press, New York, N.Y.
- 17.Dai, S., F. H. Vaillancourt, H. Maaroufi, N. M. Drouin, D. B. Neau, V. Snieckus, J. T. Bolin, and L. D. Eltis. 2002. Identification and analysis of a bottleneck in PCB biodegradation. Nat. Struct. Biol. 9:934-939. [DOI] [PubMed] [Google Scholar]
- 17a.Denef, V. J., J. Park, T. V. Tsoi, J. M. Rouillard, H. Zhang, J. A. Wibbenmeyer, W. Verstraete, E. Gulari, S. A. Hashsham, and J. M. Tiedje. 2004. Biphenyl and benzoate metabolism in a genomic context: outlining genome-wide metabolic networks in Burkholderia xenovorans LB400. Appl. Environ. Microbiol. 70:4961-4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ellman, G. L. 1959. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 82:70-77. [DOI] [PubMed] [Google Scholar]
- 19.Gilmartin, N., D. Ryan, O. Sherlock, and D. Dowling. 2003. BphK shows dechlorination activity against 4-chlorobenzoate, an end product of bph-promoted degradation of PCBs. FEMS Microbiol. Lett. 222:251-255. [DOI] [PubMed] [Google Scholar]
- 20.Habig, W. H., and W. B. Jakoby. 1981. Glutathione S-transferases (rat and human). Methods Enzymol. 77:218-231. [DOI] [PubMed] [Google Scholar]
- 21.Haddock, J. D., and D. T. Gibson. 1995. Purification and characterization of the oxygenase component of biphenyl 2,3-dioxygenase from Pseudomonas sp. strain LB400. J. Bacteriol. 177:5834-5839. (Erratum, 178:258, 1996.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hofer, B., S. Backhaus, and K. N. Timmis. 1994. The biphenyl/polychlorinated biphenyl-degradation locus (bph) of Pseudomonas sp. LB400 encodes four additional metabolic enzymes. Gene 144:9-16. [DOI] [PubMed] [Google Scholar]
- 23.Kiefer, P. M., Jr., and S. D. Copley. 2002. Characterization of the initial steps in the reductive dehalogenation catalyzed by tetrachlorohydroquinone dehalogenase. Biochemistry 41:1315-1322. [DOI] [PubMed] [Google Scholar]
- 24.Kiefer, P. M., Jr., D. L. McCarthy, and S. D. Copley. 2002. The reaction catalyzed by tetrachlorohydroquinone dehalogenase does not involve nucleophilic aromatic substitution. Biochemistry 41:1308-1314. [DOI] [PubMed] [Google Scholar]
- 25.Maltseva, O. V., T. V. Tsoi, J. F. Quensen, M. Fukuda, and J. M. Tiedje. 1999. Degradation of anaerobic reductive dechlorination products of Aroclor 1242 by four aerobic bacteria. Biodegradation 10:363-371. [DOI] [PubMed] [Google Scholar]
- 26.McKay, D. B., M. Seeger, M. Zielinski, B. Hofer, and K. N. Timmis. 1997. Heterologous expression of biphenyl dioxygenase-encoding genes from a gram-positive broad-spectrum polychlorinated biphenyl degrader and characterization of chlorobiphenyl oxidation by the gene products. J. Bacteriol. 179:1924-1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagata, Y., A. Futamura, K. Miyauchi, and M. Takagi. 1999. Two different types of dehalogenases, LinA and LinB, involved in γ-hexachlorocyclohexane degradation in Sphingomonas paucimobilis UT26 are localized in the periplasmic space without molecular processing. J. Bacteriol. 181:5409-5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nerdinger, S., R. Marchhart, P. Riebel, C. Kendall, M. R. Johnson, C. F. Yin, V. Snieckus, and L. D. Eltis. 1999. Directed ortho-metalation and Suzuki-Miyaura cross-coupling connections: regiospecific synthesis of all isomeric chlorodihydroxybiphenyls for microbial degradation studies of PCBs. Chem. Commun. 22:2259-2260. [Google Scholar]
- 29.Nishio, T., T. Watanabe, A. Patel, Y. Wang, P. C. K. Lau, P. Grochulski, Y. Li, and M. Cygler. Unpublished data.
- 30.Pieper, D. H. 2005. Aerobic degradation of polychlorinated biphenyls. Appl. Microbiol. Biotechnol. 67:170-191. [DOI] [PubMed] [Google Scholar]
- 31.Romine, M. F., L. C. Stillwell, K.-K. Wong, S. J. Thurston, E. C. Sisk, C. Sensen, T. Gaasterland, J. K. Fredrickson, and J. D. Saffer. 1999. Complete sequence of a 184-kilobase catabolic plasmid from Sphingomonas aromaticivorans F199. J. Bacteriol. 181:1585-1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seah, S. Y. K., G. Labbe, S. R. Kaschabek, F. Reifenrath, W. Reineke, and L. D. Eltis. 2001. Comparative specificities of two evolutionarily divergent hydrolases involved in microbial degradation of polychlorinated biphenyls. J. Bacteriol. 183:1511-1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seah, S. Y. K., G. Labbe, S. Nerdinger, M. R. Johnson, V. Snieckus, and L. D. Eltis. 2000. Identification of a serine hydrolase as a key determinant in the microbial degradation of polychlorinated biphenyls. J. Biol. Chem. 275:15701-15708. [DOI] [PubMed] [Google Scholar]
- 34.Seeger, M., K. N. Timmis, and B. Hofer. 1995. Conversion of chlorobiphenyls into phenylhexadienoates and benzoates by the enzymes of the upper pathway for polychlorobiphenyl degradation encoded by the bph locus of Pseudomonas sp. strain LB400. Appl. Environ. Microbiol. 61:2654-2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sheehan, D., G. Meade, V. M. Foley, and C. A. Dowd. 2001. Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem. J. 360:1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suenaga, H., M. Mitsuoka, Y. Ura, T. Watanabe, and K. Furukawa. 2001. Directed evolution of biphenyl dioxygenase: emergence of enhanced degradation capacity for benzene, toluene, and alkylbenzenes. J. Bacteriol. 183:5441-5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tabor, S., and C. C. Richardson. 1985. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc. Natl. Acad. Sci. USA 82:1074-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taira, K., J. Hirose, S. Hayashida, and K. Furukawa. 1992. Analysis of bph operon from the polychlorinated biphenyl-degrading strain of Pseudomonas pseudoalcaligenes KF707. J. Biol. Chem. 267:4844-4853. [PubMed] [Google Scholar]
- 39.Tiedje, J. M., J. F. Quensen, J. Chee-Sanford, J. P. Schimel, and S. A. Boyd. 1993. Microbial reductive dechlorination of PCBs. Biodegradation 4:231-240. [DOI] [PubMed] [Google Scholar]
- 40.Tiirola, M. A., H. Wang, L. Paulin, and M. S. Kulomaa. 2002. Evidence for natural horizontal transfer of the pcpB gene in the evolution of polychlorophenol-degrading sphingomonads. Appl. Environ. Microbiol. 68:4495-4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tocheva, E. I., P. D. Fortin, L. D. Eltis, and M. E. P. Murphy. Submitted for publication.
- 42.Vaillancourt, F. H., G. Labbe, N. M. Drouin, P. D. Fortin, and L. D. Eltis. 2002. The mechanism-based inactivation of 2,3-dihydroxybiphenyl 1,2-dioxygenase by catecholic substrates. J. Biol. Chem. 277:2019-2027. [DOI] [PubMed] [Google Scholar]
- 43.Vuilleumier, S. 1997. Bacterial glutathione S-transferases: what are they good for? J. Bacteriol. 179:1431-1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vuilleumier, S., and M. Pagni. 2002. The elusive roles of bacterial glutathione S-transferases: new lessons from genomes. Appl. Microbiol. Biotechnol. 58:138-146. [DOI] [PubMed] [Google Scholar]
- 45.Vuilleumier, S., Z. Ucurum, S. Oelhafen, T. Leisinger, J. Armengaud, R.-M. Wittich, and K. N. Timmis. 2001. The glutathione S-transferase OrfE3 of the dioxin-degrading bacterium Sphingomonas sp. RW1 displays maleylpyruvate isomerase activity. Chem.-Biol. Interact. 133:265-267. [Google Scholar]
- 46.Warner, J. R., S. L. Lawson, and S. D. Copley. 2005. A mechanistic investigation of the thiol-disulfide exchange step in the reductive dehalogenation catalyzed by tetrachlorohydroquinone dehalogenase. Biochemistry 44:10360-10368. [DOI] [PubMed] [Google Scholar]
- 47.Zylstra, G. J., and E. Kim. 1997. Aromatic hydrocarbon degradation by Sphingomonas yanoikuyae B1. J. Ind. Microbiol. Biotechnol. 19:408-414. [DOI] [PubMed] [Google Scholar]