

Abstract
Disruption of eshA, which encodes a 52-kDa protein that is produced late during the growth of Streptomyces coelicolor A3(2), resulted in elimination of actinorhodin production. In contrast, disruption of eshB, a close homologue of eshA, had no effect on antibiotic production. The eshA disruptant accumulated lower levels of ppGpp than the wild-type strain accumulated. The loss of actinorhodin production in the eshA disruptant was restored by expression of a truncated relA gene, which increased the ppGpp level to the level in the wild-type strain, indicating that the reduced ppGpp accumulation in the eshA mutant was solely responsible for the loss of antibiotic production. Antibiotic production was also restored in the eshA mutant by introducing mutations into rpoB (encoding the RNA polymerase β subunit) that bypassed the requirement for ppGpp, which is consistent with a role for EshA in modulating ppGpp levels. EshA contains a cyclic nucleotide-binding domain that is essential for its role in triggering actinorhodin production. EshA may provide new insights and opportunities to unravel the molecular signaling events that occur during physiological differentiation in streptomycetes.
One of the most intriguing challenges in biology today is elucidation of the mechanisms by which cells detect and respond to extracellular nutritional conditions. Among the prokaryotes, Bacillus subtilis and Streptomyces spp. provide tractable experimental systems for studying such mechanisms because they exhibit a wide range of adaptations to extreme nutrient limitation, including the production and secretion of antibiotics and enzymes and the formation of aerial mycelium (Streptomyces spp.) and endospores (Bacillus spp.) (12). Nutritional status and sporulation have been successfully linked in B. subtilis (45), in which CodY detects and responds to nutrient limitation by monitoring the level of the intracellular GTP pool as an overall indicator of cellular physiology. Recent work by Inaoka and Ochi (20) supported this proposal. The stringent response, a general and ubiquitous response to nutrient limitation in prokaryotes, plays a central role in responding to nutrient stress, mediating its effect through the nucleotide guanosine 5′-diphosphate 3′-diphosphate (ppGpp) (5). By analyzing mutants with impaired abilities to elicit the stringent response, we have shown that ppGpp plays a role in triggering the onset of antibiotic production in both B. subtilis (21) and Streptomyces spp. (6, 7, 15, 29, 37, 39, 41, 52, 55), whereas morphological differentiation is triggered by reduced levels of GTP. Streptomycetes are gram-positive, filamentous soil bacteria that have a complex process of morphological differentiation and the ability to produce a wide variety of secondary metabolites (referred to as physiological differentiation) that include antibiotics and other useful medicinal compounds. Morphological differentiation and physiological differentiation in streptomycetes often coincide and occur in response to environmental signals that include nutrient limitation. Streptomyces coelicolor A3(2), the streptomycete that has been genetically characterized most, is particularly appropriate for studying the regulation of morphological and physiological differentiation. This strain produces at least four antibiotics, two of which, the blue-pigmented polyketide actinorhodin and the red-pigmented prodiginine (formerly called prodigiosin) complex, are usually produced in the stationary phase (9, 10). There has been much progress in elucidating not only the organization of antibiotic biosynthetic gene clusters in many Streptomyces species but also the roles of pathway-specific regulatory genes that are required for the activation of their cognate biosynthetic genes (3, 19). In S. coelicolor A3(2), ActII-ORF4 (a positively acting regulatory protein) is crucially important for the expression of all of the biosynthetic genes that encode the enzymes of the actinorhodin pathway (1, 14).
Kwak et al. (31) and we (28) independently discovered a novel 52-kDa protein that is required for initiating several developmental processes in streptomycetes. Disruption of the corresponding gene, eshA, eliminated antibiotic production in S. coelicolor A3(2) and in Streptomyces griseus (28, 47). At the same time, Kwak et al. (31) reported that EshA is required for extension of sporogenic hyphal branches (thus the origin of the gene designation eshA) in S. griseus. We found later that disruption of eshA causes a defect in aerial mycelium formation in S. griseus that appears to result from an aberrant accumulation of deoxynucleoside triphosphates that accompanies the arrest of DNA synthesis in the late growth phase (47). In contrast, disruption of eshA in S. coelicolor did not result in aberrant accumulation of deoxynucleoside triphosphates and did not affect the formation of aerial hyphae, implying that there are qualitative differences between the EshA proteins of the two species (47). Abundant EshA accumulates during sporulation induced by phosphate starvation and nutritional downshift (31) and also when cells reach the middle to late growth phase (28, 47). In S. griseus at least, EshA exists as multimers (∼20-mers) with a diameter of 27 nm (47). Homologues of EshA in two other bacterial species are also induced in a stress- or growth-phase-dependent manner; SrpI of the cyanobacterium Synechococcus sp. is induced under sulfur deprivation conditions (34, 35), while MMPI is induced during infection by Mycobacterium leprae (57, 61). These observations led Triccas et al. (57) and Kwak et al. (31) to propose that EshA, SrpI, and MMPI constitute a new family of bacterial stress response proteins. The EshA proteins of both S. coelicolor and S. griseus have a central domain that exhibits considerable amino acid sequence identity (30% to 32%) to eukaryotic-type cyclic nucleotide (cNMP)-binding domains (25, 56). Although the biochemical function of EshA is not known, it was conceivable that the protein exerts its influence via this putative nucleotide-binding domain. In this study, we used both genetic and biochemical approaches to demonstrate that EshA has a role in secondary metabolism in S. coelicolor. We found that EshA is a cyclic AMP (cAMP)-binding protein and that it is required for sustaining levels of ppGpp during the late growth phase that are sufficient for activation of actinorhodin biosynthesis.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The bacterial strains used in this study are listed in Table 1. S. coelicolor A3(2) strain 1147, a prototrophic wild-type strain, was used as the parental strain. Escherichia coli DH5α was used for plasmid construction. E. coli GM2163 (dam dcm) was used for preparation of nonmethylated DNA for transformation of S. coelicolor. S. coelicolor strains were grown at 30°C on GYM agar (37) or R5− agar (17) without sucrose (R5MS agar). Strains of S. griseus were grown on TSB agar (47). Spontaneous rifampin-resistant (rif [rpoB]) mutants were obtained from colonies that grew within 7 days after spores were spread on R5MS agar containing rifampin (100 μg/ml). The mutants were used for studies after single-colony isolation. Mutations in the rpoB gene were detected by DNA sequencing as described previously (62).
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristics | Source or reference |
|---|---|---|
| Streptomyces strains | ||
| S. coelicolor A3(2) strain 1147 | SCP1+ SCP2+ (prototrophic wild type) | Laboratory stock |
| S. coelicolor A3(2) strain KO-350 | eshA::tsr (eshA null mutant) | 28 |
| S. coelicolor A3(2) strain KO-351 | eshA::tsr rpoB | This study |
| S. coelicolor A3(2) strain KO-541 | eshB (eshB null mutant) | This study |
| S. coelicolor A3(2) strain KO-542 | eshA::tsr eshB | This study |
| S. griseus IFO13189 | Prototrophic streptomycin-producing wild type | Laboratory stock |
| S. griseus KO-390 | eshA disruptant derived from 13189 | 47 |
| Plasmids | ||
| E. coli replicons | ||
| pBluescript SK(+) | Ampr; cloning vector | Toyobo (Osaka, Japan) |
| pGEM-T | Ampr; cloning vector for PCR product | Promega |
| pSCP52 | Ampr; pBluescript SK(+) with 7.1-kb PstI fragment containing S. coelicolor eshA gene | 28 |
| pBL52SG | pBluescript SK(+) with 3.4-kb BamHI fragment containing S. griseus eshA gene | This study |
| pNS80 | pBluescript SK(+) with eshA gene of S. coelicolor under control of the eshA promoter of S. griseus | This study |
| Streptomyces replicons | ||
| pV1 | Tsrr Hygr Ampr; pIJ941 carrying pBluescript SK(+) sequence | 27 |
| pV52SC | pV1 with 2.2-kb PstI fragment containing S. coelicolor eshA gene | 28 |
| pIJ8600 | Aprr Tsrr; tipAp attP | 53 |
| pIJ6084 | Aprr Tsrr; tipAp::relA attP | 52 |
| pNS66 | pV1 with 1.7-kb PstI fragment containing S. coelicolor eshA lacking cyclic nucleotide-binding domain sequence | This study |
| pNS82 | pV1 with 3.4-kb fragment from pNS80 | This study |
| pTYM18 | Kmr; attP | 44 |
| pTYM52SC | pTYM18 with 2.2-kb PstI fragment containing S. coelicolor eshA gene | This study |
Plasmid construction.
Plasmids used or constructed in this study are listed in Table 1, and the oligonucleotide primers used are listed in Table 2. General DNA manipulations, such as plasmid isolation and transformation of E. coli, were performed as described by Sambrook et al. (48). Plasmid pNS66, which contained an S. coelicolor eshA gene that lacked the putative cyclic nucleotide-binding domain, was constructed as follows. Two DNA fragments containing sequences upstream (820 bp) and downstream (920 bp) of the cyclic nucleotide-binding domain were amplified using primers ΔCNBD-F1 and ΔCNBD-R1 and primers ΔCNBD-F2 and ΔCNBD-R2, respectively. The PCR products were digested with PstI and ApaI and cloned into the PstI site of E. coli-Streptomyces shuttle vector pV1 by three-fragment ligation, resulting in pNS66. Plasmid pNS82, which contained S. coelicolor eshA fused to the S. griseus eshA promoter, was constructed as follows. The S. griseus eshA promoter (0.6 kb) and terminator (1.4 kb) regions were amplified from pBL52SG using primers M13 forward and f1-R and primers f2-F and M13 reverse, respectively. The S. coelicolor eshA open reading frame (1.3-kb) was amplified from pSCP52 using primers f3-F and f3-R. The three PCR-generated fragments were cloned into the BamHI site of pBluescript SK(+) to obtain plasmid pNS80. Finally, the 3.4-kb BamHI fragment from pNS80 was inserted into the BamHI site of pV1, resulting in pNS82.
TABLE 2.
Primers used in this study
| Primer | Sequencea | Relevant enzyme |
|---|---|---|
| ΔCNBD-F1 | GCTCCTCCGGTGTCACCTGCAGTCCCTTCG | PstI |
| ΔCNBD-R1 | GCAACCGTCGGTGGGCCCGGTAGGC | ApaI |
| ΔCNBD-F2 | CGGGGCCCAGCGGCAGACCAAGCAC | ApaI |
| ΔCNBD-R2 | GGGGCGGGCACGTCCTGCAGGGTTGTGGGC | PstI |
| f1-R | CGAGTCAACAGCCATGGGGC | NcoI |
| f2-F | TGCAGATCGCCAGATCTCCCAGGTGAGG | BglII |
| f3-F | CTCCCCATGGCCGTCGACAG | NcoI |
| f3-R | GTGGGCGGTGCGAGATCTCAGCG | BglII |
| eshB-F1 | GAGCAACTCCTATTACTTGTTCG | |
| eshB-R1 | TACTCCCAGATGGCGTCA | |
| Act-F | ACCGATGCGGGATGTGTAATTCCG | |
| Act-R | GTGCGCGATATTGCTTTCGAGCAC | |
| relA/NdeI-F | CGCCTACACCATATGCGCACCATGC | NdeI |
| relA/Bam-R | CGGGATCCTCGGTCTACTTCTGTCAGTCGAGCAGC | BamHI |
| eshA-Fw | CTCGTCAACAACCGCGAGTTC | |
| eshA-Rv3 | CGGAGTGGGCGATGTGTTG | |
| eshA-G127V-F | GTCCTCGTCGCACGCGTCGGCCCCGCAGAC | |
| eshA-G127V-R | GTCTGCGGGGCCGACGCGTGCGACGAGGAC | |
| eshA-G161M-F | GGTGCTCGCGGACATGAGCCACTTCGGCGAGC | |
| eshA-G161M-R | GCTCGCCGAAGTGGCTCATGTCCGCGAGCACC | |
| eshA-G165N-F | GACGGCAGCCACTTCAACGAGCAGGCGCTGTCGGAC | |
| eshA-G165N-R | GTCCGACAGCGCCTGCTCGTTGAAGTGGCTGCCGTC | |
| eshA-F196S-F | CTCACGCGCGCCGACTCCACCGCGGTCCAG | |
| eshA-F196S-R | CTGGACCGCGGTGGAGTCGGCGCGCGTGAG |
Boldface type indicates restriction enzyme sites.
Site-directed mutagenesis of eshA.
Site-directed mutagenesis of eshA was performed with a Quickchange II site-directed mutagenesis kit (Stratagene) used according to the manufacturer's instructions. pTYM52SC was used as a template for all the mutagenesis reactions. The primers used in the reactions are shown in Table 2. After DNA sequence confirmation, each mutant eshA allele was introduced into the ΔeshA strain KO-350 for phenotypic analysis.
Construction of eshB mutant.
A 1,130-bp eshB gene fragment was amplified from genomic DNA using primers eshB-F1 and eshB-R1 and cloned into TA cloning vector pCR2.1 (Invitrogen), resulting in pXU21. This plasmid was digested at the unique SalI site in the eshB coding region, treated with the Klenow enzyme, and religated to introduce a frameshift mutation into eshB. A 1.7-kb BglII fragment from pIJ963 containing a hygromycin resistance cassette was introduced into this plasmid to generate pXU25. pXU25 was passed through E. coli GM2163 and introduced into S. coelicolor 1147 protoplasts. Single-crossover transformants were selected on R2YE medium containing hygromycin. After two rounds of nonselective growth on GYM medium, spores were plated to obtain single colonies and screened for hygromycin sensitivity. The expected frameshift mutation in eshB was confirmed by PCR and nucleotide sequencing. The eshB mutation constructed was introduced into strain KO-350 (eshA::tsr) by conjugation to generate the eshA eshB double mutant KO-542.
RNA isolation and RT-PCR analysis.
RNA was isolated from cells grown on R5MS agar covered with cellophane sheets (42). DNase I-treated RNA (0.5 μg) was used as a template for reverse transcription (RT) at 65°C with the ThermoScript RT-PCR system (Invitrogen) and a specific reverse primer, primer Act-R (62). The resulting cDNA was used for PCR amplification with primers Act-F and Act-R under the following conditions: 3 min at 96°C, followed by 25 cycles of 98°C for 10 s and 65°C for 30 s. DNase-treated RNA samples that had not been subjected to reverse transcription were used for PCR amplification as negative controls.
Western blot analysis.
Sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and Western blotting were performed as described previously (43).
Biacore assay.
The instrument used for a Biacore assay was a BIACORE 3000 (Biacore AB, Uppsala, Sweden), which is used for measurement of surface plasmon resonance in real time. CM5 sensor chips were first washed with HBS-EP buffer (10 mM HEPES [pH 7.4], 0.15 M NaCl, 3.0 mM EDTA, 0.005% surfactant P20; Biacore AB) for 7 min and then activated by mixing 70 μl of activating reagent with a mixture of 0.2 M 1-ethyl-3-(3 dimethylaminopropyl) carbodiimide and 0.05 M N-hydroxysuccinimide prior to immobilization. 8-(6-Aminohexyl)aminoadenosine 3′,5′-cyclic monophosphate (a derivative of cAMP) was diluted to obtain a concentration of 1 mM with 10 mM borate buffer (pH 8.5), and the resulting solution (70 μl) was injected at a flow rate of 10 μl/min. Excess reactive groups were deactivated with 1 M ethanolamine. The blanks were running buffer and either the CM5 sensor surface or the surface treated with 1-ethyl-3-(3 dimethylaminopropyl) carbodiimide-N-hydroxysuccinimide and ethanolamine without any added ligand. All buffers were filtered and degassed before use. Analysis of the sensorgrams was performed with the BIA-evaluation software (version 4.1), which allows a range of quantitative kinetic analyses and automatically calculates the binding parameters, taking into account negative control and experimental readings.
Expression and purification of the S. coelicolor RelA protein.
Plasmid pPROrelA, used for overexpression of the S. coelicolor RelA protein in E. coli, was constructed as follows. The relA gene was amplified from genomic DNA isolated from S. coelicolor using primers relA/NdeI-F and relA/Bam-R. The NdeI- and BamHI-digested PCR fragment was ligated into pPROEX-1. The recombinant RelA protein with an N-terminal His tag was purified on nickel-nitrilotriacetic acid matrices (QIAGEN). The RelA protein was further purified by SDS-polyacrylamide gel electrophoresis using a 10% polyacrylamide gel and was extracted with phosphate-buffered saline containing 0.1% SDS. The purified RelA protein obtained (1.8 mg) was used to raise rabbit antibodies.
Measurement of the intracellular nucleotide pools.
For plate culture assays, ca. 2 × 107 spores were spread on R5MS agar covered with a cellophane sheet. The methods used for extraction of intracellular nucleotides from mycelia which were grown on a cellophane sheet or in liquid medium have been described previously (36, 37). Nucleotide pool sizes were analyzed using a high-performance liquid chromatography system (Hitachi D-7000 HPLC series with an L-7100 pump and an L-7400 UV detector) with an ion-exchange column (Partisil-10 SAX; 4.6 mm by 25 cm; GL Science) as described previously (37). Elution was performed at a flow rate of 1 ml/min by using a gradient consisting of a low-ionic-strength buffer (7 mM KH2PO4, adjusted to pH 4.0 with H3PO4) and a high-ionic-strength buffer (0.5 M KH2PO4 and 0.5 M Na2SO4, adjusted to pH 5.4 with KOH). UV detection was performed at 260 nm. The following gradient conditions were used: the percentage of the high-ionic-strength buffer was increased during the initial 20 min from 0 to 100% and then was kept at 100% for the next 20 min, reduced from 100 to 0% in 3 min, and kept at 0% for 10 min to return to the initial conditions. The intracellular concentrations of nucleotides in cells grown on cellophane were expressed in pmol per mg (dry weight) of cells. A wet weight of 1 g was equivalent to a dry weight of 134 mg.
RESULTS
Disruption of eshA but not disruption of eshB eliminates actinorhodin production.
Although we previously reported that eshA disruption suppressed antibiotic production in S. coelicolor A3(2) (28), we found recently that the effect was more evident when cells were grown on R5MS agar instead of R2YE agar (Fig. 1A); production of the blue-pigmented antibiotic actinorhodin was eliminated when eshA was disrupted (throughout 10 days of incubation), which also resulted in reduced formation of aerial mycelium. In contrast, production of the red-pigmented prodiginine antibiotic complex was not affected. Inspection of the genome sequence of S. coelicolor revealed a gene encoding EshB, a close homologue of EshA with 64% amino acid sequence identity. A frameshift mutation was introduced into eshB to assess its role in morphological and physiological differentiation. On R5MS medium, the eshB mutant produced both antibiotics at levels comparable to the levels produced by the wild-type strain (Fig. 1A). The eshA eshB double mutant KO-542 did not produce actinorhodin, but it appeared to produce the red-pigmented antibiotic at wild-type levels (lack of pigmentation in the middle of the KO-542 streak reflected lower levels of mycelial growth). Thus, eshA, but not eshB, is required for actinorhodin production on R5MS agar. Similar results were obtained when strains were grown in R5MS liquid medium (Fig. 2). Actinorhodin production is regulated positively by a pathway-specific regulatory protein, ActII-ORF4 (1, 14). Analysis of actII-ORF4 expression by RT-PCR and Western blotting revealed a dramatic decrease in the level of the actII-ORF4 transcript (Fig. 1B, left panel) and a marked decrease in the level of the ActII-ORF4 protein (Fig. 1B, right panel) in the eshA mutant compared to the wild-type strain, presumably accounting for the loss of actinorhodin production.
FIG. 1.

(A) Effects of disruption of eshA, eshB, and eshA plus eshB on antibiotic production. Spores were inoculated onto R5MS agar plates, and this was followed by 4 days of incubation. The reverse sides of the plates (lower plates) are shown to better illustrate the amounts of antibiotic produced. The blue and red compounds are actinorhodin and undecylprodigiosin, respectively. (B) RT-PCR analysis (left panel) and Western blotting (right panel) for the actII-ORF4 transcript and ActII-ORF4 protein. rRNA indicates the position of 5 μg of total RNA. RNA and crude cell extracts were prepared from wild-type and mutant cells grown on R5MS agar for the times indicated. WT, wild type.
FIG. 2.

Time course of actinorhodin production in R5MS liquid medium. The total actinorhodin produced within and outside cells was measured after treatment of the culture broth with 1 M NaOH. Symbols: •, wild-type strain 1147; ○, eshA disruptant KO-350; □, eshB mutant KO-541; ▪, eshA eshB double mutant KO-542. OD633, optical density at 633 nm.
eshA disruptant accumulates lower levels of ppGpp.
ppGpp plays an essential role in initiating the onset of antibiotic production in several Streptomyces spp., including S. coelicolor A3(2) (24, 38, 52). We therefore postulated that the eshA disruptant KO-350 might have an impaired ability to accumulate intracellular ppGpp. To test this hypothesis, the parental strain (strain 1147) and a disruptant strain (strain KO-350) were grown on R5MS agar, and changes in the levels of the intracellular nucleotide pool were monitored (Fig. 3A). While the growth of the two strains was virtually identical, the eshA disruptant KO-350 accumulated lower levels of ppGpp than parental strain 1147 accumulated through the early, middle, and late phases of growth. The GTP pool size decreased sharply in both strains as growth progressed, presumably reflecting curtailment of purine nucleotide synthesis due to the reduced availability of substrates from the medium.
FIG. 3.
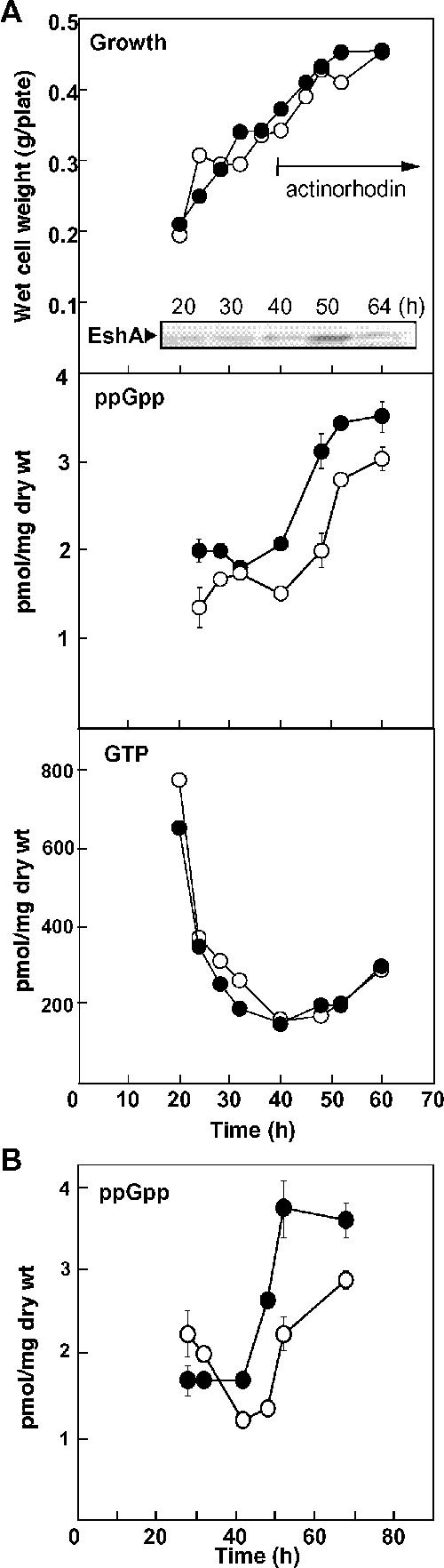
Changes in the intracellular concentrations of ppGpp and GTP and levels of the EshA protein during growth on agar medium. (A) Strains of S. coelicolor A3(2) were grown on R5MS agar covered with a cellophane sheet. Cells were harvested at different times to determine biomass, nucleotide concentrations, and the amount of EshA produced. Nucleotides were separated by high-performance liquid chromatography, whereas EshA protein levels were determined by Western analysis. The error bars indicate the standard deviations of the means of triplicate samples. Symbols: •, wild-type strain 1147; ○, eshA disruptant KO-350. (B) Strains of S. griseus were grown on TSB agar as described above for panel A. ppGpp levels were determined by high-performance liquid chromatography. Symbols: •, wild-type strain 13189; ○, eshA disruptant KO-390.
Like disruption of eshA in S. coelicolor A3(2), disruption of eshA in S. griseus resulted in the loss of antibiotic (streptomycin) production (47). Thus, it was possible that an S. griseus eshA disruptant would also exhibit an impaired ability to accumulate ppGpp. Growth of wild-type S. griseus and growth of a congenic eshA mutant on TSB agar (a medium that supports streptomycin production) demonstrated that the eshA disruptant, like its S. coelicolor A3(2) counterpart, did indeed have a reduced ability to accumulate ppGpp during the middle and late growth phases (40 to 70 h) (Fig. 3B). Thus, eshA disruptants of both S. coelicolor and S. griseus accumulate lower levels of ppGpp, which in turn might result in loss of antibiotic production. To assess whether accumulation of reduced levels of ppGpp might reflect lower levels of the ppGpp synthetase RelA, we also determined the level of RelA in S. coelicolor 1147 (wild type) and KO-350 (eshA mutant) by Western blotting of proteins derived from a P100 precipitate (ribosomal) fraction (RelA exists predominantly in a ribosome-associated form [5]). RelA was detected throughout growth; while the level of RelA decreased in the late growth phase (i.e., 40 h), there were no significant differences between 1147 and the eshA disruptant (data not shown).
Induction of ppGpp synthesis in the eshA mutant restores actinorhodin production.
To assess whether reduced levels of ppGpp were primarily responsible for the eshA phenotype, we used the method of Sun et al. (52) to induce elevated levels of ppGpp during growth. pIJ6084 contains the truncated S. coelicolor relA gene (and thus produces a ribosome-independent RelA) under control of the thiostrepton-inducible tipA promoter. pIJ6084 and the pIJ8600 vector control were introduced into eshA disruptant KO-350, and the strains were grown on R5MS agar lacking or containing 0.05 μg/ml of thiostrepton. Production of actinorhodin was completely restored in the presence of pIJ6084 but not in the presence of pIJ8600, but this occurred only when cells were grown in the presence of thiostrepton (Fig. 4A). Lower or higher concentrations of thiostrepton were less effective for restoring actinorhodin production (data not shown). We next determined ppGpp levels in mycelium harboring pIJ6084 or pIJ8600 that had been grown in the presence of thiostrepton. Cells harboring pIJ6084 had a higher level of ppGpp than cells containing pIJ8600 in both early-growth-phase (20 h) and late-growth-phase (50 h) cultures (Fig. 4B); the levels observed in induced cultures containing pIJ6084 were comparable to the levels seen in wild-type strain 1147 (Fig. 3A). Thus, although relatively low, the reduced level of ppGpp in the eshA mutant appeared to be solely responsible for the observed defect in antibiotic production. Further evidence that supports this conclusion was derived from introduction of particular rpoB mutations into the eshA disruptant. Recent X-ray crystallography of the RNA polymerase-ppGpp complex demonstrated unambiguously that there is binding of ppGpp adjacent to the RNA polymerase active center (2). We reported previously that the loss of actinorhodin production observed in relA or relC mutants (which are deficient in ppGpp synthesis) could be suppressed by introducing particular mutations into the rpoB gene that encodes the β subunit of RNA polymerase, which is believed to be a primary target of ppGpp in eliciting changes in gene expression (40, 62). Therefore, we assumed that introduction of certain rpoB mutations into KO-350 should restore actinorhodin production to levels that exceed the level in the wild-type strain. To assess this assumption, we isolated 25 rifampin-resistant (rif) mutants of KO-350, which developed spontaneously on the plates containing rifampin (see Materials and Methods), since rpoB mutations have been found frequently among rif mutants (16, 22, 23, 32, 49, 51, 59). We found that the rif mutants examined all had a point mutation or a deletion mutation in the rpoB gene. Of the 25 isolates, 12 exhibited complete restoration of actinorhodin production (or produced even higher levels than the parental strain produced), and this was accompanied by restoration of actII-ORF4 transcription, as determined by RT-PCR. The effective rpoB mutations were 416F→L, 427D→N, 440R→C, 475P→L, deletion 410V, deletion 414K-416F, and deletion 426M, whereas other mutations (427D→G, 427D→A, 433S→T, and 485S→F) did not restore actinorhodin production. One of the effective mutations, the mutation in KO-351 (427D→ N), is shown in Fig. 5A and B.
FIG. 4.
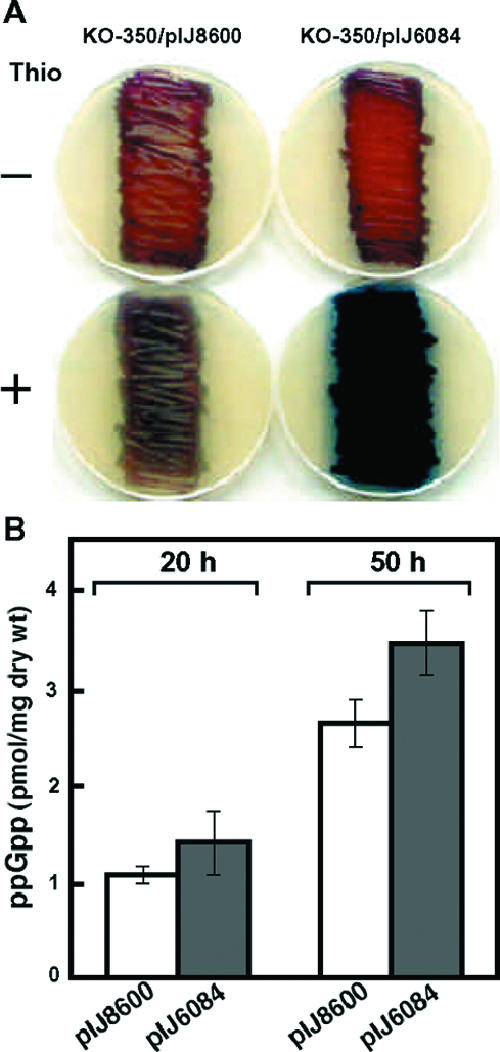
Effect of inducing the truncated relA gene on antibiotic production (A) and intracellular ppGpp pool size (B). Plasmid pIJ6084 harbors a truncated (ribosome-independent) relA gene under control of the tipA promoter, while pIJ8600 is the vector control. Cells were grown on R5MS agar plates to the early growth phase (20 h) or late growth phase (50 h) with or without thiostrepton (0.05 μg/ml) (Thio), as described in the legend to Fig. 1. The ppGpp pool size was determined as described in the legend to Fig. 3. The error bars indicate the standard deviations based on data from six independent experiments.
FIG. 5.

Effect on actinorhodin production of introducing an rpoB mutation into the S. coelicolor eshA mutant (A) and RT-PCR analysis of the actII-ORF4 transcript (B). Strains were grown on R5MS agar as described in the legend to Fig. 1. rRNA indicates the position of 5 μg of total RNA. RNA was isolated from cells grown for different times. WT, wild type.
EshA exerts its influence via a nucleotide-binding domain.
The EshA proteins of S. coelicolor and S. griseus exhibit 76% amino acid identity and are characterized by the presence of a putative cyclic nucleotide-binding domain (28, 47) that is absent from the otherwise homologous proteins MMPI and SrpI (35, 61). To investigate if the EshA domain is essential for restoration of antibiotic production, the putative cNMP-binding domain of S. coelicolor A3(2) was deleted (Fig. 6A), yielding pNS66. pNS66 and pV52SC (harboring the entire eshA gene) were introduced separately into the eshA disruptant KO-350, and the resulting strains were grown on R5MS agar. While pV52SC restored actinorhodin production to wild-type levels, pNS66 did not, despite the production of wild-type levels of the deleted EshA derivative (delEshA) (35 kDa compared to the 52-kDa wild-type protein) (Fig. 6B, C). Thus, the putative cNMP-binding domain is essential for EshA functioning. As observed for the native EshA protein, the majority of the domain-deleted EshA was recovered in the P100 precipitate fraction when it was examined by Western analysis (data not shown), probably reflecting the formation of multimers, as shown previously for S. griseus EshA (47). Neither EshA nor the deleted derivative delEshA was detected in the S100 supernatant or membrane fraction.
FIG. 6.

Effect of deleting the cyclic nucleotide-binding domain of EshA on actinorhodin production in S. coelicolor A3(2). (A) pV52SC contains the entire eshA gene, while pNS66 lacks the nucleotide-binding domain. (B) Growth of KO-350 harboring pV52SC or pNS66 on R5MS agar after incubation for 3 days. (C) Western analysis of EshA and a deleted derivative (delEshA). Cells were grown to the late growth phase (50 h) on R5MS agar plates. Western blotting was performed using the whole-cell extract.
To demonstrate further the functional importance of the cNMP-binding domain of EshA, we used site-directed mutagenesis to replace several highly conserved amino acids (28). We created four mutant alleles, G127V, G161M, G165N, and F196S. One of the mutants, the F196S mutant, failed to complement the ΔeshA mutation, as assessed by actinorhodin production (Fig. 7), demonstrating that the cNMP-binding domain is essential for EshA functioning. The other three mutants produced actinorhodin at the same level as the eshA wild-type allele (data not shown).
FIG. 7.

Effect of the eshAF196S mutation on actinorhodin production. ΔeshA cells complemented by either the wild-type or F196S mutant eshA allele were grown for 5 days on the R5MS agar plate. Wild-type and ΔeshA strains harboring the empty vector were included as controls. WT, wild type.
Kinetic study of the interaction between EshA and cAMP.
To study the kinetics of binding of cAMP to EshA, surface plasmon resonance was used. The method of Wilchek and Selinger (60) was employed with a cAMP derivative and EshA monomer (purified by gel filtration) (see Materials and Methods and reference 47 for details). The cAMP derivative bound to EshA (Fig. 8A) with an association rate constant of 6.9 M−1 s−1 and a dissociation rate constant of 0.0002 s−1, as estimated by fitting the binding curves to the Langmuir binding model. The dissociation equilibrium constant was 28.9 μM.
FIG. 8.

Biacore analysis of the interaction between EshA and an immobilized cAMP derivative (A) and inhibition of EshA binding by externally added cAMP (B). The data were fitted globally to a 1:1 Lagmuir interaction model. The arrow indicates the time of external addition of cAMP at the concentrations indicated on the right. RU, resonance unit.
The association of EshA with the cAMP derivative was eliminated by external addition of 400 to 800 μM cAMP (Fig. 8B), whereas this association was not affected by any other nucleoside triphosphate, diphosphate, or monophosphate, including cyclic GMP and ppGpp (each added at a concentration of 1 mM). AMP had a slight effect when it was added at a concentration greater than 500 μM. Thus, EshA appears to bind specifically to cAMP.
EshA from S. coelicolor does not function in S. griseus.
Previously, we failed to detect phenotypic expression of S. coelicolor eshA in S. griseus and speculated that this might reflect inactivity of the S. coelicolor eshA promoter in S. griseus (47). To address this issue further, we constructed a plasmid in which the coding sequence of S. coelicolor eshA was located downstream of the S. griseus eshA promoter. The resulting plasmid, pNS82, was introduced into the S. griseus eshA disruptant KO-390. As expected, the pNS82 transformant of KO-390 produced abundant amounts of EshA, as determined by Western analysis (Fig. 9C). Despite the marked accumulation of EshA, neither aerial mycelium formation (Fig. 9A) nor streptomycin production (Fig. 9B) was restored in the S. griseus eshA disruptant (47). In contrast, introduction of pV52SG (harboring the S. griseus eshA gene) into KO-390 resulted in the formation of abundant aerial mycelium (Fig. 9A) and extensive production of streptomycin (47; data not shown). Thus, despite the high level of amino acid sequence identity to its homologue and assumed orthologue, EshA from S. coelicolor is not able to function in S. griseus, at least in the context of morphological and physiological differentiation.
FIG. 9.
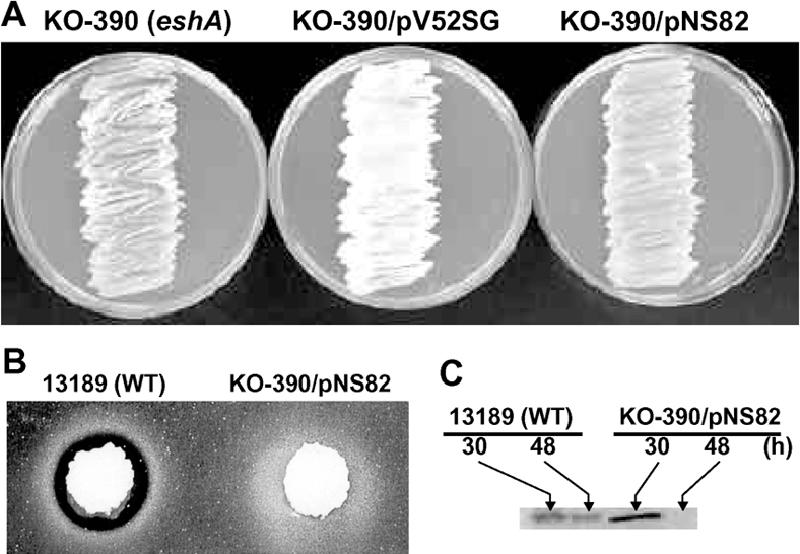
Expression of S. coelicolor A3(2) eshA in the S. griseus eshA disruptant KO-390. pV52SG harbors S. griseus eshA, while pNS82 harbors S. coelicolor eshA expressed from the promoter of S. griseus eshA. (A) Strains were grown on TSB agar for 2 days. The white appearance of the growth in the middle plate reflects the development of aerial mycelium. (B) Streptomycin production by S. griseus wild-type strain 13189 and the eshA disruptant KO-390 harboring pNS82. Agar plugs (diameter, 8 mm) were cut from each of the TSB agar plates after 48 h of incubation and placed on the assay plate, and this was followed by 12 h of incubation. (C) Western analysis of EshA. The strains were grown on TSB agar plates for 30 h or 48 h. A Western analysis was performed as described in the legend to Fig. 6 using whole-cell extracts. WT, wild type.
Temperature-induced deletion of eshA in S. coelicolor results in loss of actinorhodin production.
Chromosome instability is a characteristic of many Streptomyces species, in which deletions of up to 2 Mb have been observed (11), and it is believed to account for at least some instances of loss of antibiotic production. The apparent absence of essential genes in the less conserved terminal regions of the linear chromosomes of streptomycetes may well predispose these regions to deletion and rearrangement, events that may be induced by a variety of physiological stresses. eshA is located close to one of the ends of the S. coelicolor A3(2) chromosome (130 kb from the end; located on cosmid 1A4 [46]). To examine whether eshA was prone to such a deletion event, we grew S. coelicolor at 42°C (the highest temperature that allowed growth) for 7 days on GYM agar. A total of 150 randomly selected colonies were assayed for the presence of eshA by PCR using primers eshA-Fw and eshA-Rv3, and 18 of these isolates (12%) lacked eshA. Of the smaller-colony types found in these 150 colonies, 55% had lost eshA. All of the deletion mutants, like the eshA disruptant KO-350, were totally or severely impaired for actinorhodin production on R5MS agar. Moreover, actinorhodin production was restored upon introduction of pV52SC harboring eshA (data not shown), suggesting that the loss of antibiotic production in the temperature-induced deletion mutants was attributable solely to deletion of eshA and not to deletion of flanking genes.
DISCUSSION
We demonstrated that EshA plays a crucial role in establishing an intracellular level of ppGpp sufficient to trigger actinorhodin production in S. coelicolor A3(2) at least under some nutrient conditions and that the cyclic nucleotide-binding domain of EshA is essential for this activity. Thus, as demonstrated in this study (Fig. 1) and in previous work (28, 47), EshA functions as a positive regulator for antibiotic production in both S. coelicolor and S. griseus. Strikingly, relatively small differences in ppGpp pool sizes resulted in dramatic changes in antibiotic productivity (Fig. 4). This suggests that there is a threshold level of ppGpp that is required for triggering secondary metabolism and that ppGpp concentrations in the cell can be finely tuned by factors such as EshA. Thus, at least some of the physiological instability that is sometimes associated with antibiotic production in streptomycetes could be attributable to factors that influence EshA activity. Despite a high level of amino acid sequence identity, EshA of S. coelicolor was unable to complement an EshA mutant of S. griseus. However, there are other functional differences between the two proteins. While EshA plays a role in sustaining DNA synthesis during the late growth phase in S. griseus, no such function is evident for its homologue in S. coelicolor (47).
Disruption of eshA resulted in the elimination of antibiotic production in both S. coelicolor and S. griseus, but only in some culture conditions. This conditional phenotype is reminiscent of that observed for ppGpp itself, which is required for triggering antibiotic production under nitrogen limitation conditions but not under phosphate limitation conditions (6, 41). Interestingly, a relA null mutant of S. coelicolor also does not produce actinorhodin on R5MS agar (unpublished data), mimicking the phenotype of the eshA mutant, which is consistent with our interpretation that EshA has a role in modulating ppGpp levels. Although not required for antibiotic production under any of the growth conditions used in these experiments, we cannot rule out the possibility that EshB plays a role in triggering antibiotic production under other conditions. A recent proteomic analysis of S. coelicolor demonstrated that the level of SCO5249 (= EshB) was dramatically reduced in a bldA mutant (30).
eshA is located close to one end of the linear S. coelicolor A3(2) chromosome, in a region of the genome predicted to be more susceptible to deletion and rearrangement. Indeed, when organisms were subjected to elevated growth temperatures, no fewer than 12% of the resulting colonies had lost eshA and did not produce actinorhodin, suggesting that loss of eshA (if it is located near the end of the chromosome of other streptomycetes) may account for the genetic instability of antibiotic production observed in many different streptomycetes.
EshA appears to be a member of a protein superfamily (31, 57) whose activities are modulated by binding cyclic nucleotides (33, 50), and the cyclic nucleotide-binding domain of EshA is clearly required for activity (Fig. 6 and 7). Moreover, we successfully demonstrated binding of cAMP to EshA with high specificity (Fig. 8). In prokaryotes, only Crp and DnaA are known to bind cAMP (4, 8, 18). dnaA of E. coli is essential for the initiation of DNA replication at the chromosomal origin, oriC (58), and interaction of DnaA with oriC is stimulated by binding of cAMP (13). Recent studies with Streptomyces spp. (26, 54) have demonstrated that extracellular levels of cAMP are high during the late growth phase, prompting speculation that cAMP is involved, directly or indirectly, in the onset of secondary metabolism. Moreover, Kang et al. (26) reported that addition of a low level of cAMP enhanced the formation of aerial mycelium by the S. griseus mutant HO2 (although accumulation of very high levels of cAMP, mediated through a cloned copy of the adenylate cyclase gene cyaA, eliminated both formation of aerial mycelium and production of streptomycin during the late growth phase, accompanied by severely reduced accumulation of ppGpp). The stimulatory effect on the formation of aerial mycelium by a low level of exogenous cAMP may be accounted for, at least partially, by activation of EshA via cAMP binding to the cAMP-binding domain.
eshA (or its homologue) genes are widely distributed and occur in streptomycetes (28), including S. griseus and Strepotmyces avermitilis, the actinobacterium Frankia sp. strain EAN1pec, and the cyanobacterium Nostoc punctiforme PCC73102, as determined by searching the nucleotide sequence database, although the eshA gene was not found in E. coli, Bacillus subtilis, and Thermus thermophilus. Therefore, EshA may offer new opportunities for uncovering and analyzing the unknown regulatory events that occur at the onset of the stationary phase in these physiologically and morphologically complex organisms.
Acknowledgments
This work was supported by grants from the Organized Research Combination System and the Effective Promotion of Joint Research of Special Coordination Funds (Ministry of Education, Culture, Sports, Science and Technology of the Japanese Government).
We are grateful to Guo-Jun Wang and Norimasa Tamehiro for preliminary work performed in several of the experiments.
REFERENCES
- 1.Arias, P., M. A. Fernandez-Moreno, and F. Malpartida. 1999. Characterization of the pathway-specific positive transcriptional regulator for actinorhodin biosynthesis in Streptomyces coelicolor A3(2) as a DNA-binding protein. J. Bacteriol. 181:6958-6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Artsimovitch, I., V. Patlan, S. Sekine, M. N. Vassylyeva, T. Hosaka, K. Ochi, S. Yokoyama, and D. G. Vassylyev. 2004. Structural basis for transcription regulation by alarmone ppGpp. Cell 117:299-310. [DOI] [PubMed] [Google Scholar]
- 3.Bibb, M. J. 2005. Regulation of secondary metabolism in streptomycetes. Curr. Opin. Microbiol. 8:208-215. [DOI] [PubMed] [Google Scholar]
- 4.Botsford, J. L., and J. G. Harman. 1992. Cyclic AMP in prokaryotes. Microbiol. Rev. 56:100-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cashel, M., D. R. Gentry, V. J. Hernandez, and D. Vinella. 1996. The stringent response, p. 1458-1496. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and E. Umbarger (ed.), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. American Society for Microbiology, Washington, D. C.
- 6.Chakraburtty, R., and M. Bibb. 1997. The ppGpp synthetase gene (relA) of Streptomyces coelicolor A3(2) plays a conditional role in antibiotic production and morphological differentiation. J. Bacteriol. 179:5854-5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chakraburtty, R., J. White, E. Takano, and M. Bibb. 1996. Cloning, characterization and disruption of a (p)ppGpp synthetase gene (relA) of Streptomyces coelicolor A3(2). Mol. Microbiol. 19:357-368. [DOI] [PubMed] [Google Scholar]
- 8.Chandler, M. S. 1992. The gene encoding cAMP receptor protein is required for competence development in Haemophilus influenzae Rd. Proc. Natl. Acad. Sci. USA 89:1626-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chater, K. F., and M. J. Bibb. 1997. Regulation of bacterial antibiotic production, p. 57-105. In H. Kleinkauf and H. Von Dohren (ed.), Products of secondary metabolism. Biotechnology, vol. 6. VCH, Weinheim, Germany. [Google Scholar]
- 10.Chater, K. F., and D. A. Hopwood. 1993. Streptomyces, p. 83-89. In A. L. Sonenshein, J. A. Hoch, and R. Losick (ed.), Bacillus subtilis and other gram-positive bacteria: biochemistry, physiology, and molecular genetics. American Society for Microbiology, Washington, D. C.
- 11.Chen, C. W., C. H. Huang, H. H. Lee, H. H. Tsai, and R. Kirby. 2002. Once the circle has been broken: dynamics and evolution of Streptomyces chromosomes. Trends Genet. 18:522-529. [DOI] [PubMed] [Google Scholar]
- 12.Dworkin, J., and R. Losick. 2001. Linking nutritional status to gene activation and development. Genes Dev. 15:1051-1054. [DOI] [PubMed] [Google Scholar]
- 13.Fuller, R. S., B. E. Funnell, and A. Kornberg. 1984. The dnaA protein complex with the E. coli chromosomal replication origin (oriC) and other DNA sites. Cell 38:889-900. [DOI] [PubMed] [Google Scholar]
- 14.Gramajo, H. C., E. Takano, and M. J. Bibb. 1993. Stationary-phase production of the antibiotic actinorhodin in Streptomyces coelicolor A3(2) is transcriptionally regulated. Mol. Microbiol. 7:837-845. [DOI] [PubMed] [Google Scholar]
- 15.Hesketh, A., J. Sun, and M. Bibb. 2001. Induction of ppGpp synthesis in Streptomyces coelicolor A3(2) grown under conditions of nutritional sufficiency elicits actII-ORF4 transcription and actinorhodin biosynthesis. Mol. Microbiol. 39:136-144. [DOI] [PubMed] [Google Scholar]
- 16.Hu, H., Q. Zhang, and K. Ochi. 2002. Activation of antibiotic biosynthesis by specified mutations in the rpoB gene (encoding the RNA polymerase β subunit) of Streptomyces lividans. J. Bacteriol. 184:3984-3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang, J., C. J. Lih, K. H. Pan, and S. N. Cohen. 2001. Global analysis of growth phase responsive gene expression and regulation of antibiotic biosynthetic pathways in Streptomyces coelicolor using DNA microarrays. Genes Dev. 15:3183-3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hughes, P., A. Landoulsi, and M. Kohiyama. 1988. A novel role for cAMP in the control of the activity of the E. coli chromosome replication initiator protein, DnaA. Cell 55:343-350. [DOI] [PubMed] [Google Scholar]
- 19.Hunter, I. S., and S. Baumberg. 1989. Molecular genetics of antibiotic formation, p. 121-162. In S. Baumberg, I. S. Hunter, and P. M. Rhodes (ed.), Microbial products: new approaches. Cambridge University, Cambridge, England.
- 20.Inaoka, T., and K. Ochi. 2002. RelA protein is involved in induction of genetic competence in certain Bacillus subtilis strains by moderating the level of intracellular GTP. J. Bacteriol. 184:3923-3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inaoka, T., K. Takahashi, M. Ohnishi-Kameyama, M. Yoshida, and K. Ochi. 2003. Guanine nucleotides guanosine 5′-diphosphate 3′-diphosphate and GTP co-operatively regulate the production of an antibiotic bacilysin in Bacillus subtilis. J. Biol. Chem. 278:2169-2176. [DOI] [PubMed] [Google Scholar]
- 22.Inaoka, T., K. Takahashi, H. Yada, M. Yoshida, and K. Ochi. 2004. RNA polymerase mutation activates the production of a dormant antibiotic 3, 3′-neotrehalosadiamine via an autoinduction mechanism in Bacillus subtilis. J. Biol. Chem. 279:3885-3892. [DOI] [PubMed] [Google Scholar]
- 23.Jin, D. J., and C. A. Gross. 1988. Mapping and sequencing of mutations in the Escherichia coli rpoB gene that lead to rifampicin resistance. J. Mol. Biol. 202:45-58. [DOI] [PubMed] [Google Scholar]
- 24.Jin, W., Y. G. Ryu, S. G. Kang, S. K. Kim, N. Saito, K. Ochi, S. H. Lee, and K. J. Lee. 2004. Two relA/spoT homologous genes are involved in the morphological and physiological differentiation of Streptomyces clavuligerus. Microbiology 150:1485-1493. [DOI] [PubMed] [Google Scholar]
- 25.Kalderon, D., and G. M. Rubin. 1989. cGMP-dependent protein kinase genes in Drosophila. J. Biol. Chem. 264:10738-10748. [PubMed] [Google Scholar]
- 26.Kang, D. K., X. M. Li, K. Ochi, and S. Horinouchi. 1999. Possible involvement of cAMP in aerial mycelium formation and secondary metabolism in Streptomyces griseus. Microbiology 145:1161-1172. [DOI] [PubMed] [Google Scholar]
- 27.Kawamoto, S., H. Watanabe, A. Hesketh, J. C. Ensign, and K. Ochi. 1997. Expression analysis of the ssgA gene product, associated with sporulation and cell division in Streptomyces griseus. Microbiology 143:1077-1086. [DOI] [PubMed] [Google Scholar]
- 28.Kawamoto, S., M. Watanabe, N. Saito, A. Hesketh, K. Vachalova, K. Matsubara, and K. Ochi. 2001. Molecular and functional analyses of the gene (eshA) encoding the 52-kilodalton protein of Streptomyces coelicolor A3(2) required for antibiotic production. J. Bacteriol. 183:6009-6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawamoto, S., D. Zhang, and K. Ochi. 1997. Molecular analysis of the ribosomal L11 protein gene (rplK = relC) of Streptomyces griseus and identification of a deletion allele. Mol. Gen. Genet. 255:549-560. [DOI] [PubMed] [Google Scholar]
- 30.Kim, D.-W., K. F. Chater, K.-J. Lee, and A. Hesketh. 2005. Effect of growth phase and the developmentally significant bldA-specified tRNA on the membrane-associated proteome of Streptomyces coelicolor. Microbiology 151:2707-2720. [DOI] [PubMed] [Google Scholar]
- 31.Kwak, J., L. A. McCue, K. Trczianka, and K. E. Kendrick. 2001. Identification and characterization of a developmentally regulated protein, EshA, required for sporogenic hyphal branches in Streptomyces griseus. J. Bacteriol. 183:3004-3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lai. C., J. Xu, Y. Tozawa, Y. Okamoto-Hosoya, X. Yao, and K. Ochi. 2002. Genetic and physiological characterization of rpoB mutations that activate antibiotic production in Streptomyces lividans. Microbiology 148:3365-3373. [DOI] [PubMed] [Google Scholar]
- 33.McCue, L. A., K. A. McDonough, and C. E. Lawrence. 2000. Functional classification of cNMP-binding proteins and nucleotide cyclases with implications for novel regulatory pathways in Mycobacterium tuberculosis. Genome Res. 10:204-219. [DOI] [PubMed] [Google Scholar]
- 34.Nicholson, M. L., M. Gaasenbeek, and D. E. Laudenbach. 1995. Two enzymes together capable of cysteine biosynthesis are encoded on a cyanobacterial plasmid. Mol. Gen. Genet. 247:623-632. [DOI] [PubMed] [Google Scholar]
- 35.Nicholson, M. L., and D. E. Laudenbach. 1995. Genes encoded on a cyanobacterial plasmid are transcriptionally regulated by sulfur availability and CysR. J. Bacteriol. 177:2143-2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ochi, K. 1987. Change in nucleotide pools during sporulation of Streptomyces griseus in submerged culture. J. Gen. Microbiol. 133:2787-2795. [Google Scholar]
- 37.Ochi, K. 1987. Metabolic initiation of differentiation and secondary metabolism by Streptomyces griseus: significance of the stringent response (ppGpp) and GTP content in relation to A factor. J. Bacteriol. 169:3608-3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ochi, K. 1990. A relaxed (rel) mutant of Streptomyces coelicolor A3(2) with a missing ribosomal protein lacks the ability to accumulate ppGpp, A-factor and prodigiosin. J. Gen. Microbiol. 136:2405-2412. [DOI] [PubMed] [Google Scholar]
- 39.Ochi, K. 1990. Streptomyces relC mutants with an altered ribosomal protein ST-L11 and genetic analysis of a Streptomyces griseus relC mutant. J. Bacteriol. 172:4008-4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ochi, K., S. Okamoto, Y. Tozawa, T. Inaoka, T. Hosaka, J. Xu, and K. Kurosawa. 2004. Ribosome engineering and secondary metabolite production. Adv. Appl. Microbiol. 56:155-184. [DOI] [PubMed] [Google Scholar]
- 41.Ochi, K., D. Zhang, S. Kawamoto, and A. Hesketh. 1997. Molecular and functional analysis of the ribosomal L11 and S12 protein genes (rplK and rpsL) of Streptomyces coelicolor A3(2). Mol. Gen. Genet. 256:488-498. [DOI] [PubMed] [Google Scholar]
- 42.Okamoto, S., A. Lezhava, T. Hosaka, Y. Okamoto-Hosoya, and K. Ochi. 2003. Enhanced expression of S-adenosylmethionine synthetase causes overproduction of actinorhodin in Streptomyces coelicolor A3(2). J. Bacteriol. 185:601-609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okamoto, S., and K. Ochi. 1998. An essential GTP-binding protein functions as a regulator for differentiation in Streptomyces coelicolor. Mol. Microbiol. 30:107-119. [DOI] [PubMed] [Google Scholar]
- 44.Onaka, H., S. Taniguchi, H. Ikeda, Y. Igarashi, and T. Furumai. 2003. pTOYAMAcos, pTYM18, and pTYM19, actinomycete-Escherichia coli integrating vectors for heterologous gene expression. J. Antibiot. (Tokyo) 56:950-956. [DOI] [PubMed] [Google Scholar]
- 45.Ratnayake-Lecamwasam, M., P. Serror, K. W. Wong, and A. L. Sonenshein. 2001. Bacillus subtilis CodY represses early-stationary-phase genes by sensing GTP levels. Genes Dev. 15:1093-1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Redenbach, M., H. M. Kieser, D. Denapaite, A. Eichner, J. Cullum, H. Kinashi, and D. A. Hopwood. 1996. A set of ordered cosmids and a detailed genetic and physical map for the 8 Mb Streptomyces coelicolor A3(2) chromosome. Mol. Microbiol. 21:77-96. [DOI] [PubMed] [Google Scholar]
- 47.Saito, N., K. Matsubara, M. Watanabe, F. Kato, and K. Ochi. 2003. Genetic and biochemical characterization of EshA, a protein that forms large multimers and affects developmental processes in Streptomyces griseus. J. Biol. Chem. 278:5902-5911. [DOI] [PubMed] [Google Scholar]
- 48.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 49.Severinov, K., M. Soushko, A. Goldfarb, and V. Nikiforrov. 1994. Rif mutations in the beginning of the Escherichia coli rpoB gene. Mol. Gen. Genet. 244:120-126. [DOI] [PubMed] [Google Scholar]
- 50.Shabb, J. B., and J. D. Corbin. 1992. Cyclic nucleotide-binding domains in proteins having diverse functions. J. Biol. Chem. 267:5723-5726. [PubMed] [Google Scholar]
- 51.Singer, M., D. J. Jin, W. A. Walter, and C. A. Gross. 1993. Genetic evidence for the interaction between cluster I and cluster III rifampicin resistant mutations. J. Mol. Biol. 231:1-5. [DOI] [PubMed] [Google Scholar]
- 52.Sun, J., A. Hesketh, and M. Bibb. 2001. Functional analysis of relA and rshA, two relA/spoT homologues of Streptomyces coelicolor A3(2). J. Bacteriol. 183:3488-3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun, J., G. H. Kelemen, J. M. Fernandez-Abalos, and M. J. Bibb. 1999. Green fluorescent protein as a reporter for spatial and temporal gene expression in Streptomyces coelicolor A3(2). Microbiology 145:2221-2227. [DOI] [PubMed] [Google Scholar]
- 54.Susstrunk, U., J. Pidoux, S. Taubert, A. Ullmann, and C. J. Thompson. 1998. Pleiotropic effects of cAMP on germination, antibiotic biosynthesis and morphological development in Streptomyces coelicolor. Mol. Microbiol. 30:33-46. [DOI] [PubMed] [Google Scholar]
- 55.Takano, E., H. C. Gramajo, E. Strauch, N. Andres, J. White, and M. J. Bibb. 1992. Transcriptional regulation of the redD transcriptional activator gene accounts for growth-phase-dependent production of the antibiotic undecylprodigiosin in Streptomyces coelicolor A3(2). Mol. Microbiol. 6:2797-2804. [DOI] [PubMed] [Google Scholar]
- 56.Takio, K., R. D. Wade, S. B. Smith, E. G. Krebs, K. A. Walsh, and K. Titani. 1984. Guanosine cyclic 3′,5′-phosphate dependent protein kinase, a chimeric protein homologous with two separate protein families. Biochemistry 23:4207-4218. [DOI] [PubMed] [Google Scholar]
- 57.Triccas, J. A., N. Winter, P. W. Roche, A. Gilpin, K. E. Kendrick, and W. J. Britton. 1998. Molecular and immunological analyses of the Mycobacterium avium homolog of the immunodominant Mycobacterium leprae 35-kilodalton protein. Infect. Immun. 66:2684-2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.von Meyenbyrg, K., and F. G. Hansen. 1996. Regulation of chromosome replication, p. 1555-1577. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. American Society for Microbiology, Washington, D. C.
- 59.Wichelhaus, T. A., V. Schafer, V. Brade, and B. Boddinghaus. 1999. Molecular characterization of rpoB mutations conferring cross-resistance to rifamycins on methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 43:2813-2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wilchek, M., and Z. Selinger. 1974. The preparation and use of cyclic AMP Sepharose. Methods Enzymol. 38:385-387. [DOI] [PubMed] [Google Scholar]
- 61.Winter, N., J. A. Triccas, B. Rivoire, M. C. Pessolani, K. Eiglmeier, E. M. Lim, S. W. Hunter, P. J. Brennan, and W. J. Britton. 1995. Characterization of the gene encoding the immunodominant 35 kDa protein of Mycobacterium leprae. Mol. Microbiol. 16:865-876. [DOI] [PubMed] [Google Scholar]
- 62.Xu, J., Y. Tozawa, C. Lai, H. Hayashi, and K. Ochi. 2002. A rifampicin resistance mutation in the rpoB gene confers ppGpp-independent antibiotic production in Streptomyces coelicolor A3(2). Mol. Genet. Genomics 268:179-189. [DOI] [PubMed] [Google Scholar]