

Abstract
The role of adjuvant on the Th1 and Th2 immune responses to Aβ-immunotherapy (Aβ42 peptide) was examined in wild-type mice. Fine epitope analysis with overlapping oligomers of the Aβ42 sequence identified the 1–15 region as a dominant B cell epitope. The 6–20 peptide was recognized only weakly by antisera from mice administrated with Aβ42 peptide formulated in complete Freund’s adjuvant (CFA), alum or TiterMax Gold (TMG). However, mice immunized with Aβ42 mixed with QS21 induced a significant antibody response to the 6–20 peptide. The only T cell epitope found was within the 6–28 sequence of Aβ42. QS21 and CFA induced the strongest humoral response to Aβ, alum was intermediate, and TMG the weakest adjuvant. Analysis of antibody isotypes specific for Aβ indicates that alum induces primarily Th2-type immune response, whereas TMG, CFA and QS21 shift the immune responses toward a Th1 phenotype. Stimulation of splenocytes from Aβ-immunized mice with Aβ40 peptide induced strikingly different cytokine expression profiles. QS21 and CFA induced significant IFN-γ, IL-4 and tumor necrosis factor-α expression, whereas alum induced primarily IL-4 production. As Th1-type immune responses have been implicated in many autoimmune disorders, whereas Th2-type responses have been shown to inhibit autoimmune disease, the choice of adjuvant may be critical for the design of a safe and effective immunotherapy for Alzheimer’s disease.
Keywords: antibody, B/T cell response, cytokine, epitope, peptide
Abbreviations: AD Alzheimer’s disease, CFA complete Freund’s adjuvant, HMAT half-maximal antibody titer, HRP horseradish peroxidase, IFA incomplete Freund’s adjuvant, PBST PBS containing 0.1% Tween 20, TMG TiterMax Gold, TNF tumor necrosis factor, TTBS Tween 20/Tris buffer solution
Introduction
Alzheimer’s disease (AD) is purported to account for >50% of the dementia observed in the elderly. It is estimated that by age 65, ~17% of the population will have AD and this percentage will increase progressively to ~47% by 85 years of age. AD neuropathology usually includes both extracellular plaques containing the β-amyloid peptide (Aβ) and intra-cellular neurofibrillary tangles composed of hyperphosphorylated tau protein. In addition, neuronal loss in specific regions of the brain, degeneration of pericytes and leptomeningeal smooth muscle cells, as well as marked gliosis are common in AD (1–3). Currently, treatments for AD include the use of drugs to enhance cognitive function, antioxidants, non-steroidal anti-inflammatory drugs and hormone replacement therapy (4). However, these treatments have produced only a modest delay in disease progression or a small reduction in the risk of developing AD. Thus new prophylactic and/or therapeutic approaches for treating AD are essential.
Recently, several groups demonstrated that immunization of APP transgenic mouse (APP/Tg) models of AD with human Aβ42 peptide blocks the deposition of Aβ in the central nervous system and can at least partially promote the clearance of established plaque material from the brain. A reduction in the development of dystrophic neurites and astrogliosis was also observed in the brains of immunized mice (5). In other studies, immunization of APP/Tg with Aβ42 induced significant antibody production (6) and reduced deposition of Aβ without altering the total level of Aβ in the brain (7). Additional studies demonstrated a correlation between learning deficits and Aβ plaque deposition (8), and immunization with Aβ42 protected APP/Tg mice from the Aβ-associated learning and memory deficits (6). Importantly, both protective and therapeutic vaccinations with fibrillar Aβ42 were effective in transgenic mice and there was no evidence of an autoimmune or inflammatory response in peripheral tissues or the brain (5,6).
Two potential clearance mechanisms for the anti-Aβ antibody induced by Aβ immunotherapy have been proposed. The first is an FcR-mediated clearance within the brain by microglial cells (5,9). The second mechanism involves the binding of Aβ by antibody in the blood rather than in the brain (9–11). This ‘sequestering of Aβ’ in an immune complex might lower blood levels of free Aβ, contributing to a net efflux of Aβ from the brain. Although the mechanism(s) of clearance of Aβ from the brain following immunization with Aβ42 remains to be determined, the remarkable results obtained with Aβ immunotherapy on APP/Tg mice suggest that this novel therapeutic approach is worthy of further investigation.
An effective vaccine typically includes a strong immunogen and a potent adjuvant. In the case of Aβ, a self-antigen, it is necessary to also break the tolerance. To accomplish this task the current immunization protocols rely on powerful adjuvant systemsĐcomplete Freund’s adjuvant (CFA)/incomplete Freund’s adjuvant (IFA) in mice and QS21 in humans (5– 7,12,13). In light of the recent announcement of a halt in the Aβ immunotherapy trials in AD patients because of meningoencephalomyelitis in some patients (12,13), a careful analysis of the role of adjuvants and the immunogen in the overall immune response is warranted. We have analyzed the role of different adjuvants in the generation of B and T cell immune responses to immunization with fibrillar Aβ42. Both the magnitude and the type of the immune response were affected by the choice of adjuvant. Currently it is unclear what caused the meningoencephalomyelitis in the patients; however, the antibody titers did not correlate with the presence or severity of the meningoencephalomyelitis (12,13). Moreover, there has been speculation that the meningoencephalomyelitis observed in the clinical trial may be due to the QS21 adjuvant (13). In our study we observed significant differences in the level of T and B cell immune responses to Aβ immunotherapy depending on whether alum or QS21 was used as the adjuvant.
Methods
Mice
Eight- or 10-week-old female BALB/c mice were purchased from Jackson Laboratories (Bar Harbor, ME) and housed in the animal facility at the Institute for Brain Aging and Dementia, UCI. All animals were housed in a temperature- and light-cycle controlled facility, and their care was under the guidelines of the National Institutes of Health and the Institute for Brain Aging and Dementia, UCI.
Preparation of Aβ peptides
Overlapping peptides for mapping of antigenic determinants of murine B and T cell were synthesized at the Peptide Core Facility at the Institute for Brain Aging and Dementia, UCI. We routinely obtain very high quality Aβ synthetic peptides (>95% purity). Peptides were synthesized by solid-phase Fmoc amino acids substitution, purified by reverse-phase HPLC and underwent rigorous quality control, as we described earlier (14). Aβ peptides 1–42 (Aβ42), 1–40 (Aβ40) and 1–28 (Aβ28), as well as 15 and 17mer overlapping small peptides spanning amino acids 1–15 (Aβ1–15), 6–20 (Aβ6–20), 11–25 (Aβ11–25), 16–30 (Aβ16–30), 21–35 (Aβ21–35) and 26–42 (Aβ26–42) of Aβ were prepared.
Preparation of antigen and immunization
The protocol for preparation of immunogen was adapted from Schenk et al. (5). Briefly, lyophilized Aβ42 was mixed prior to the injections with sterile low-endotoxin deionized water (2 mg in 0.9 ml H2O) and mixed to generate a uniform suspension. The peptide solution was then put through three freeze– thaw cycles and allowed to assemble overnight at 4°C. Subsequently, 100 μl of 10 × PBS (where 1 × PBS is 0.15 M NaCl and 0.01 M sodium phosphate, pH7.5) was added to the peptide (to yield 2 mg/ml) before mixing with the appropriate adjuvant.
In this study we analyzed four different adjuvants: alum (aluminum salt; Sigma, St Louis, MO), TiterMax Gold (TMG; Sigma), QS21 (a purified fraction of Saponin extracted from Quillarja saponaria; Antigenics, New York, NY) or CFA/IFA (Sigma). The Aβ42 peptide was mixed with the adjuvants to bring the concentration of the immunogen to 1 mg/ml. For CFA/IFA and TMG, equal volumes of antigen and adjuvant were emulsified immediately prior to immunization, and each mouse was injected s.c. with 100 μl of this mixture. For QS21 and alum, each mouse was injected with 100 μg antigen and 20 μg of QS21 or 1 mg of alum. Control groups of mice were immunized either with PBS or PBS mixed with adjuvant. After immunization, three boosts were performed at 2-week intervals. All groups of mice (eight to 12 animals per group/two or three experiments) were then bled 9–11 days after each injection and sera was prepared for ELISA. In addition, 10 days after the last boost animals were sacrificed and spleens were excised for T cell proliferation assay.
Detection of the titer of anti-Aβ42 antibody and mapping of B cell epitopes
ELISA methodology was used to detect binding of polyclonal sera to immunogen as well as to small overlapping peptides (mapping of B cell antigenic determinants). Briefly, wells of 96-well plates (Immulon II; Dynatech, Chantilly, VA) were coated with 2.5 μM of Aβ42 or overlapping small peptides in bicarbonate coating buffer (pH 9.7) and incubated overnight at 4°C. They were then washed and blocked with 3% non-fat dry milk in Tween 20/Tris buffer solution (TTBS) (1–2 h at 37°C). After washing of the wells, primary sera from experimental and control mice were added in duplicate at an initial dilution of 1:500 or as indicated and diluted serially in TTBS to 1:64,000. After incubation (overnight at 4°C) and washing, anti-mouse IgG conjugated with horseradish peroxidase (HRP) was added as recommended by manufacturer (Jackson Laboratories). Plates were incubated for 1 h at 37°C, washed and freshly prepared OPD substrate solution (o-phenylenediamine in 0.05 M phosphate citrate buffer, pH 5.0; Sigma) was added to develop reaction. All plates were analyzed spectrophotometrically at 405 nm. Half-maximal antibody titers (HMAT) were determined by dividing the highest OD405 value in the dilution range of each serum sample by two. All HMAT data was determined for each mouse separately.
Antibody subclass determination
To determine the specific isotypes, sera from four mice of the same group were pooled, serially diluted from 1:500 to 1:64,000 and tested in duplicate as described above. To detect mouse IgM, IgG1, IgG2a and IgG2b isotypes, anti-mouse Ig subclass-specific HRP-conjugated secondary antibodies (Zymed, San Francisco, CA) were used. The ratio of IgG1 and IgG2a isotypes was calculated by dividing OD values for IgG1 by OD values for IgG2a.
Peptide competition assay
Plates were coated with Aβ42, washed and blocked as described above. Sera from immunized and control mice were diluted 1:4000, or as indicated, in TTBS buffer with 0.3% non-fat dry milk and mixed with equal volume of small overlapping peptides or with control Aβ42 peptide at different concentrations. After 1 h incubation at 37°C the peptide-treated sera were added in duplicate to Aβ42-coated plates. The plates were analyzed for binding as described above. The percent of inhibition by small peptides as well as by control full-length peptide was calculated considering the binding of sera without competing peptides to Aβ42 as 100%.
Mapping of T cell epitopes
Ten days after the last boost, mice were sacrificed, spleens were excised and single viable cell suspensions were isolated. Erythrocytes were lysed by commercial red blood cell lysis buffer (8.3 g/l ammonium chloride in 0.01 M Tris–HCl, pH 7.5; Sigma) and splenocytes cultures from individual animals were prepared. After washing in media (RPMI 1640), lymphocytes were resuspended to a concentration of 5 × 106 cells/ml in 100 μl of culture media R5 (RPMI 1640 supplemented with 5% FBS, 2 mM glutamine, 1 mM sodium pyruvate, 100 U/ml penicillin and 100 μg/ml streptomycin) (Invitrogen, Carlsbad, CA). To map antigenic determinant(s) of T cells we analyzed the proliferation of individual cultures of splenocytes after stimulation with small overlapping peptides or with Aβ40 peptide. Briefly, to induce lymphocyte proliferation, 100 μl of each peptide solubilized directly in R5 at a concentration of 20 μg/ml was added to triplicate wells with 5 × 105 splenocytes (100 μl). To ensure the viability of cells, we detected proliferation of splenocytes to polyclonal mitogen phytohemagglutinin (2 μg/ml) (Sigma, St Louis, MO). Cells were incubated in flat-bottom 96-well plates (Costar, Corning, NY) at 37°C (5% CO2, 72 h). After incubation, 1 μCi [3H]thymidine (Amersham Pharmacia, Piscataway, NJ) was added to each well and plates were incubated for another 16– 18 h. Cells were harvested using Tomtec Mach III (Tomtec, Hamden, CT) harvester and 3[H]thymidine uptake (c.p.m.) was counted on a MicroBeta 1450 Trilux scintillation counter (Wallac, Turku, Finland). The stimulation index was calculated as we described earlier (15).
Detection of cytokines production by ELISPOT
To detect production of IFN-γ (Th1) or IL-4 (Th2) lymphokines, as well as tumor necrosis factor (TNF)-α (pro-inflammatory) cytokine we used re-stimulated splenocytes from experimental mice. Briefly, 96-well ELISPOT plates (PharMingen, San Diego, CA) were coated with purified anti-mouse IFN-γ, TNF-α or IL-4 mAb as recommended by the manufacturer (PharMingen, San Diego, CA). After overnight incubation and washing with PBS, wells were first blocked with RPMI 1640 supplemented with 10% FBS (2 h) and then washed with PBS. Splenocytes from individual immunized mice were seeded in eight wells (2 × 105/well) and four cultures were re-stimulated by Aβ40 peptide (final concentration 15 μg/ml) and the other four cultures were incubated with R5 only. After incubation for 24 h (37°C, 5% CO2), plates were washed 10 times with deionized water and PBS containing 0.1% Tween 20 (PBST). Then the wells were incubated with biotinylated monoclonal anti-mouse IFN-γ, TNF-α or IL-4 antibody as recommended by manufacturer (PharMingen) and washed with PBST. Avidin– HRP complex was added (100 μl) for 1 h and then wells were washed with PBST and 100 μl of the AEC substrate solution (Sigma) was added to develop the reaction. After 10–15 min the plates were washed with deionized water, air-dried overnight and colored spots were counted using a dissecting microscope (Olympus, Tokyo, Japan). The results were first examined for differences between stimulated and non-stimulated conditions for each of the adjuvants using ANOVA. When significant differences between groups were observed further post-hoc analyses with a Bonferroni–Dunn test for multiple between comparisons was computed to determine specific between-group differences. The ELISPOT data for stimulated and non-stimulated conditions for each adjuvant was then normalized to the percent change and between-group differences examined using ANOVA and a post-hoc Bonferroni–Dunn test when appropriate.
Results
Generation of anti-Aβ42 antibody responses in BALB/c mice
Differences in the humoral responses generated by various adjuvants were analyzed, using fibrillar Aβ42 as a common immunogen. The adjuvants selected, CFA/IFA, QS21, alum and TMG, provide a wide spectrum of the commercially available adjuvants. Of these adjuvants, alum is the only one approved for use in humans; however, QS21 has shown promise in several human clinical trials and has been used in the first AD clinical trial (13). TMG contains a block copolymer (CRL-8300), squalene and a sorbitan monooleate, and is an example of the more modern approach to adjuvants where they are designed to reduce the unwanted side effects of adjuvants such as CFA, but still induce an adequate immune response. As negative controls, mice were injected with PBS and adjuvant only (mock).
Mice were injected s.c. with a standard prime-boost regimen using fibrillar Aβ42 as the antigen in combination with the indicated adjuvants. Serum was collected following each boost, and the humoral immune response was evaluated. All of the adjuvants induced a detectable antibody response to fibrillar Aβ42 after the first and second boosts (Fig. 1A and B). Mice immunized and boosted 3 times with Aβ42 and TMG adjuvant induced the lowest titer of anti-Aβ42 antibody (HMAT = 6700–7100), whereas mice injected with the Aβ42 and QS21 induced the highest titer (HMAT = 25,300–53,200). QS21 adjuvant was even more potent than CFA (HMAT = 21,900 to 28,600) and it was definitely superior to alum (HMAT = 12,400–21,300) in induction of anti-Aβ42 antibody (Fig. 1C). Thus, CFA and QS21 were the most effective adjuvants, followed by alum and then TMG.
Fig. 1.
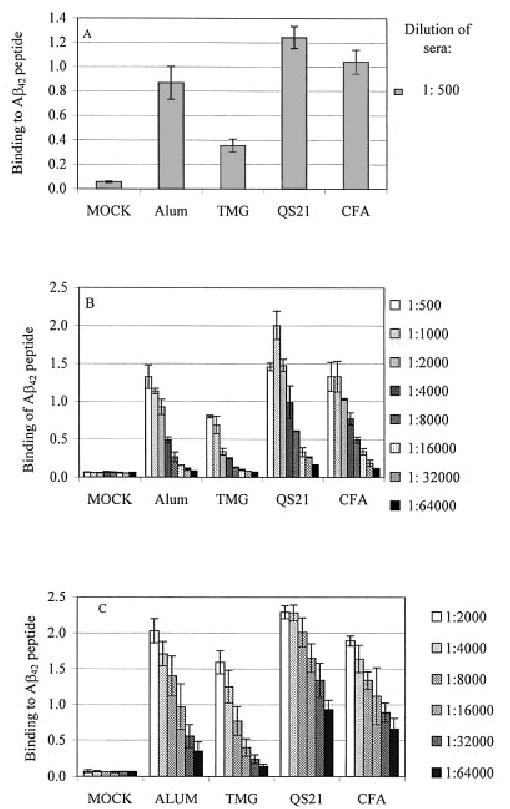
Aβ42-specific immune responses in mice immunized with fibrillar Aβ42 peptide and different adjuvants. Mice were boosted 1 (A), 2 (B) or 3 (C) times with immunogen formulated in specified adjuvants. Control mice (mock) were immunized with PBS + CFA and boosted with PBS + IFA. Pooled sera (A and B, each with n = 12) or individual sera (C, SD represent n = 8) were used in ELISA.
Isotypic profiles of humoral immune responses
The subclass of Ig that is induced after immunization is an indirect measure of the relative contribution of Th2-type cytokines versus Th1-type cytokines (16). More specifically, the production of IgG1-type antibodies is primarily induced by Th2-type cytokines, whereas production of IgG2a-type anti-bodies reflects the involvement of Th1-type cytokines. Given that immunization with Aβ42 can reduce the AD-like pathology in rodents (5–8), we analyzed the effect of different adjuvants on the Th1 and Th2 profile of humoral immune responses by determining the ratio of IgG1 to IgG2a anti-Aβ42 antibody. Serum was collected after the second and third boosts from experimental and mock-immunized animals, and the IgG1 and IgG2a responses were measured by ELISA (dilution of pooled sera = 1:500). From these data we calculated the ratio of IgG1 to IgG2a isotypes (Fig. 2). We observed significant differences in the humoral responses generated against Aβ42 when different adjuvants were used in BALB/c mice. The ratio of IgG1 to IgG2a antibodies in mice immunized with Aβ42 and alum in one particular experiment was >20 (data not shown). CFA, TMG and QS21 shifted the immune response toward a Th1 phenotype. The same results were generated with antisera diluted from 1:500 to 1:64,000 (data not shown). Thus, the choice of adjuvant plays a significant role in the level of involvement of the Th1 and Th2 responses in mice immunized with fibrillar Aβ42.
Fig. 2.
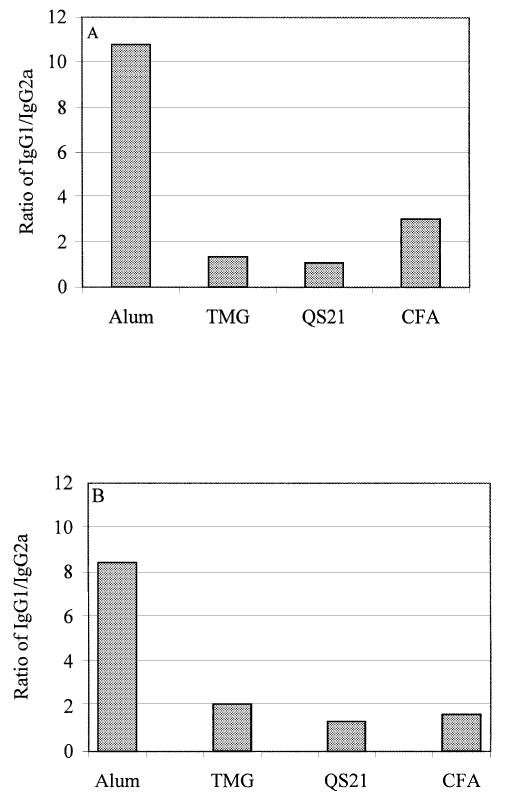
IgG2a (Th1) and IgG1 (Th2) profiles of humoral immune responses in mice immunized with fibrillar Aβ42 formulated in different adjuvants. Sera for this assay were pooled and diluted 1:500 prior to the detection of IgG1 and IgG2a isotypes. Mean values were detected spectrophotometrically and the IgG1:IgG2a ratio was calculated as described in Methods. Sera were collected from mice boosted 2 (A) or 3 (B) times.
Previously it was demonstrated that Aβ immunization might generate different isotypes and subtypes of anti-Aβ42 antibody (11,17–21). In our experiments we also measured production of IgM and IgG2b anti-Aβ antibody, along with IgG1 and IgG2a (see above). All four adjuvants induced a significant level of IgG2b (at a dilution of 1:500 OD = 1.62 ± 0.24, 1.26 ± 0.17, 1.09 ± 0.12 and 1.18 ± 0.13 for QS21, CFA/IFA, TMG and alum respectively) that was comparable to the IgG1 response. The IgM response was approximately equivalent for three of the adjuvants, QS21, CFA/IFA and alum (at a dilution of 1:500 OD = 0.667 ± 0.14, 0.501 ± 0.09 and 0.551 ± 0.11), whereas the TMG-induced response was significantly lower (OD = 0.125 ± 0.14).
Mapping of B cell epitopes
In order to generate the safest and most cost-effective immunotherapy for use in humans, the overall humoral and cellular immune responses need to be investigated. Thus, we decided to identify the B cell antigenic determinant(s) within Aβ42 using six overlapping peptides for epitope mapping, which include five 15mer peptides and one 17mer peptide. Previously, several authors have detected B cell epitope(s) in the N-terminal region of Aβ42 (amino acids 1–28, 1–16 and 1–15), using different small peptides (17,18,20,21). We analyzed the relative binding of antisera (at a dilution of 1:2000) from mice immunized with immunogen formulated in four different adjuvants, using six overlapping peptides. The ELISA plates were coated with small peptides or Aβ42 to detect antibody binding (Fig. 3). All four adjuvants induced significant antibody response to peptide Aβ1–15, which spans the first 15 amino acids from the N-terminal region of Aβ42. However, QS21 was unique in that it also induced a significant humoral response to peptide Aβ6–20, which spans amino acids 6–20. In contrast, alum, CFA and TMG induced only a low-level antibody response against peptide Aβ6–20. Surprisingly, no antibody were detected that recognized the remaining four overlapping linear peptides that span amino acids 11–42 (Fig. 3) even when antisera were used at a dilution 1:500 (data not shown). Even so, overall, the titer of antibody that bound to peptides Aβ1–15 and Aβ6–20 was much lower than the titer of antibody that bound to the full-length Aβ42 (Fig. 1 and data not shown). These results indicate that full-length Aβ42 possesses some conformational antigenic determinant(s) that is not found in the shorter overlapping peptides used for the fine epitope mapping of the B cell epitopes of Aβ42.
Fig. 3.
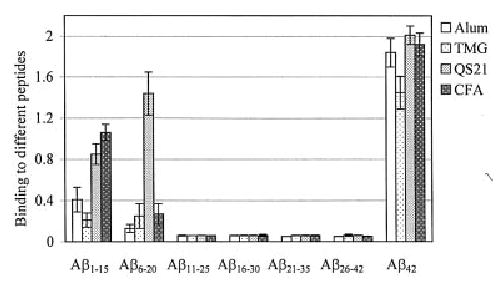
Mapping of B cell epitopes recognized by anti-Aβ42 antibodies generated in mice immunized and boosted 3 times with immunogen and four different adjuvants. The relative binding of 1:2000 diluted antisera to six overlapping peptides and Aβ42 are presented. Two independent experiments were performed (SD represent n = 8).
Because adsorption of a peptide to an ELISA plate may mask some of the peptide’s epitopes, we confirmed the presence of conformational antigenic determinants in Aβ42 by using a competition assay where the peptides are in solution and the Aβ42 is bound to the ELISA plate. The sera from immune mice were diluted and pre-incubated with the mixture of peptides Aβ1–15 + 6–20 or Aβ11–25 + 16–30 + 21–35 + 26–42 (2.5 μM final concentration of each peptide) before reaction with Aβ42 peptide. Pre-incubation with full-length Aβ42 peptide resulted in complete loss of antibody binding to the bound Aβ42, whereas only a mixture of Aβ1–15 and Aβ6–20 peptides was effective at inducing a partial inhibition of binding (Fig. 4A). Notably, a mixture of Aβ11–25 + 16–30 + 21–35 + 26–42 peptides was practically ineffective. To confirm this data, we performed additional inhibition studies with different concentrations of Aβ1–15, Aβ11–25 or Aβ42 peptides (Fig. 4B). Peptide Aβ1–15 at 5 μM blocked binding of serum antibody from all adjuvant groups to Aβ42 antigen up to 70–85%. As expected the peptide Aβ11–25 even at 5 μM was ineffective. Interestingly, Aβ42 was still a very potent inhibitor (inhibition >85%) even at concentrations as low as 0.078 μM, while peptide Aβ1–15 was practically inactive at this concentration. The results from the binding and competition assays indicate for the presence of conformational antigenic determinant(s) in Aβ42 whether attached to the ELISA plate or in solution.
Fig. 4.
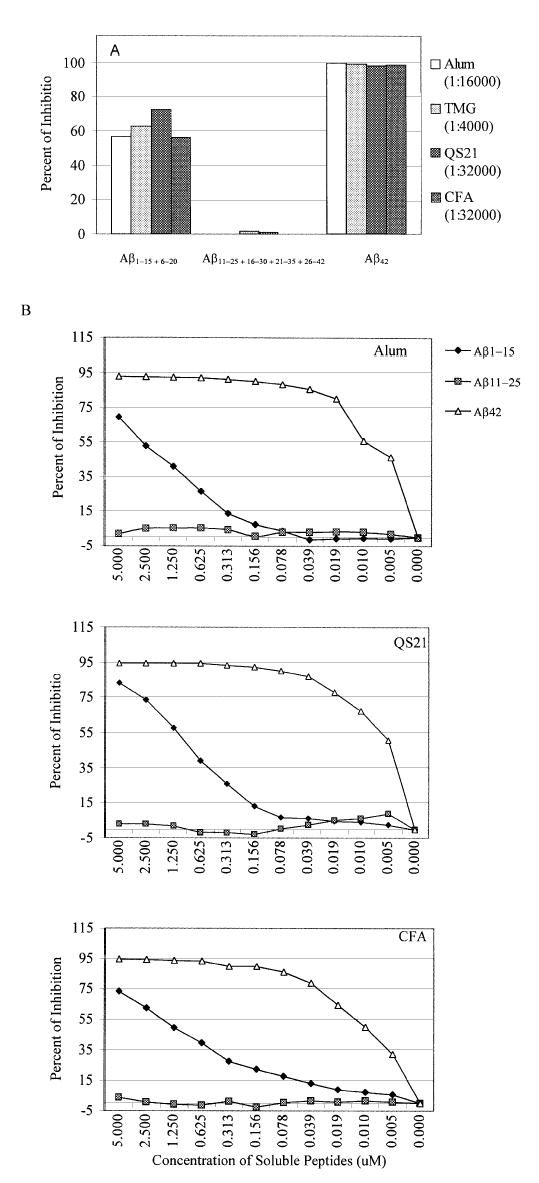
Mapping of B cell epitopes by competition assay. (A) Pooled sera from immune mice (after third boost) were collected and pre-incubated with a mixture of Aβ1–15 + 6–20, Aβ11–25 + 16–30 + 21–35 + 26–42 or Aβ42 peptides (concentration of each peptide 2.5 μM) before binding to Aβ42-coated wells. In order to standardize anti-Aβ42 antibody level we used different dilutions of antisera. (B) Inhibition studies with indicated concentrations of Aβ1–15, Aβ11–25 and Aβ42 peptides. The sera from mice immunized with fibrillar Aβ42 and different adjuvants were diluted 1:4000 (final dilution 1:8000) and pre-incubated with peptides Aβ1–15, Aβ11–25 or Aβ42 before binding to Aβ42-coated wells. The percent of inhibition by small peptides as well as by control full-length peptide was calculated considering the binding of sera without competing peptides to Aβ42 as a 100%. These experiments were repeated twice with similar results.
Mapping of T cell epitopes
To analyze T cell responses in mice immunized with Aβ42 formulated in different adjuvants the splenocytes from individual animals were isolated (after the final boost) and activated in vitro with soluble Aβ40. In these experiments QS21 induced the highest T cell proliferation, followed by alum, TMG and CFA. Splenocytes from mock-immunized animals did not induce significant T cell proliferation to any of the tested peptides (Fig. 5A). Thus, all adjuvants induced antigen-specific T cell responses. The presence of a T cell-reactive antigenic epitope in Aβ42 has not been previously documented. To map Th responses to Aβ42 we stimulated splenocytes with overlapping peptides described above. Only T cells activated in vitro with peptide Aβ6–20 (spans amino acids 6–20) induced a significant T cell proliferation. QS21 induced the greatest T cell response to peptide Aβ6–20, followed by CFA, TMG and alum, which promoted a moderate response to this linear peptide. Importantly, animals from all groups responded significantly stronger to in vitro stimulation by Aβ40 peptide than to stimulation by the short overlapping peptides (Fig. 5A). In separate experiments, we tested T cell proliferation of immune splenocytes (from the CFA/IFA group) after in vitro stimulation by Aβ28 or Aβ40 peptides. Both peptides stimulated T cell proliferation equally well, which indicates that a major T cell antigenic epitope may be expressed within the 6–28 region of Aβ42 (Fig. 5B). Interestingly, in vitro re-stimulation of immune splenocytes with fibrillar Aβ42 demonstrates that the assembled peptide is less effective in T cell activation than either soluble Aβ40 or soluble Aβ28 (data not shown). In sum, smaller overlapping peptides (10–11mers) should be used to map more precisely the T cell epitope of Aβ42.
Fig. 5.
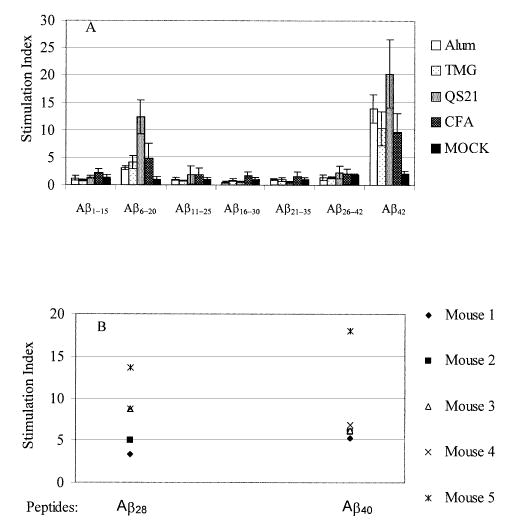
Mapping of T cell epitopes in mice immunized and boosted 3 times with immunogen mixed with alum, TMG, QS21 or CFA/IFA. (A) Proliferation of T cells (stimulation index) was determined after in vitro activation of individual mouse splenocyte cultures (SD represent n = 8) with 10 μg/ml of overlapping peptides or Aβ40. (B) Proliferation of T cells was determined in five mice immunized with Aβ42 formulated in CFA/IFA. The stimulation index was determined after in vitro stimulation of individual mouse splenocyte cultures (n = 5) with 10 μg/ml of Aβ28 or Aβ40.
Production of IFN-γ (Th1) and IL-4 (Th2) lymphokines, and TNF-α (pro-inflammatory) cytokine
To detect Th1 and Th2 types of T cell response we analyzed the production of IFN-γ (Th1) and IL-4 (Th2) lymphokines in splenocytes of individual mice immunized and boosted 3 times with fibrillar Aβ42 formulated in alum, QS21 or CFA (Fig. 6). Results from the ELISPOT test for stimulated versus non-stimulated cells was computed independently for IL-4 and IFN-γ. ANOVA for IL-4 indicated a significant differences between the adjuvants and stimulus condition [F(2,18) = 4.154, P = 0.0328]. Post-hoc analyses with Bonferroni–Dunn test indicated significant differences between the stimulated versus non-stimulated conditions for all three adjuvants (P < 0.05). ANOVA for IFN-γ also indicated a significant difference between the adjuvant and stimulus conditions [F(2,18) = 4.807, P < 0.0212]. Post-hoc analyses with Bonferroni–Dunn test for specific between-group differences indicated a significant differences between the stimulated versus non-stimulated conditions for QS21 and CFA, but not alum. Thus mice from all three experimental groups produced significant amounts of IL-4, a Th2 lymphokine. However, only splenocytes from mice immunized with immunogen and QS21 or CFA produced significant amounts of IFN-γ, a Th1 lymphokine.
Fig. 6.
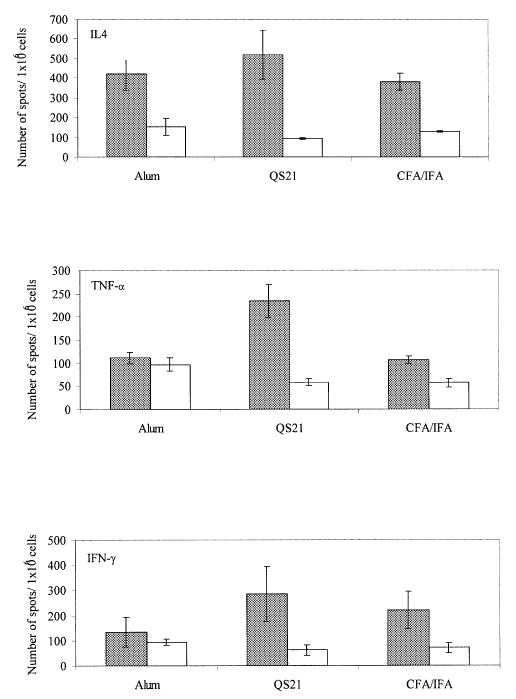
Detection of IFN-γ (Th1) and IL-4 (Th2) lymphokines, and TNF-α pro-inflammatory cytokine in mice immunized and boosted 3 times with Aβ42 formulated in alum, QS21 or CFA/IFA. Splenocytes from immunized mice (2 × 105/well) were incubated with Aβ40 peptide or medium alone in ELISPOT plates as described in Methods. Colored spots were counted after 24 h incubation. ANOVA has been used to compare production of each cytokine in the culture of splenocytes incubated with (experiment) or without (control) Aβ40 peptide (see Methods). Results are shown as mean number of spots/106 cells ± SD for stimulated (closed bars) versus unstimulated (open bars).
TNF-α is expressed by activated macrophages, monocytes, neutrophils, activated lymphocytes and NK cells, and has been suggested to play a pivotal role in regulating the synthesis of other pro-inflammatory cytokines (22). Thus, we analyzed production of this important pro-inflammatory cytokine by splenocytes from mice immunized with Aβ42 mixed with alum, QS21 or CFA (Fig. 6). Results from the ELISPOT test for stimulated versus non-stimulated cells were computed independently for TNF-α. ANOVA for these results indicated a significant differences between the adjuvant and stimulus conditions [F(2,18) = 47.336, P < 0.0001]. Post-hoc analyses with Bonferroni–Dunn test for specific between-group differences indicated a significant differences between the stimulated versus non-stimulated conditions for the QS21 and CFA groups, but not for mice immunized with immunogen mixed with alum. Thus, only splenocytes from the QS21 and CFA groups produced TNF-α after in vitro re-stimulation; however, a significantly higher number of cells produced this pro-inflammatory cytokine after immunization of mice with fibrillar Aβ42 formulated in QS21, rather than in CFA.
Discussion
Active immunization of APP/Tg mice with fibrillar Aβ42, as well as passive transfer (immunization) of anti-Aβ antibody, significantly reduces amyloid plaque deposition, neuritic dystrophy and astrogliosis in APP/Tg mouse models of AD (5–10,17– 19,23,24). All of these reports have implicated antibody in the clearance of Aβ from the brains of APP/Tg mice. In order to achieve a high antibody titer using the Aβ immunotherapy approach, a strong adjuvant will have to be incorporated into the protocol. In the absence of an adjuvant, there is an inadequate T cell response. In this case, the few antigen-presenting cells that actually get loaded with antigen and migrate to the lymph nodes are poorly stimulatory, and occasionally tolerogenic (25,26). In contrast, in the presence of both antigen and adjuvant many antigen-presenting cell precursors will be continuously recruited to the site of immunization, and from these inflamed tissues they will migrate to the draining lymph nodes and stimulate T cells. Thus, T cell stimulation (rapid proliferation of antigen-specific T cell clones) and antibody production is critically dependent on the adjuvant and continuous presence of antigen (27). Since Aβ peptide is a ‘self protein’, it is even more crucial to identify the best adjuvant for Aβ42 immunotherapy.
To investigate the role of adjuvant in the humoral and cell-mediated immune response to Aβ42, four different adjuvants (CFA/IFA, QS21, alum and TMG) were selected and the overall immune responses in BALB/c mice were analyzed. Of these four adjuvants, only alum is at present licensed for use in humans. However, QS21 is currently being tested in several clinical trials, which have provided some evidence that QS21 might be superior to alum and this adjuvant was used in the recently halted Aβ immunotherapy clinical trial (13). The powerful classical adjuvant system, CFA/IFA, was selected as the standard by which to compare the other adjuvants because it has been used in the other Aβ immunotherapy studies in APP/Tg mice (5–7). TMG adjuvant was selected based on our preliminary studies on canines, which have the human Aβ42 sequence and deposit significant amounts of Aβ in the brain.
All four adjuvants induced a substantial anti-Aβ42 antibody response following the first boost, and the antibody titers increased considerably after the second and third boosts with fibrillar Aβ42 (Fig. 1). However, there was a significant difference in the magnitude of the antibody response to Aβ42 immunization with the different adjuvants. The highest titers of antibody were generated in mice immunized and boosted with Aβ42 formulated in QS21 adjuvant followed by CFA/IFA > alum > TMG. Therefore, at least two adjuvants (QS21 and alum) that can be used in humans induced a strong antibody response after Aβ42 immunization.
However, because the type of immune response generated may be critical to the effectiveness and the safety of a potential vaccine, a careful examination of the overall immune response is necessary. For example, the generation of cytotoxic T lymphocytes induced by Th1 cytokines may be highly effective at inhibiting tumor growth, but may also increase the risk of inducing an autoimmune response. On the other hand, Th2 lymphokines promote the production of antibody, suppress the activation of Th1 cells and can inhibit experimental autoimmune encephalomyelitis (28–30). More importantly, Th1 response and the Th1 pro-inflammatory cytokines and chemokines have been linked with autoimmune disease progression and severity (31,32). In the central nervous system, the clinical course of experimental autoimmune encephalomyelitis and inflammation is controlled by the expression levels of CD40 on microglia and macrophages (33). Importantly, CD40 expression is induced by Th1 lymphokine IFN-γ and pro-inflammatory cytokine TNF-α (34). We, therefore, first examined the ratios of IgG1 to IgG2a antibody generated in response to Aβ immunization to provide a relative measure of the contribution of Th2- and Th1-type humoral immune responses (16). All mice immunized with Aβ42 formulated in alum had IgG1:IgG2a ratios >1, indicating that this adjuvant induced primarily Th2-type antibody response against Aβ42. On the other hand, CFA, TMG and QS21 shifted the humoral immune responses toward a Th1 phenotype (Fig. 2). This data correlated with detection of cells producing IFN-γ, IL-4 and TNF-α cytokines by ELISPOT (Fig. 6). We found that mice from groups immunized with alum, QS21 and CFA generated significant numbers of splenocytes that produced IL-4. However, immunization with QS21 and CFA also induced cells producing IFN-γ and TNF-α. These results indicate that both the overall magnitude of the humoral and cellular immune responses are significantly affected by the choice of adjuvant used for Aβ42 immunizations.
Aβ42 can exist both in soluble and fibrillar forms; therefore, antibodies generated against this antigen may recognize different immunogenic structures within Aβ42. Thus, it is important to identify within Aβ42 antigenic determinants for B and T cells in order to design the most effective vaccine. Previously, using various immunization approaches, monoclonal anti-Aβ42 antibody against different regions of human Aβ42 have been generated. At least two mAb raised against the N-terminal region of Aβ were shown to partially inhibit fibrillar assembly of Aβ42 peptide in vitro (35–37). Several authors have also endeavored to map B cell epitopes using antisera from mice immunized with Aβ peptide. For example, Lemere et al. (17,18) described intranasal vaccination (without adjuvant) of PDAPP mice and mentioned that from six overlapping peptides, only Aβ1–15 abolished immunoreactivity of experimental sera with AD plaques in tissue sections. Town et al. (21), after in vitro stimulation of immune splenocytes from C57Bl/6 mice, were able to generate low levels of antibody against Aβ42, which recognized Aβ28, Aβ40 and Aβ42, but not Aβ17–40 or Aβ13–40 peptides. Another group (20), using four peptides spanning amino acids 1–16, 10–20, 20–29 and 29– 40 of Aβ40, demonstrated by competition ELISA that antisera from PS1/Tg mice recognize Aβ1–16 along with Aβ40 and Aβ42 peptides. These data were generated after immunization of mice with Aβ42 formulated in CFA/IFA. However, no study of mapping B cell epitopes has been performed with antisera generated after immunization of mice with Aβ42 formulated in alum, QS21 or TMG adjuvants. Accordingly, we have performed fine epitope mapping with polyclonal anti-Aβ42 sera by utilizing overlapping linear peptides derived from Aβ42. The Aβ1–15 and Aβ6–20 peptides were the only ones recognized by antibody raised against fibrillar Aβ42. However, alum and TMG were less potent for generation of antibody against peptide Aβ1–15 than QS21 and CFA. Surprisingly, only QS21 induced a significant level of antibody against peptide Aβ6–20 (Fig. 3). Since this is the region of the human Aβ42 that differs from the rodent sequence (human Aβ42 differs from rodent Aβ42 by 3 amino acids at positions 5, 10 and 13) (38), at least some of the immune response may be due to recognition of the non-self human residues. However, it was surprising that immune sera collected from BALB/c mice immunized with different adjuvants did not recognize any of the other overlapping peptides spanning the rest of Aβ42 (Figs 3 and 4). More importantly, the titer of antibody that bound to Aβ1–15 and Aβ6–20 pentadecapeptides was much lower than the titer of antibody that bound to full-length Aβ42 (data not shown). These results indicate that the full-length Aβ42 contains some conformational antigenic determinant(s) that is not present in the shorter peptides. These data were further confirmed by competition assays using both the overlapping oligomers and Aβ42 (Fig. 4). These results indicate that murine anti-Aβ42 polyclonal antibody could identify a bigger region of human Aβ42 (spans amino acids 1–16 and 6–20) and recognize a conformational epitope(s) in the Aβ42 antigen. Gaskin et al. (39) previously showed that four human monoclonal IgM antibodies derived from one AD patient also demonstrated conformational antigenic determinant(s) localized in Aβ42. This antigenic epitope(s) was conserved in the amyloid plaques in situ, which indicates that humans may recognize this unique conformational antigenic determinant(s) in Aβ42 as non-self. Our data suggest that immunization of mice may also induce IgG antibodies, which recognize non-self conformational epitope(s). Interestingly, two conformation-specific mouse mAb against Aβ40 have recently been generated (40).
In previous reports of Aβ42 immunotherapy in APP/Tg mice, cellular immune responses were not investigated. Only recently has it been demonstrated that wild-type mice, but not APP/Tg animals, immunized once with Aβ40 and CFA induced T cell proliferation (41). In the present study, we established that wild-type mice, immunized and boosted with Aβ42 mixed with four different adjuvants, were also capable of a robust T cell proliferation in vitro after re-stimulation with soluble Aβ40 (Fig. 5). Importantly, all the adjuvants supported the generation of Th responses in immune splenocytes after in vitro re-stimulation with peptide Aβ6–20. QS21 induced the most potent re-stimulation in the cultured of splenocytes. However, the T cell responses to peptide Aβ6–20 were much weaker than the response to Aβ40. The stronger response to Aβ40 could not be explained by higher concentrations of Aβ40, because we used 6.6 μM (10μg/ml) of peptide Aβ6–20 and 2.4 μM (10 μg/ml) of Aβ40 for in vitro stimulation. Further experiments with smaller peptides (10–11mers) are necessary to map more precisely the major T cell epitope(s), since both Aβ28 and Aβ40 stimulate in vitro T cell proliferation equally well (Fig. 5B).
The generation of an effective vaccine, in general, and against a self-antigen like Aβ, in particular, is a very difficult task. In order to develop a safe and effective immunotherapy to treat AD it is critical to vigorously analyze both the innate and acute immune responses (activation of antigen-presenting cells, mapping of B and T cell epitopes, detection of Th1 and Th2 phenotypes, and production of cytokines and chemokines, measuring of lymphoproliferation as well as cytotoxic T lymphocyte activity) after vaccination. Because data with APP/Tg mice generated by several laboratories were very promising, ELAN/Wyeth Inc. moved to human clinical trials after 2.5 years of preclinical animal studies. Unfortunately, 15 volunteers that received multiple doses of the Aβ42 immunogen formulated in QS21 adjuvant developed central nervous system inflammation (13,42). It is unclear at the present time what was the actual cause of the meningoencephalomyelitis; however, the anti-Aβ antibody titers did not correlate with the severity of symptoms in the AN-1792 clinical trial (13,42). As mentioned above, Th1 responses have been implicated in many autoimmune disorders, whereas Th2-type responses induce IL-4, IL-10 and transforming growth factor-β, attenuate cell-mediated immunity, and have been shown to inhibit autoimmune disease (30). Our data indicate that using QS21 as an adjuvant in mice induces primarily Th1-type, whereas alum induces primarily Th2-type immune responses. Interestingly, intranasal immunization with Aβ40 in PBS also induces primarily a Th2-type humoral immune response in mice and significantly reduces Aβ deposition in the brain of PDAPP mice (19). As the available experimental evidence indicates that the clearance of Aβ from the brain is dependent on anti-Aβ antibody and not on T cell-mediated mechanisms, the induction of the Th2 response may be more beneficial and safer than a Th1 response. In addition, the possibility that other factors besides the Aβ immunogen or the adjuvant may have triggered the encephalomyelitis in the AD patients needs to be investigated. Among them, the level of vascular pathology, changes in blood–brain barrier permeability induced by immunization and the effects of aging on the response to Aβ immunotherapy in AD patients. A complete analysis of the results of the first clinical trial of Aβ immunotherapy and data obtained from other pre-clinical trials in animal models may help to establish the feasibility of an immunotherapeutic approach for treating AD.
Acknowledgments
This work was supported by R01 grants from the NIH (AG20241 to D. H. C. and M. G. A., AG00538 to C. W. C. and D. H. C., and AI 44809 to M.G. A.).
Footnotes
The senior authors (D. H. C. and M. G. A.) contributed equally to this work
Transmitting editor: R. Medzhitov
References
- 1.Price DL, Sisodia SS. Cellular and molecular biology of Alzheimer’s disease and animal models. Annu Rev Med. 1994;45:435. doi: 10.1146/annurev.med.45.1.435. [DOI] [PubMed] [Google Scholar]
- 2.Verbeek MM, de Waal RM, Schipper JJ, Van Nostrand WE. Rapid degeneration of cultured human brain pericytes by amyloid beta protein. J Neurochem. 1997;68:1135. doi: 10.1046/j.1471-4159.1997.68031135.x. [DOI] [PubMed] [Google Scholar]
- 3.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 4.Helmuth L. New Alzheimer’s treatment that may ease the mind. Science. 2002;297:1260. doi: 10.1126/science.297.5585.1260. [DOI] [PubMed] [Google Scholar]
- 5.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse [see Comments] Nature. 1999;400:173. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 6.Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, WIL-cock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature. 2000;408:982. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- 7.Janus C, Chishti MA, Westaway D. Transgenic mouse models of Alzheimer’s disease. Biochim Biophys Acta. 2000;1502:63. doi: 10.1016/s0925-4439(00)00033-8. [DOI] [PubMed] [Google Scholar]
- 8.Chen G, Chen KS, Knox J, Inglis J, Bernard A, Martin SJ, Justice A, McConlogue L, Games D, Freedman SB, Morris RGM. A learning deficit related to age and beta-amyloid plaques in a mouse model of Alzheimer’s disease. Nature. 2000;408:975. doi: 10.1038/35050103. [DOI] [PubMed] [Google Scholar]
- 9.Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 10.DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2001;98:8850. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vehmas AK, Borchelt DR, Price DL, McCarthy D, Wills-Karp M, Peper MJ, Rudow G, Luyinbazi J, Siew LT, Troncoso JC. Beta-amyloid peptide vaccination results in marked changes in serum and brain Abeta levels in APPswe/PS1DeltaE9 mice, as detected by SELDI-TOF-based ProteinChip technology. DNA Cell Biol. 2001;20:713. doi: 10.1089/10445490152717578. [DOI] [PubMed] [Google Scholar]
- 12.Sigurdsson EM, Scholtzova H, Mehta PD, Frangione B, Wisniewski T. Immunization with a nontoxic/nonfibrillar amyloid-beta homologous peptide reduces Alzheimer’s disease-associated pathology in transgenic mice. Am J Pathol. 2001;159:439. doi: 10.1016/s0002-9440(10)61715-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Birmingham K, Frantz S. Set back Alzheimer vaccine studies. Nat Med. 2002;8:199. doi: 10.1038/nm0302-199b. [DOI] [PubMed] [Google Scholar]
- 14.Cribbs DH, Pike CJ, Weinstein SL, Velazquez P, Cotman CW. β-Amyloid stereoisomers exhibit similar structural and biological properties: implications for mechanisms of toxicity. J Biol Chem. 1997;272:7431. doi: 10.1074/jbc.272.11.7431. [DOI] [PubMed] [Google Scholar]
- 15.Agadjanyan MG, Trivedi NN, Kudchodkar S, Bennett M, Levine W, Lin A, Boyer J, Levy D, Ugen KE, Kim JJ, Weiner DB. An HIV type 2 DNA vaccines induces cross-reactive immune responses against HIV type 2 and SIV. AIDS Res Human Retroviruses. 1997;13:1561. doi: 10.1089/aid.1997.13.1561. [DOI] [PubMed] [Google Scholar]
- 16.Finkelman FD, Holmes J, Katona IM, Urban JF, Beckmann MP, Park LS, Schooley KA, Coffman RL, Mossmann TR, Paul WE. Lymphokine control of in vivo immunoglobulin isotype selection. Annu Rev Immunol. 1990;8:303. doi: 10.1146/annurev.iy.08.040190.001511. [DOI] [PubMed] [Google Scholar]
- 17.Lemere CA, Maron R, Selkoe DJ, Weiner HL. Nasal vaccination with beta-amyloid peptide for the treatment of Alzheimer’s disease. DNA Cell Biol. 2001;20:705. doi: 10.1089/10445490152717569. [DOI] [PubMed] [Google Scholar]
- 18.Lemere CA, Maron R, Spooner ET, Grenfell TJ, Mori C, Desai R, Hancock WW, Weiner HL, Selkoe DJ. Nasal Aβ treatment induces anti-Aβ antibody production and decreases cerebral amyloid burden in PD-APP mice. Ann NY Acad Sci. 2000;920:328. doi: 10.1111/j.1749-6632.2000.tb06943.x. [DOI] [PubMed] [Google Scholar]
- 19.Weiner HL, Lemere CA, Maron R, Spooner ET, Grenfell TJ, Mori C, Issazadeh S, Hancock WW, Selkoe DJ. Nasal administration of amyloid-beta peptide decreases cerebral amyloid burden in a mouse model of Alzheimer’s disease. Ann Neurol. 2000;48:567. [PubMed] [Google Scholar]
- 20.Dickey CA, Morgan DG, Kudchodkar S, Weiner DB, Bai Y, Cao C, Gordon MN, Ugen KE. Duration and specificity of humoral immune responses in mice vaccinated with the Alzheimer’s disease-associated beta-amyloid 1±42 peptide. DNA Cell Biol. 2001;20:723. doi: 10.1089/10445490152717587. [DOI] [PubMed] [Google Scholar]
- 21.Town T, Tan J, Sansone N, Obregon D, Klein T, Mullan M. Characterization of murine immunoglobulin G antibodies against human amyloid-b 1. Neurosci Lett. 2001;307:101. doi: 10.1016/s0304-3940(01)01951-6. [DOI] [PubMed] [Google Scholar]
- 22.Arend WP, Dayer JM. Inhibition of the production and effects of interleukin-1 and tumor necrosis factor α in rheumatoid arthritis. Arthritis Rheum. 1995;38:151. doi: 10.1002/art.1780380202. [DOI] [PubMed] [Google Scholar]
- 23.DeMattos RB, Bales KR, Parsadanian M, O’Dell MA, Foss EM, Paul SM, Holtzman DM. Plaque-associated disruption of CSF and plasma amyloid-b (Ab) equilibrium in a mouse model of Alzheimer’s disease. J Neurochem. 2002;81:229. doi: 10.1046/j.1471-4159.2002.00889.x. [DOI] [PubMed] [Google Scholar]
- 24.Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Immunization reverse memory deficits without reducing brain Ab burden in Alzheimer’s disease model [Advanced online publication] Nat Neurosci. 2002;5:452. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- 25.Suss G, Shortman K. A subclass of dendritic cells kills CD4 T cells via Fas/Fas-ligand-induced apoptosis. J Exp Med. 1996;183:1789. doi: 10.1084/jem.183.4.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurts C, Kosaka H, Carbone FR, Miller JFAP, William R. Heath, class I-restricted cross-presentation of exogenous self-antigens leads to deletion of autoreactive CD8+ T cells. J Exp Med. 1997;186:239. doi: 10.1084/jem.186.2.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lanzavecchia A, Sallusto F. Dynamics of T lymphocyte responses: intermediates, effectors, and memory cells. Science. 2000;290:92. doi: 10.1126/science.290.5489.92. [DOI] [PubMed] [Google Scholar]
- 28.Aharoni R, Teitelbaum D, Leitner O, Meshorer A, Sela M, Arnon R. Specific Th2 cells accumulate in the central nervous system of mice protected against experimental autoimmune encephalomyelitis by copolymer 1. Proc Natl Acad Sci USA. 2000;97:11472. doi: 10.1073/pnas.97.21.11472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shaw MK, Lorens JB, Dhawan A, DalCanto R, Tse HY, Tran AB, Bonpane C, Eswaran SL, Brocke S, Sarvetnick N, Steinman L, Nolan GP, Fathman CG. Local delivery of interleukin 4 by retrovirus-transduced T lymphocytes ameliorates experimental autoimmune encephalomyelitis. J Exp Med. 1997;185:1711. doi: 10.1084/jem.185.9.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Shea JJ, Ma A, Lipsky P. Cytokines and autoimmunity. Nat Rev Immuol. 2001;2:37. doi: 10.1038/nri702. [DOI] [PubMed] [Google Scholar]
- 31.Smeltz RB, Swanborg RH. Concordance and contradiction concerning cytokines and chemokines in experimental demyelinating disease. J Neurosci Res. 1998;51:147. doi: 10.1002/(SICI)1097-4547(19980115)51:2<147::AID-JNR3>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 32.Swanborg RH. Experimental autoimmune encephalomyelitis in the rat: lessons in T-cell immunology and autoreactivity. Immunol Rev. 2001;184:129. doi: 10.1034/j.1600-065x.2001.1840112.x. [DOI] [PubMed] [Google Scholar]
- 33.Becher B, Durell BG, Miga AV, Hickey WF, Noelle RJ. The clinical course of experimental autoimmune encephalomyelitis and inflammation is controlled by the expression of CD40 within the central nervous system. J Exp Med. 2001;193:967. doi: 10.1084/jem.193.8.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen VT, Benveniste EN. Critical role of tumor necrosis factor-alpha and NF-kappa B in interferon-gamma-induced CD40 expression in microglia/macrophages. J Biol Chem. 2002;277:13796. doi: 10.1074/jbc.M111906200. [DOI] [PubMed] [Google Scholar]
- 35.Solomon B, Koppel R, Hanan E, Katzav T. Monoclonal antibodies inhibit in vitro fibrillar aggregation of the Alzheimer beta-amyloid peptide. Proc Natl Acad Sci USA. 1996;93:452. doi: 10.1073/pnas.93.1.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Solomon B, Koppel R, Frankel D, Hanan-Aharon E. Disaggregation of Alzheimer beta-amyloid by site-directed mAb. Proc Natl Acad Sci USA. 1997;94:4109. doi: 10.1073/pnas.94.8.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frenkel D, Balass M, Katchalski-Katzir E, Solomon B. High affinity binding of monoclonal antibodies to the sequential epitope EFRH of beta-amyloid peptide is essential for modulation of fibrillar aggregation. J Neuroimmunol. 1999;95:136. doi: 10.1016/s0165-5728(99)00003-x. [DOI] [PubMed] [Google Scholar]
- 38.Hilbich C, Kisters-Woike B, Reed J, Masters CL, Beyreuther K. Human and rodent sequence analogs of Alzheimer’s amyloid beta A4 share similar properties and can be solubilized in buffers of pH 7.4. Eur J Biochem. 1991;201:61. doi: 10.1111/j.1432-1033.1991.tb16256.x. [DOI] [PubMed] [Google Scholar]
- 39.Gaskin F, Finley J, Fang Q, Xu S, Fu SM. Human antibodies reactive with beta-amyloid protein in Alzheimer’s disease. J Exp Med. 1993;177:1181. doi: 10.1084/jem.177.4.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Nuallain B, Wetzel R. Conformational Abs recognizing a generic amyloid fibril epitope. Proc Natl Acad Sci USA. 2002;99:1485. doi: 10.1073/pnas.022662599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Monsonego A, Maron R, Zota V, Selkoe DJ, Weiner HL. Immune hyporespobnsivness to amyloid b-peptide in amyloid precursor protein transgenic mice: implications for the pathogenesis and treatment of Alzheimer’s disease. Proc Natl Acad Sci USA. 2001;98:10273. doi: 10.1073/pnas.191118298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steinberg D. Companies halt first Alzheimer’s vaccine trial. Scientist. 2002;16:22. [Google Scholar]