

Abstract
Bacterial primases are essential for DNA replication due to their role in polymerizing the formation of short RNA primers repeatedly on the lagging-strand template and at least once on the leading-strand template. The ability of recombinant Staphylococcus aureus DnaG primase to utilize different single-stranded DNA templates was tested using oligonucleotides of the sequence 5′-CAGA (CA)5 XYZ (CA)3-3′, where XYZ represented the variable trinucleotide. These experiments demonstrated that S. aureus primase synthesized RNA primers predominately on templates containing 5′-d(CTA)-3′ or TTA and to a much lesser degree on GTA-containing templates, in contrast to results seen with the Escherichia coli DnaG primase recognition sequence 5′-d(CTG)-3′. Primer synthesis was initiated complementarily to the middle nucleotide of the recognition sequence, while the third nucleotide, an adenosine, was required to support primer synthesis but was not copied into the RNA primer. The replicative helicases from both S. aureus and E. coli were tested for their ability to stimulate either S. aureus or E. coli primase. Results showed that each bacterial helicase could only stimulate the cognate bacterial primase. In addition, S. aureus helicase stimulated the production of full-length primers, whereas E. coli helicase increased the synthesis of only short RNA polymers. These studies identified important differences between E. coli and S. aureus related to DNA replication and suggest that each bacterial primase and helicase may have adapted unique properties optimized for replication.
Bacterial genomes contain multiple RNA polymerase enzymes, of which dnaG encodes the sole primase involved in replication. As shown by temperature-dependent mutation studies, DnaG primase is essential for DNA replication and bacterial survival, leading to its identification as a possible target for antibiotic development (9, 14). However, little is known about the differences in primase structure and function among eubacteria, since only the primases from Escherichia coli and Geobacillus (formerly Bacillus) stearothermophilus have been studied in detail (22, 23).
The RNA polymer synthesized by primase is essential for providing a free 3′ hydroxyl group from which DNA polymerase can elongate. DNA polymerase cannot synthesize a nucleotide polymer de novo. Primase functions at least once during leading-strand synthesis and multiple times during lagging-strand synthesis, where the RNA primers are extended by DNA polymerase to form Okazaki fragments that are processed and ligated together into progeny genomes.
Replicative primases from viruses and phages have been shown to require a specific recognition sequence in order to synthesize an RNA polymer de novo. However, several studies suggested that the preferred primase binding sequences may differ among different bacteria genera (2, 6). Bacterial primases show approximately 39% sequence homology over the three major domains: the zinc-binding domain, the RNA polymerase domain, and the carboxy-terminal domain. The sequencing of many different bacteria has allowed comparison of primase regions, and although the proposed DNA-binding domain is conserved overall, a number of residues potentially involved in molecular interactions are variable among different species. We noted that certain residues are divergent within the zinc-binding domain that may interact with single-stranded DNA (ssDNA), as suggested by X-ray crystallography (16). Therefore, we hypothesized that these amino acid differences might affect primase initiation specificity.
The domain of primase with greatest variability in amino acid composition and the lowest homology among species is the carboxy-terminal domain (7). Biochemical and structural studies have demonstrated that the carboxy-terminal domain of E. coli DnaG is an important mediator of the primase-helicase interaction (15). E. coli DnaB helicase has been shown to modulate DnaG primase by stimulating the kinetics of primer synthesis, shortening the length of the primers, and releasing the initiation specificity of primase such that almost any ssDNA supports primer synthesis (2, 11).
The potential for a correlation between primase DNA binding specificity and genome content was first suggested by Blattner et al., who demonstrated that the most overrepresented 8-bp sequences in the E. coli genome contain the trinucleotide binding site for DnaG primase and are preferentially located on the strand of DNA that serves as the lagging-strand template (3). Bioinformatic analysis has since demonstrated the presence of overrepresented sequences that are biased toward one of the replicating strands, termed skewed oligomers, in every eubacterial genome analyzed (19). The conservation of the skewed oligomers throughout many unrelated genomes suggests that they contribute to the overall fitness of eubacteria. However, it is unknown whether the overrepresented sequences correlate with the DNA binding site for DnaG primase in other eubacteria.
Given its substantial increase in antibiotic resistance, S. aureus is of significant biological and medical importance. In contrast with the gram-negative bacterium E. coli, whose genome has a GC content of 50.8%, the gram-positive bacterium S. aureus has an AT-rich genome with only 32.8% GC content. Therefore, S. aureus was chosen for this investigation to determine whether primase binding specificity and activity are restricted at the genus level as well as to assess whether a correlation exists with genome content. We have cloned, expressed, and purified both S. aureus DnaG primase and DnaC helicase for biochemical analyses. The ability of S. aureus primase to utilize different ssDNA templates was examined using denaturing high-pressure liquid chromatography (HPLC) to analyze primer composition and quantity. Here we report that S. aureus DnaG primase utilizes three initiation sequences that are distinct from that of E. coli, the best-characterized bacterial primase, but similar to that of another gram-positive bacteria, G. stearothermophilus (23). S. aureus DnaC helicase was found to stimulate a substantial increase in S. aureus DnaG primase activity that resulted in the synthesis of RNA primers that were predominately full length. In addition, experiments were performed assessing the ability of replicative helicases from both S. aureus and E. coli to stimulate either S. aureus or E. coli primase. These studies demonstrated that the helicase of one bacterial species could not stimulate the primase of the other bacterial species.
MATERIALS AND METHODS
Generation of the ssDNA templates.
Deoxyribonucleotides of the sequence 5′-CAGA (CA)5XYZ(CA)3-3′, where XYZ was AAA, AAG, AAT, ATA, ATC, ATT, CAG, CAT, CGA, CTA, CTC, CTG, CTT, GAA, GTA, TAA, TAG, TAT, TTA, TTC, TTG, or TTT, were generated by the University of Nebraska Core Facility or Integrated DNA Technologies (Coralville, IA). The oligonucleotides contained a 3′ C3 spacer that is required to prevent primase from elongating from a stabilized 3′ hairpin (2, 12). Purification of the ssDNA templates was performed using urea-polyacrylamide gel electrophoresis, UV shadowing, and elution of the oligonucleotide into Tris-EDTA buffer. Quantitation was performed using spectrophotometry at 260 nm with the respective extinction coefficients.
Construction of plasmid for the expression of recombinant S. aureus DnaG.
The open reading frame of dnaG from S. aureus sp. strain N315 was amplified by PCR using primers dnaG-For (5′-CATGCCATGGGGAGATTTAATTTGCGAATATGATC-3′) and dnaG-Rev (5′-GGAATTCAAATCACATGCTACATGCGTTC-3′). The primers contained NcoI and EcoRI restriction enzyme cloning sites in their 5′ and 3′ regions, respectively (underlined). The gene was cloned downstream of the lac promoter and glutathione S-transferase (GST)-tag coding sequence in vector pET41a+ (Novagen, Madison, WI) and transformed into E. coli BL21(DE3) (Novagen, Madison, WI). The cloned insert was sequenced in both directions by the University of Nebraska Medical Center Eppley Molecular Biology Core Facility to verify content.
Construction of plasmid for the expression of recombinant S. aureus DnaC.
The coding sequence for dnaC from S. aureus sp. strain N315 was amplified by PCR using primers dnaC-For (5′-AAGAGGTGTAACCATCCATGGATAG-3′) and dnaC-Rev (5′-TGCAAATAAAACTCGAGCATTGATTTTC-3′). The primers contained NcoI and XhoI restriction enzyme cloning sites in their 5′ and 3′ regions, respectively (underlined). The gene was cloned downstream of the lac promoter in vector pET19b+ (Novagen, Madison, WI) and transformed into E. coli BL21(DE3) (Novagen, Madison, WI). The cloned insert was sequenced in both directions by the University of Nebraska Medical Center Eppley Molecular Biology Core Facility to verify content.
Expression and purification of S. aureus and E. coli DnaG primases.
For the production of recombinant S. aureus DnaG, E. coli BL21(DE3) (pET41a+::dnaG) was grown in 4 liters of 2×YT medium containing 50 μg of kanamycin per ml at 30°C, with shaking at 200 rpm. When the culture reached an optical density at 600 nm of 1.0, 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside) was added, and the induced cells were grown for 2 h. Cultures were centrifuged at 5,000 × g for 15 min at 4°C, and the cell pellets (15.5 g wet weight) were resuspended in 46.5 ml of lysis buffer containing 50 mM Tris, 5 mM EDTA, 0.4 mM phenylmethylsulfonyl fluoride, 0.5% Triton X (pH 8.0), and 15 mg of lysozyme. Cells were broken by four freeze-thaw cycles followed by homogenization on ice. Soluble cell extracts were obtained by centrifugation at 12,000 × g for 30 min at 4°C, and lysates were applied to a 5-ml bed volume of glutathione Sepharose column (Novagen, Madison, WI). The column was washed three times with 15 ml of 50 mM Tris (pH 8.0), and the enzyme was eluted from the column with 15 ml of 50 mM Tris containing 5 mM reduced glutathione (pH 8.0). The enzyme was then loaded onto a 5 ml MonoQ anion exchange column (Bio-Rad, Hercules, CA). A 0 to 1 M NaCl gradient was used to elute the enzyme, and the fractions containing primase were concentrated using a Vivaspin 15R concentrator (Vivascience, Hannover, Germany). E. coli DnaG was isolated as previously described (8) from a primase-overproducing strain kindly supplied by Roger McMacken (John Hopkins University). Enzyme purity and subunit molecular mass were estimated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The concentrations of S. aureus and E. coli primase were determined using monomer extinction coefficients of 87,840 M−1 cm−1 and 47,800 M−1 cm−1, respectively, at 280 nm.
Expression and purification of S. aureus and E. coli replicative helicases.
For the production of recombinant S. aureus DnaC helicase, E. coli BL21(DE3) (pET19b+::dnaC) was grown at 30°C with shaking at 250 rpm in 2 liters of LB medium containing 50 μg of ampicillin, 20 μg thiamine, and 5 mg glucose per ml. When the culture reached an optical density at 600 nm of 0.8, 0.4 mM IPTG (isopropyl-β-d-thiogalactopyranoside) was added and the induced cells were grown for 2 h. Cultures were centrifuged at 5,000 × g for 15 min at 4°C, and the cell pellets (16.2 g wet weight) were resuspended in 48.6 ml of lysis buffer containing 50 mM Tris, 5 mM EDTA, 0.4 mM phenylmethylsulfonyl fluoride, 0.5% Triton X, 250 mM NaCl (pH 8.0), and 16 mg lysozyme. Cells were broken by three cycles of sonication, and the insoluble fraction was collected by centrifugation at 12,000 × g for 30 min at 4°C. The DnaC was made soluble by incubating the pellet with 5 ml of 50 mM Tris-2 mM MgCl2-2 mM ATP (pH 8.0) for 20 min on ice. The suspension was cleared by centrifugation at 12,000 × g for 30 min at 4°C. The protein solution was then loaded onto a 5 ml MonoQ anion exchange column (Bio-Rad, Hercules, CA), and a 0 to 1 M NaCl gradient was used to elute the enzyme. Fractions containing helicase were concentrated using a Vivaspin 15R concentrator (Vivascience, Hannover, Germany). E. coli DnaB helicase was isolated using a procedure similar to that described for E. coli DnaG primase (11); pRLM1038, the E. coli helicase expression plasmid, was generously provided by Roger McMacken. Enzyme purity and subunit molecular mass were estimated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The concentration of S. aureus helicase was determined using the method of Bradford (4), and the E. coli helicase concentration was determined using the hexamer extinction coefficient of 185,000 M−1 cm−1 at 280 nm.
RNA primer synthesis.
Reactions were carried out using previously optimized conditions. Briefly, the reaction mixtures were incubated at 30°C in nuclease-free buffer (50 mM HEPES [pH 7.5], 5 mM potassium glutamate, 10 mM dithiothreitol) containing 10 mM magnesium acetate, a 200 or 400 μM concentration of each ribonucleotide, and the indicated amount of ssDNA template and primase either without or with helicase (800 nM). RNA primer synthesis was quenched by desalting the reaction in a Sephadex G-25 spin column.
RNA primer analysis.
The primase reaction products were subjected to thermally denaturing HPLC, as previously described (12). Briefly, the reaction products were analyzed using an HPLC column that separated the nucleic acids by size and hydrophobicity. Nucleic acids were detected and quantitated spectrophotometrically at 260 nm. Control single-stranded oligoribonucleotides and deoxyribonucleotides were used to correlate retention time on the column with the sequence of the nucleic acid species. All experiments were performed in triplicate.
Bioinformatic analysis of the S. aureus and E. coli genomes.
The full-length nucleotide sequences of S. aureus N315 (NC002745) and E. coli K-12 (NC000913) were obtained from the GenBank database. The algorithm for identification and quantitation of skewed octamers was applied as described previously (19). The output included the 10 most overrepresented and skewed octamers along with their frequencies and percentages of skew toward the strand of DNA that serves as the lagging-strand template.
RESULTS
The first goal of this study was to determine S. aureus primase initiation specificity and whether it correlated with the S. aureus genome signature. The second goal of this study was to determine whether one bacterial replication fork helicase was capable of stimulating another bacteria's primase activity.
Expression of S. aureus DnaG primase and DnaC helicase.
The putative S. aureus primase dnaG gene was identified within the genome of N315 (NC002745) and was cloned using standard PCR methods. All attempts to overexpress DnaG in E. coli using a variety of conditions resulted in an insoluble product. Therefore, since GST fusion proteins have been reported to improve recombinant protein solubility and have been used successfully in studies of other DNA-binding proteins (10), dnaG was cloned and expressed as a GST fusion protein. The N-terminal GST tag on S. aureus primase allowed for purification using affinity chromatography followed by ion-exchange chromatography. The replicative helicase dnaC gene from S. aureus sp. strain N315 was also amplified and cloned using standard procedures. Expression of S. aureus DnaC in E. coli resulted in an insoluble product that was then solubilized with magnesium ion and ATP and purified using ion-exchange chromatography. Enzyme purity and subunit molecular mass for both S. aureus DnaG and DnaC were estimated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The molecular masses for the GST-tagged DnaG and untagged DnaC were 102 kDa and 53 kDa, respectively (Fig. 1). Both of these values correspond to the predicted molecular masses. After isolation, both proteins were more than 98% pure. The final yield was 2 mg of S. aureus primase and 10 mg of S. aureus helicase per liter of culture.
FIG. 1.
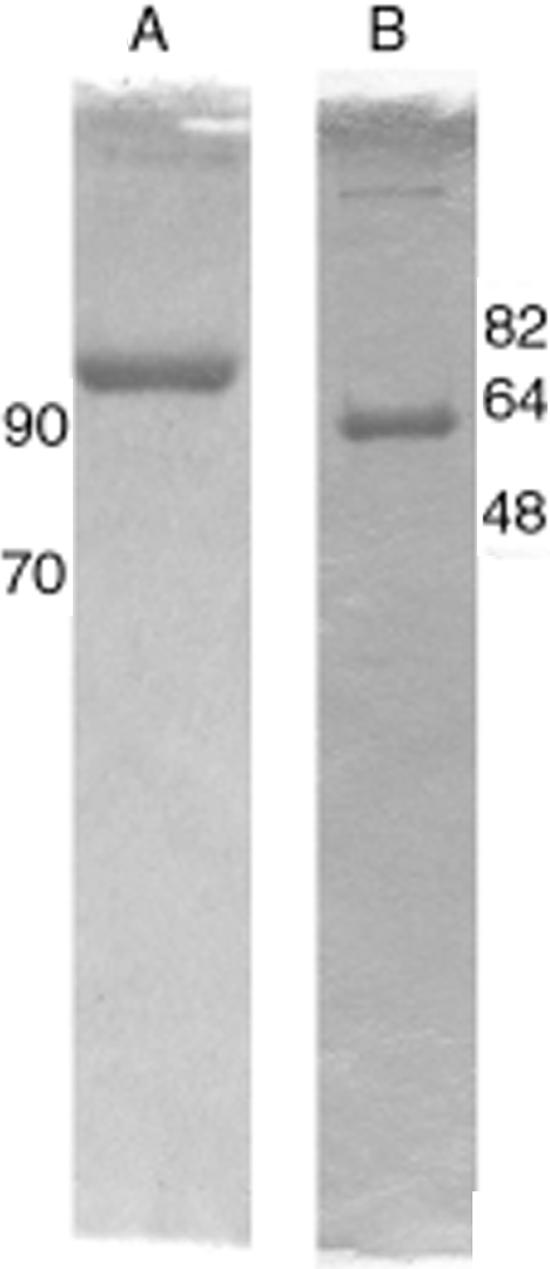
Purity analysis of recombinant S. aureus DnaG primase and DnaC helicase. Sodium dodecyl sulfate-10% polyacrylamide gel electrophoresis analysis of 5 μg of purified primase (A) or helicase (B). The locations of the molecular mass markers (in kilodaltons) are indicated next to the respective gel photograph.
Proteolytic cleavage of the GST tag from primase, as well as expression of an untagged protein, resulted in an insoluble protein under a wide variety of conditions. Therefore, the primase fusion protein was tested for in vitro activity using a previously developed RNA primer synthesis assay in conjunction with an HPLC detection method (12). The assay was developed to determine the quantity and length of all RNA primers. The ability of the purified enzyme to generate primers de novo was determined using a 23-mer synthetic ssDNA template in a reaction mixture containing ribonucleoside triphosphates (rNTPs) and magnesium ion. For uniformity of analysis, the ssDNA template backbone used in previous studies with E. coli primase was utilized.
Bioinformatic analysis of overrepresented octamers in S. aureus.
Computational analysis of the complete DNA sequences of S. aureus N315 and E. coli K12 was performed using a previously defined algorithm that tabulated the nonrandom overrepresentation of sequences eight nucleotides in length (octamers) (19). The most abundant octamers and their skew toward the lagging-strand DNA template were calculated. The nine most frequently occurring octamers in S. aureus contained the trinucleotide TTA (Table 1A), whereas in E. coli eight of the skewed octamers contained the trinucleotide CTG (Table 1B). None of the overrepresented S. aureus octamers contained any known chi sequences. In contrast, the E. coli octamers were found to be permutations of the chi octamer (GCTGGTGG), as previously described (3).
TABLE 1.
Computational analysis of overrepresented and skewed octamers found in the genomes of S. aureus strain N315 and E. coli strain K-12a
| Rank |
S. aureus strain N315 result
|
E. coli strain K-12 result
|
||||
|---|---|---|---|---|---|---|
| Octamer | No. of occurrences | % Skew | Octamer | No. of occurrences | % Skew | |
| 1 | tTTAaaat | 1,494 | 59.2 | tgCTGgcg | 1,511 | 58.4 |
| 2 | tTTAtttt | 1,486 | 59.8 | ggcgCTGg | 1,388 | 60.3 |
| 3 | tTTAattt | 1,319 | 61.8 | tgCTGgcg | 1,317 | 56.5 |
| 4 | TTAatttt | 1,315 | 57.7 | gCTGgcgg | 1,275 | 57.7 |
| 5 | tttTTAtt | 1,283 | 57.2 | gCTGgcgc | 1,180 | 56.5 |
| 6 | ttTTAttt | 1,268 | 58.3 | gcgCTGgc | 1,179 | 58.9 |
| 7 | tttTTAat | 1,233 | 57.9 | tggcgCTG | 1,176 | 58.6 |
| 8 | ttTTAatt | 1,225 | 58.9 | TGcgccag | 1,020 | 56.8 |
| 9 | TTAatttt | 1,223 | 56.6 | Gccgccag | 1,019 | 58.3 |
| 10 | atatttTT | 1,221 | 60.2 | gCTGgtgg | 1,008 | 75.7 |
The 10 most frequently occurring skewed octamers are listed in the order of frequency of occurrence as calculated using a previously described algorithm. The total count for each octamer and the skew percentage for each octamer are listed. The octamers are aligned according to the TTA motif in S. aureus and the CTG motif in E. coli.
Trinucleotide initiation specificity of S. aureus primase.
To investigate a possible relationship between genome content and the preferred primase recognition sequence, the ssDNA templates listed in Table 2 were synthesized. These templates contained the previously defined minimal structural elements necessary to support primer synthesis by E. coli primase and provided for testing of all the possible trinucleotide motifs contained within the overrepresented octamers listed in Table 1 (22). In addition, since leucine is the most abundant amino acid found in proteins expressed by both E. coli and S. aureus, all trinucleotides that encode for leucine were assessed. Several trinucleotides present in the proposed OriC of S. aureus (13) as well as remaining trinucleotides that complete the NTA series were also examined in the S. aureus primase activity assays.
TABLE 2.
ssDNA oligonucleotides (templates) showing their ability to support DNA-dependent RNA synthesis activity by recombinant S. aureus primasea
| Template | Primer synthesis | Template | Primer synthesis |
|---|---|---|---|
| AAA | − | CTG | − |
| AAG | − | CTT | − |
| AAT | − | GAA | − |
| ATA | − | GTA | + |
| ATC | − | TAA | − |
| ATT | − | TAG | − |
| CAG | − | TAT | − |
| CAT | − | TTA | ++ |
| CGA | − | TTC | − |
| CTA | +++ | TTG | − |
| CTC | − | TTT | − |
Templates of the format 5′-CAGA(CA)5 XYZ (CA)3-3′, where XYZ is the trinucleotide listed in the table were tested for their ability (+++, ++, or +) or inability (−) to support RNA primer synthesis. The reactions were performed using 200 nM template, 2 μM primase, and 200 μM rNTPs during a 1 h incubation at 30°C.
Primase, magnesium ion, and rNTPs were incubated with the various templates, and the reactions were analyzed by HPLC to determine whether any of the ssDNA templates shown in Table 2 supported RNA primer synthesis. E. coli primase was used in control reactions with an oligonucleotide template containing the initiation trinucleotide CTG (2). Unlike the findings for E. coli, S. aureus primase was capable of producing specific and appropriately sized RNA primers from templates containing either the trinucleotide CTA or TTA (Table 2 and Fig. 2). Templates containing the trinucleotide GTA produced only a small amount of RNA primer product (Table 2 and Fig. 2). The area of the RNA polymer peaks was calculated to determine the relative amounts of primers synthesized. The template containing CTA resulted in the largest amount of primer product (131.1 mV·min), with the template containing TTA being the next best trinucleotide sequence to result in a high quantity of RNA polymers (57.3 mV·min). The GTA-containing template resulted in the least amount of primer production by S aureus DnaG (16.3 mV·min). None of the other templates tested produced a detectable amount of primer product (Table 2).
FIG. 2.

Representative chromatograms of primase activity on ssDNA templates. Denaturing HPLC analysis was performed on the RNA products derived from S. aureus primase activity on templates containing the trinucleotide CTA (A), TTA (B), GTA (C), ATA (D), or CTG (E). The reactions were performed using 2 μM template, 2 μM primase, and 400 μM rNTPs during a 1 -h incubation at 30°C. Abs, absorbance at 260 nm.
The major RNA polymers synthesized by S. aureus primase eluted at 8.05 min and 8.48 min with ssDNA templates containing CTA and TTA, respectively (Fig. 2). The elution time for these two RNA primers, both 16 nucleotides in length but differing in composition by a single nucleotide, corresponded to the expected elution time based on our previous studies, which were determined using control oligomers with various hydrophobicity (12). Less-abundant and shorter RNA primers were also generated that eluted at 7.72, 7.44, 7.27, 6.89, 6.05, and 5.90 min with the CTA template and 8.17, 7.90, 7.75, and 7.43 min with the TTA template, corresponding to a series of smaller products that progressively differed by one fewer nucleotide.
Additional templates containing mutations of the second or third base within the CTA or TTA trinucleotide were generated and tested in the primase activity assay. Use of these templates resulted in undetectable levels of primer products (Table 2). These data showed that both the second and third nucleotides in the recognition sequence were essential for S. aureus primer initiation. As an important control for these studies, S. aureus primase activity exhibited no activity in a reaction using the template containing CTG, the trinucleotide initiation sequence for E. coli primase (Fig. 2). Overall, these experiments demonstrated that the observed activity of S. aureus primase was specific and not due to a contaminant derived from the E. coli strain used for recombinant protein expression.
Identification of the initial ribonucleotide in the primers synthesized by S. aureus DnaG.
Previous studies showed that RNA primers generated by E. coli primase are initiated complementarily to the middle T in the CTG template (11). To identify whether the RNA primer synthesized by the S. aureus DnaG initiated complementarily to the middle T in the TTA embedded within the ssDNA template, a series of reactions were performed using progressively lower amounts of rCTP. Since the template contained only one guanosine located in the antepenultimate position, primer synthesis initiating complementarily to the middle T of the trinucleotide TTA should generate a 16-mer RNA primer eluting at 8.48 min, as illustrated in Fig. 3A. However, in the absence of rCTP, primer synthesis would stall before the antepenultimate position and result in a 13-mer RNA primer (Fig. 3A).
FIG. 3.

Determination of the RNA primer initiation site on the TTA template for S. aureus primase. (A) Schematics of the predicted RNA primer product (italics) in either the presence or absence of rCTP (bold) on the ssDNA template. (B) Denaturing HPLC analysis of RNA primer synthesis in the presence of 0, 25, 50, 100, 200, and 400 μM rCTP. The reactions were performed using 2 μM ssDNA template, 2 μM primase, and 400 μM rNTPs during a 1 -h incubation at 30°C. Abs, absorbance at 260 nm.
In the absence of rCTP, S. aureus primase created one major primer species that eluted at 7.75 min., corresponding with a 13-mer RNA species (Fig. 3B) (12). As rCTP was titrated into the reaction, the 13-mer peak disappeared and a new 8.48 min peak consistent with a 16-mer became the dominant primer species. A similar experiment was performed using S. aureus primase and the template containing the trinucleotide CTA. These results also demonstrated that the full-length primer was a 16-mer in the presence of rCTP and that, in the absence of rCTP, a 13-mer primer product was the major RNA polymer synthesized (data not shown). In the absence of rATP, no RNA product was observed for either the CTA- or TTA-containing templates. These results confirmed that the RNA primer initiates complementarily to the middle T in both of these trinucleotide templates and that the third nucleotide in the initiation sequence serves an essential but cryptic function.
Helicase stimulation of primase.
The replicative E. coli helicase (DnaB) has been shown to stimulate the synthesis of RNA primers by E. coli primase and to release E. coli primase specificity for the trinucleotide CTG (11). Thus, in the presence of helicase, E. coli primase is capable of generating primers from a variety of templates. To determine whether DnaC (an E. coli DnaB ortholog), the S. aureus replicative helicase, would also stimulate S. aureus primase DnaG activity, S. aureus primase activity assays in the presence or absence of S. aureus DnaC were performed using a low concentration of primase (400 nM) and either the CTA or TTA templates. Note that this primase concentration was much less than the 2.0 μM used as described above for S. aureus primase (Fig. 2). The concentration was intentionally reduced to a level that would generate a visible product when helicase was added at 800 nM for helicase monomer (see Fig S1 in the supplemental material). The addition of S. aureus helicase DnaC stimulated S. aureus primase DnaG activity by 2.7-fold with the CTA template and by 1.6-fold with the TTA template (Fig. 4). The predominant RNA polymer for both templates was the full-length 16-mer. Moreover, the stimulatory effect of the helicase on S. aureus primase activity did not release primase initiation specificity, since templates other than those containing CTA or TTA did not result in a detectable primer product (data not shown). These results demonstrated that the interaction of S. aureus helicase with the S. aureus primase resulted in stimulation of predominately full-length primer synthesis in the presence of the appropriate initiation template.
FIG. 4.

Stimulatory effect of S. aureus replicative helicase on S. aureus primase activity. Denaturing HPLC analysis was performed on the RNA products derived from S. aureus primase activity on templates containing the trinucleotide CTA with helicase (A) and without helicase (B) or the trinucleotide TTA with helicase (C) and without helicase (D). S. aureus DnaG primase (400 nM) was incubated for 1 h with 400 μM rNTPs and 2 μM ssDNA without or with S. aureus DnaC helicase (800 nM). Abs, absorbance at 260 nm.
Restricted helicase stimulation of primase.
To determine whether helicase stimulation of primase was restricted at the genus level, the ability of E. coli DnaB helicase to stimulate S. aureus primase, as well as the ability of S. aureus DnaC helicase to stimulate E. coli primase, was tested. In reactions using a low concentration of primase (400 nM), S. aureus primase produced a detectable amount of RNA primers whereas E. coli primase did not (Fig. 5). The amount of E. coli primase used in this assay was much lower than in an earlier study (12). It was reduced to a level that would generate a visible product when helicase was added. The addition of E. coli DnaB helicase stimulated E. coli DnaG activity that resulted in primers shorter than the expected 16-mer that averaged 12 bases in length (Fig. 5), similar to the findings in previous studies (11). S. aureus DnaC helicase stimulated the production by S. aureus DnaG of full-length RNA polymers 16 bases in length but did not produce an observable change in E. coli primase activity. Similarly, E. coli DnaB helicase stimulated only E. coli primase activity and not S. aureus primase activity. Notably, the addition of E. coli DnaB helicase inhibited the basal level of S. aureus DnaG activity. Collectively, these results showed that S. aureus DnaC substantially stimulated S. aureus primase activity and that this stimulatory effect differed from that observed for E. coli primase and helicase interactions. More specifically and in contrast to the findings for E. coli, S. aureus helicase stimulation of S. aureus DnaG did not shorten the length of the RNA primers synthesized and primase initiation specificity was not released. In addition, these data demonstrated that for both E. coli and S. aureus, the helicase interaction with primase was not cross-reactive.
FIG. 5.

Cross-species effect of replicative helicase stimulation on primase activity. S. aureus or E. coli primase (400 nM) was incubated for 30 min with 400 μM rNTPs, 2 μM ssDNA, and S. aureus or E. coli helicase (800 nM). The reaction products were analyzed by denaturing HPLC. The ssDNA template contained the TTA trinucleotide in S. aureus primase reactions and the CTG trinucleotide in the E. coli primase reactions. Abs, absorbance at 260 nm.
DISCUSSION
S. aureus DnaG primase recognition sequence and stimulation by helicase.
Bacterial genomes are constantly being exposed to a variety of plasmids and other foreign DNA. A mechanism to protect the genome from replicating nonnative DNA may convey a selective advantage to the species. The studies described here identified a number of important distinctions between E. coli and S. aureus related to DNA replication, including different initiation sequences and primase-helicase interactions restricted to each bacterial species. Unlike E. coli primase, which initiates de novo primer synthesis on templates containing the trinucleotide CTG, S. aureus primase synthesized an RNA primer predominately on templates containing CTA or TTA and to a much lesser degree on GTA-containing templates. The results of the cytosine titration studies in S. aureus were consistent with previous findings for E. coli with the RNA primer initiating complementarily to the middle T in the trinucleotide-containing template and the third nucleotide in the recognition trinucleotide serving as a cryptic nucleotide that is essential for efficient primer formation (6).
A second level of restriction at the genus level was found in the interaction between primase and helicase. More specifically, helicase from E. coli was unable to stimulate S. aureus primase activity and S. aureus helicase was incapable of stimulating E. coli primase in our system. These findings contrast with findings obtained with the subunits of the S. aureus PolC and E. coli polymerase III holoenzymes, which have been demonstrated to be generally interchangeable (5).
Interestingly, most bacteriophages encode their own primase and helicase, indicating that the host primase and helicase are unable to support replication of the phage genome. Despite the likelihood that eubacterial primases and helicases from different species have the same overall structure, differences in key residues may have evolved to prevent the primase and/or helicase from functioning on nonnative replication forks, such as those found with invading bacteriophages. For example, several residues in G. stearothermophilus DnaB helicase that are not conserved in E. coli DnaB have been recently identified to have an important role in mediating protein-protein interactions required for helicase modulation of primase activity (23).
Correlation of genome content and primase initiation specificity.
For over 10 years bacterial genomes have been known to contain certain oligonucleotides that are overrepresented or skewed (18). The biologic significance of this phenomenon, however, remains a matter of significant debate. A variety of asymmetrical pressures could contribute to overrepresentation of a particular sequence on either DNA strand, including recombination and repair, composition strand bias, DNA replication, and codon bias. In agreement with other studies (1, 24), our data do not support the recombination-repair hypothesis as a basis for generation of overrepresented oligonucleotides. Our studies demonstrated that the preferred recognition sequence for S. aureus primase was different from the recognition sequence for E. coli. However, in both genomes a functional primase recognition sequence was present within the most frequently occurring octamers.
Codon bias may partially explain overrepresentation of the TTA-containing octamers in S. aureus and the CTG-containing octamers in E. coli. In both organisms, leucine is the most abundant amino acid within synthesized proteins and the most biased codons for leucine are TTA in S. aureus and CTG in E. coli (see the codon usage table for Staphylococcus aureus N315 and Escherichia coli K-12 at the website for The Institute for Genomic Research [http://www.tigr.org]). However, the skewed octamers are not preferentially located in coding versus noncoding regions of the genome (19, 20). Therefore, codon bias and amino acid usage cannot fully explain the basis for the genomic signatures observed.
While TTA was overrepresented on the lagging-strand template, the other significant S. aureus primase initiation trinucleotide, CTA, was not. One possible explanation is that the evolution of an AT-rich genome in S. aureus required DnaG primase to adapt accordingly, with TTA representing a more biologically appropriate initiation sequence given the abundance of this trinucleotide in the genome. Another possibility is that TTA is the most-utilized primase initiation sequence during routine DNA synthesis and that CTA serves as the primase template during specialized conditions such as during the initiation of the replication fork or during DNA repair. Alternatively, the initial two ribonucleotides in the primer product, specifically 5′-pppApG-3′, and/or the pyrimidine-pyrimidine-purine recognition sequence on the template has been conserved due to common structural or conformational constraints within the recognition-active site of bacterial primase. Ongoing structural studies should facilitate determination of the precise mechanism for S. aureus primase recognition of specific initiation trinucleotides and subsequent de novo synthesis of short RNA polymers.
Bidirectional replication from a single origin produces a finite limit on prokaryotic DNA replication time (17). When all other features are constant, long genomes take longer to replicate. If cell duplication rates are an important component of fitness, then single origins favor small genomes and the elimination of nonuseful DNA. We propose here that single origins of bidirectional replication also favor a uniform overabundance of primase initiation sequences on the lagging-strand template. When the primase initiation sequences are sufficiently abundant, the DNA replication rate is limited by other factors, such as deoxynucleoside triphosphate pools, DNA polymerase subunit concentrations, fork helicase concentration, SSB, rNTP pools, and primase concentration (25, 26). However, in an exponentially growing cell none of these factors varies. So, for those portions of the genome in which the primase initiation sequence is rare, the replication rate will slow down and will be limited by the rate of primase initiation, the rate-limiting step for primer synthesis. In this way, primase specificity exerts selective pressure on the genome sequence to maintain an abundance of initiation sequences on the lagging-strand template.
In a search for new sequence elements within a single strand of the E. coli genome, the Blattner laboratory counted the abundance of every octanucleotide sequence (3). They found that the overabundant sequences were GC rich, crossed from one strand to the other at the replication origin and terminus, and occurred with higher frequency than the average Okazaki fragment length. Most of these sequences contained the CTG trinucleotide and were preferentially found on the strand that is the lagging-strand template. Since the overrepresented sequences cross over at the origin and terminus, these sequences are related to the replication process. Since E. coli primase preferentially initiates from CTG, they suggested that the function of these sequences was to promote primer initiation. This correlation provides a mechanism for primase to select for the maintenance of these overrepresented sequences and further suggests another mechanism to constrain lateral gene transfer between distantly related species.
Primase and helicase interactions in other bacteria and genomic signatures.
Our study of S. aureus primase initiation specificity and modulation by helicase has identified at least two issues that will require further investigation. Since E. coli and S. aureus are representatives of the distantly related families of gram-negative and gram-positive bacteria, further studies with other bacterial enzymes will establish whether the relationship between genome content and primase recognition sequence is universal. Although the majority of bacterial genomes are expected to have overrepresented and strand-skewed oligonucleotides, the primase recognition sequence has only been determined for a few bacteria (18, 19). The recognition sequences for G. stearothermophilus primase have been recently reported to be CTA and TTA, as with our findings for S. aureus, but the initiating nucleotide appears to be different (23). Since the genomic sequence of G. stearothermophilus has not been determined, a correlation between the G. stearothermophilus DnaG initiation sequences and genome content cannot currently be evaluated. The second question requiring further study is whether the specific stimulatory effect of helicase on primase will be restricted at the species level or will have a general effect among all gram-positive organisms. These issues are important because it may be possible to exploit the close relationship between primase and helicase and the potential differences in activity at the species or group level for new therapeutic drug discovery.
Specific strategies for antibiotic development have been proposed based on the essentiality and divergence of the eubacterial primases from their eukaryotic counterparts and the key interactions between primase and replicative helicases (21). X-ray crystallography of portions of the enzymes believed to be involved in the stimulation of primase by helicase has identified a variety of sites for targeting inhibitory chemicals. Our findings suggest that inhibitors of primase-helicase interactions will probably have narrower activity than inhibitors of DNA binding by primase.
Supplementary Material
Acknowledgments
This work was supported in part by a grant from the Department of Defense, Defense Advanced Research Program Agency (award W911NF0510275).
We thank Dhundy Bastola and Khalid Sayood for assistance with genome sequence analysis.
Footnotes
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Bell, S. J., Y. C. Chow, J. Y. Ho, and D. R. Forsdyke. 1998. Correlation of chi orientation with transcription indicates a fundamental relationship between recombination and transcription. Gene 216:285-292. [DOI] [PubMed] [Google Scholar]
- 2.Bhattacharyya, S., and M. A. Griep. 2000. DnaB helicase affects the initiation specificity of Escherichia coli primase on single-stranded DNA templates. Biochemistry 39:745-752. [DOI] [PubMed] [Google Scholar]
- 3.Blattner, F. R., G. Plunkett III, C. A. Bloch, N. T. Perna, V. Burland, M. Riley, J. Collado-Vides, J. D. Glasner, C. K. Rode, G. F. Mayhew, J. Gregor, N. W. Davis, H. A. Kirkpatrick, M. A. Goeden, D. J. Rose, B. Mau, and Y. Shao. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453-1474. [DOI] [PubMed] [Google Scholar]
- 4.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 5.Bruck, I., R. E. Georgescu, and M. O'Donnell. 2005. Conserved interactions in the Staphylococcus aureus DNA PolC chromosome replication machine. J. Biol. Chem. 280:18152-18162. [DOI] [PubMed] [Google Scholar]
- 6.Frick, D. N., and C. C. Richardson. 2001. DNA primases. Annu. Rev. Biochem. 70:39-80. [DOI] [PubMed] [Google Scholar]
- 7.Griep, M. A. 1995. Primase structure and function. Indian J. Biochem. Biophys. 32:171-178. [PubMed] [Google Scholar]
- 8.Griep, M. A., and E. R. Lokey. 1996. The role of zinc and the reactivity of cysteines in Escherichia coli primase. Biochemistry 35:8260-8267. [DOI] [PubMed] [Google Scholar]
- 9.Grompe, M., J. Versalovic, T. Koeuth, and J. R. Lupski. 1991. Mutations in the Escherichia coli dnaG gene suggest coupling between DNA replication and chromosome partitioning. J. Bacteriol. 173:1268-1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Howell, L. D., R. Borchardt, J. Kole, A. M. Kaz, C. Randak, and J. A. Cohn. 2004. Protein kinase A regulates ATP hydrolysis and dimerization by a CFTR (cystic fibrosis transmembrane conductance regulator) domain. Biochem. J. 378:151-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson, S. K., S. Bhattacharyya, and M. A. Griep. 2000. DnaB helicase stimulates primer synthesis activity on short oligonucleotide templates. Biochemistry 39:736-744. [DOI] [PubMed] [Google Scholar]
- 12.Koepsell, S., D. Bastola, S. H. Hinrichs, and M. A. Griep. 2004. Thermally denaturing high-performance liquid chromatography analysis of primase activity. Anal. Biochem. 332:330-336. [DOI] [PubMed] [Google Scholar]
- 13.Kuroda, M., T. Ohta, I. Uchiyama, T. Baba, H. Yuzawa, I. Kobayashi, L. Cui, A. Oguchi, K. Aoki, Y. Nagai, J. Lian, T. Ito, M. Kanamori, H. Matsumaru, A. Maruyama, H. Murakami, A. Hosoyama, Y. Mizutani-Ui, N. K. Takahashi, T. Sawano, R. Inoue, C. Kaito, K. Sekimizu, H. Hirakawa, S. Kuhara, S. Goto, J. Yabuzaki, M. Kanehisa, A. Yamashita, K. Oshima, K. Furuya, C. Yoshino, T. Shiba, M. Hattori, N. Ogasawara, H. Hayashi, and K. Hiramatsu. 2001. Whole genome sequencing of methicillin-resistant Staphylococcus aureus. Lancet 357:1225-1240. [DOI] [PubMed] [Google Scholar]
- 14.Liu, J., M. Dehbi, G. Moeck, F. Arhin, P. Bauda, D. Bergeron, M. Callejo, V. Ferretti, N. Ha, T. Kwan, J. McCarty, R. Srikumar, D. Williams, J. J. Wu, P. Gros, J. Pelletier, and M. DuBow. 2004. Antimicrobial drug discovery through bacteriophage genomics. Nat. Biotechnol. 22:185-191. [DOI] [PubMed] [Google Scholar]
- 15.Oakley, A. J., K. V. Loscha, P. M. Schaeffer, E. Liepinsh, G. Pintacuda, M. C. Wilce, G. Otting, and N. E. Dixon. 2005. Crystal and solution structures of the helicase-binding domain of Escherichia coli primase. J. Biol. Chem. 280:11495-11504. [DOI] [PubMed] [Google Scholar]
- 16.Pan, H., and D. B. Wigley. 2000. Structure of the zinc-binding domain of Bacillus stearothermophilus DNA primase. Structure Fold. Des. 8:231-239. [DOI] [PubMed] [Google Scholar]
- 17.Poole, A. M., M. J. Phillips, and D. Penny. 2003. Prokaryote and eukaryote evolvability. Biosystems 69:163-185. [DOI] [PubMed] [Google Scholar]
- 18.Rocha, E. P. 2004. The replication-related organization of bacterial genomes. Microbiology 150:1609-1627. [DOI] [PubMed] [Google Scholar]
- 19.Salzberg, S. L., A. J. Salzberg, A. R. Kerlavage, and J. F. Tomb. 1998. Skewed oligomers and origins of replication. Gene 217:57-67. [DOI] [PubMed] [Google Scholar]
- 20.Sandberg, R., C. I. Branden, I. Ernberg, and J. Coster. 2003. Quantifying the species-specificity in genomic signatures, synonymous codon choice, amino acid usage and G+C content. Gene 311:35-42. [DOI] [PubMed] [Google Scholar]
- 21.Soultanas, P. 2005. The bacterial helicase-primase interaction: a common structural/functional module. Structure (Cambridge) 13:839-844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swart, J. R., and M. A. Griep. 1993. Primase from Escherichia coli primes single-stranded templates in the absence of single-stranded DNA-binding protein or other auxiliary proteins. Template sequence requirements based on the bacteriophage G4 complementary strand origin and Okazaki fragment initiation sites. J. Biol. Chem. 268:12970-12976. [PubMed] [Google Scholar]
- 23.Thirlway, J., and P. Soultanas. 2006. In the Bacillus stearothermophilus DnaB-DnaG complex, the activities of the two proteins are modulated by distinct but overlapping networks of residues. J. Bacteriol. 188:1534-1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uno, R., Y. Nakayama, K. Arakawa, and M. Tomita. 2000. The orientation bias of Chi sequences is a general tendency of G-rich oligomers. Gene 259:207-215. [DOI] [PubMed] [Google Scholar]
- 25.Zechner, E. L., C. A. Wu, and K. J. Marians. 1992. Coordinated leading- and lagging-strand synthesis at the Escherichia coli DNA replication fork. II. Frequency of primer synthesis and efficiency of primer utilization control Okazaki fragment size. J. Biol. Chem. 267:4045-4053. [PubMed] [Google Scholar]
- 26.Zechner, E. L., C. A. Wu, and K. J. Marians. 1992. Coordinated leading- and lagging-strand synthesis at the Escherichia coli DNA replication fork. III. A polymerase-primase interaction governs primer size. J. Biol. Chem. 267:4054-4063. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.