

Abstract
The diffuse, extensive infiltration of malignant gliomas into the surrounding normal brain is believed to rely on modification of the proteolysis of extracellular matrix components. Our previous results clearly demonstrate that uPA, uPAR and MMP-9 concentrations increase significantly during tumor progression and that tumor growth can be inhibited with antisense stable clones of these molecules. Because antisense-mediated gene silencing does not completely inhibit the translation of target mRNA and high concentrations of antisense molecules are required to achieve gene silencing, we used the RNAi approach to silence uPA, uPAR and MMP-9 in this study. We examined a cytomegalovirus (CMV) promoter-driven DNA-template approach to induce hairpin RNA (hpRNA)-triggered RNAi to inhibit uPA, uPAR and MMP-9 gene expression with a single construct. uPAR protein levels and enzymatic activity of uPA and MMP-9 were found to significantly decrease in cells transfected with a plasmid expressing hairpin siRNA for uPAR, uPA and MMP-9. pU2M-transfected SNB19 cells significantly decreased uPA, uPAR and MMP-9 expression compared to mock and EV/SV-transfected cells, determined by immunohistochemical analysis. Furthermore, the effect of the single constructs for these molecules was a specific inhibition of their respective protein levels, as demonstrated by immunohistochemical analysis. After transfection with a plasmid vector expressing dsRNA for uPA, uPAR and MMP-9, glioma-cell invasion was retarded compared with mock and EV/SV-treated groups, demonstrated by Matrigel-invasion assay and spheroid-invasion assay. Downregulation of uPA, uPAR and MMP-9 using RNAi inhibited angiogenesis in an in vitro (co-culture) model. Direct intratumoral injections of plasmid DNA expressing hpRNA for uPA, uPAR and MMP-9 significantly regressed pre-established intracranial tumors in nude mice. In addition, cells treated with RNAi for uPAR, uPA and MMP-9 showed reduced pERK levels compared with parental and EV/SV-treated SNB19 cells. Our results support the therapeutic potential of RNAi as a method for gene therapy in treating gliomas.
Keywords: gliomas, invasion, RNA interference, angiogenesis
INTRODUCTION
Tumor progression involves modulation of tumor-cell adhesion during migration and degradation of the extracellular matrix (ECM) during invasion. An intricate balance of proteases, their activators and their inhibitors regulates both these processes during tumor invasion. Three classes of ECM-degrading proteinases are the serine proteinases, metalloproteases and cysteine proteinases. A large body of evidence assigns a key role in tumor progression and invasion to uPA by virtue of its ability to initiate a cascade of proteases that can degrade most matrix and basement membrane components and interfere with cell–cell and cell–matrix interactions (Andreasen et al., 1997; Carroll et al., 1999). uPA, bound to its cell surface receptor uPAR, is the principal participant of ECM degradation, as demonstrated by a several-fold increase in plasminogen activation (Alonso et al., 1996; Ellis et al., 1991). uPA also activates several growth factors after degradation of ECM components (Naldini et al., 1995; Werb et al., 1996). The absence of uPA negatively affects the progression of chemically induced neoplasms in mice (Shapiro et al., 1996). The inoculation of metastatic Lewis lung carcinoma cells into plasminogen-deficient mice resulted in the formation of either smaller or less hemorrhagic tumors than those observed in control, wild-type mice (Bugge et al., 1997). Regulation of either uPA or uPAR is associated with increased aggressiveness in diverse tumor types (Blasi, 1999). In murine tumor models, either decreased expression of uPAR or administration of uPAR antagonists marked inhibits the metastatic ability of cancer cells (Crowley et al., 1993), primary tumor growth (Min et al., 1996) and downregulation of uPAR, which leads to dormancy of carcinoma cells in vivo (Yu et al., 1997). Targeted deletion of the gene encoding uPAR in mice significantly reduces the uPA-mediated activation of plasminogen in peritoneal macrophages (Dewerchin et al., 1996), indicating that the principal role of uPAR is to localize plasminogen activation to the cell surface (Ellis et al., 1992). MMPs are also implicated in cancer invasion through ECM degradation and the processing of a range of molecules, including growth factors and cytokines. These processes are involved in promoting all aspects of tumor growth, such as cell proliferation, adhesion and dispersion, migration, differentiation, angiogenesis, apoptosis and host-defense evasion (Egeblad and Werb, 2002).
Malignant gliomas are characterized by rapid cell proliferation, a high level of invasiveness into the surrounding brain and increased vascularity. In humans, diffuse astrocytomas, glioblastoma multiforme in particular, invade the brain preferentially along white matter fiber tracts (Giese and Westphal, 1996; Pedersen et al., 1995), resulting in rapid infiltrative growth that prevents successful surgical resection. This invasive behavior seems to depend in part on several proteolytic enzymes. Our previous studies and those of others have reported significantly increased levels of uPA and uPAR in human glioblastoma tissue samples compared to low-grade and normal brain tissue samples (Gladson et al., 1995; Yamamoto et al., 1994a; Yamamoto et al., 1994b). Downregulation of uPAR expression using antisense and gene-therapy approaches has resulted in increased survival in animal models. The antisense, stable, uPAR glioblastoma clones result in an inability of the cells to generate tumors when transplanted into nude mice (Go et al., 1997) and reduced invasiveness in in vitro models (Mohanam et al., 1997). The infection of glioblastoma cells with antisense uPAR adenoviral vectors in cultures resulted in tumor growth in vivo (Mohan et al., 1999). Adenovirus-mediated downregulation of bicistronic constructs of uPA and uPAR expression inhibited glioma cell migration, invasion and tumor-induced capillary formation (Gondi et al., 2003). Antisense stable clones for uPA significantly reduced invasiveness and tumor formation in in vitro and in vivo models (Mohanam et al., 2001). In another study, stably transfected glioma cells expressing the amino terminal fragment (ATF) domain (residues 1–46) of uPA, which binds uPAR, did not form tumors in nude mice (Mohanam et al., 2002). Several studies report significantly increased levels of MMP-9 during the progression of human gliomas (Forsyth et al., 1999; Raithatha et al., 2000; Rao et al., 1993; Rao et al., 1996).
We have demonstrated that glioblastoma cells expressing antisense MMP-9 exhibited decreased migration and invasion in vitro and did not form tumors when injected intracranially in nude mice (Kondraganti et al., 2000). Intracranial injections of glioblastoma cells (SNB19) infected with an adenovirus that expresses antisense MMP-9 did not produce tumors in nude mice (Lakka et al., 2002a). A bicistronic Ad-construct with antisense uPAR and MMP-9 had more effect with regard to inhibition of invasion, angiogenesis and tumor growth in vivo (Lakka et al., 2003). Our recent studies demonstrat that a siRNA bicistronic construct for cathepsin B and MMP-9 completely repressed pre-established intracranial tumors (Lakka et al., 2004).
Together, these studies indicate the biological significance of uPAR, uPA and MMP-9 in glioma invasion and tumor growth. The stability/specificity of the delivery approach of Ad-vector and antisense technology to intracellular target RNA seems to be a crucial limiting factor in exerting an inhibitory effect on the targeted molecule. The siRNA duplex was significantly more stable in cells than the cognate, single-stranded sense of anti-sense RNA with transcription under the control of identical promoter in each case (Miyagishi and Taira, 2002). RNAi was shown to be superior to antisense because only a few molecules of dsRNA per cell can trigger gene silencing (Zamore, 2001). The activity of siRNA is high, even at low concentrations, and without apparent toxicity. In a recent study, siRNA was quantitatively more efficient than antisense oligonucleotides at suppressing cotransfected GFP expression in vitro and in vivo (Bertrand et al., 2002).
Therefore, RNAi might provide a more powerful strategy for the regression of pre-established intracranial tumor growth and inhibition of invasion/angiogenesis through a nucleic acid drug or a gene therapy approach. We show here that the siRNA-mediated suppression of uPAR, uPA and MMP-9 results in significant regression of pre-established intracranial tumor growth as well as inhibition of invasion and angiogenesis. Our results demonstrate that a single vector capable of expressing siRNA for three genes can block expression of the targeted proteases/receptor and indicate a potentially useful method for developing highly specific, siRNA-based, gene-silencing therapeutics for the treatment of glioma.
OBJECTIVES
The present study was conducted to determine the effectiveness of downregulating multiple targets in human glioma cells to control invasion, growth and angiogenesis. We hypothesized that the simultaneous RNAi-mediated targeting of uPAR, uPA and MMP-9, using a single CMV promoter, would retard tumor-cell invasion, growth and angiogenesis, and that the effects would be synergistic rather than additive. The study was conducted in three stages. Stage 1 dealt with the development of plasmid-based, CMV-driven, RNAi vectors to target uPAR, uPA and MMP-9 either singly or together, and the determination of their ability to effectively downregulate the target molecules. Stage 2 dealt with the in vitro behavior of SNB 19 human glioblastoma cell with respect to invasion and angiogenesis. Stage 3 dealt with the in vivo ability of the RNAi vectors to retard the growth of pre-established intracranial tumors in nude mice.
METHODS
Constructing a vector expressing siRNA for uPAR, uPA and MMP-9
pcDNA3 was used for the construction of a vector expressing siRNA for uPAR, uPA and MMP-9 downstream of the cytomegalovirus (CMV) promoter. The uPAR sequence from +77 to +98 was used as the target sequence and for convenience a self-complimentary oligo was used. The uPAR sequence 21 bases long with a 9 base loop region with BamHI sites incorporated at the ends (5′gatcctacagcagtggagagcgattatatataataatcgctctccactgctgtag3′) was used. The oligo was self-annealed in 6× SSC using standard protocols and ligated into the BamHI site of a pcDNA3 vector plasmid. Similarly, uPA complimentary sequence from +346 to +367 (5′agcttgagagccctgctggcgcgccatatataatggcgcgccagcagggctctca3′) with HindIII sites incorporated at the ends was ligated into the HindIII site and MMP-9 +360 to +381 (5′aattcaagtggcaccaccacaacaatatataattgttgtggtggtgccacttg3′) was ligated into the EcoRI site of the vector containing the siRNA sequence for uPAR and uPA. This finally resulted in a siRNA expression plasmid for uPAR, uPA and MMP-9 designated pU2M (Fig. 1). Single siRNA expression vectors for uPAR (puPAR), uPA (puPA) and MMP-9 (pMMP-9) were also constructed. The orientation of the insert in either the single or tricistronic construct was not crucial because the oligos were self-complimentary and had bilateral symmetry. BGH poly A terminator served as a stop signal for RNA synthesis for all four constructs.
Fig. 1. Schematic of the possible formation of hpRNA molecules from a single, CMV-driven, tri-inverted repeat construct for uPAR, uPA and MMP-9.
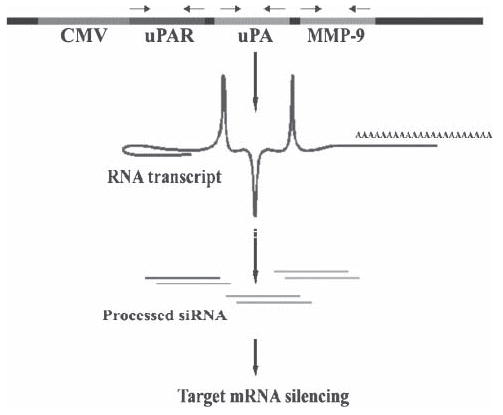
The powerful CMV viral promoter drives the formation of an RNA molecule that possesses self-complementary inverted repeats for uPA, uPAR and MMP-9.
Cell culture and transfection
The SNB19 (or SNB19 GFP) cell line, established from a human, high-grade glioma, was used for this study. Cells were grown in Dulbecco’s modified Eagle medium/F12 media (1:1, v:v) supplemented with 10% fetal calf serum in a humidified atmosphere containing 5% CO2 at 37°C. SNB19 and SNB19 GFP cells were transfected with plasmid constructs empty vector (EV), scrambled vector (SV), puPAR, puPA, pMMP-9 and pU2M using lipofectamine (Life Technologies) according to the manufacturer’s instructions.
Western blot analysis
SNB19 cells were transfected with EV/SV, puPAR, puPA, pMMP-9 and pU2M. After 48 hours, cells were collected and total cell lysates prepared in extraction buffer containing 0.1 M Tris (pH 7.5), 1% Triton-X114, 10 mM EDTA, aprotinin and phenylmethylsulfonyl fluoride as described previously (Mohan et al., 1999). The extracts were incubated at 37°C for 5 minutes and centrifuged to separate the lower (detergent) phase that contains mainly hydrophobic membrane proteins including the glycosylphosphatidylinositol-anchored uPAR. Subsequently, 20 μg of protein from these samples were separated under non-reducing conditions by 12% SDS-PAGE and transferred to nitrocellulose membranes (Schleicher & Schuell). The membranes were probed with polyclonal antibodies against uPAR (#399 American Diagnostics) and secondary antibodies (anti-rabbit-horseradish peroxidase) as required, and developed according to enhanced chemiluminescence protocol (Amersham). The membranes were stripped and probed with monoclonal antibodies for GAPDH as described above to verify that similar amounts of protein was loaded in each lane.
Gelatin zymography
The enzymatic activity and molecular weight of MMPs was determined by gelatin zymography in the conditioned medium of SNB19 cells transfected with EV/SV, puPAR, puPA, pMMP-9 and pU2M, as described previously (Lakka et al., 2003). To prepare conditioned medium, cells were washed with serum-free medium and resupplied with fresh, serum-free medium. After 24 hours, the conditioned medium was harvested, centrifuged to remove cellular debris and protein concentrations determined. Equal amounts of protein were electrophoresed under non-reducing conditions through 8% SDS-PAGE containing 0.1% gelatin. Gels were washed in 2.5% Triton X-100 and incubated overnight in Tris–CaCl2 buffer. The gels were then stained with 0.1% amido black for 1 hour and destained in 20% methanol and 10% acetic acid. The clear bands represented gelatinase activity.
Fibrin zymography
The enzymatic activity and molecular weight of electrophoretically separated forms of uPA were determined in conditioned medium of SNB19 cells transfected with EV/SV puPAR, puPA, pMMP-9 and pU2M by SDS-PAGE as described previously (Mohanam et al., 2001). Briefly the SDS-PAGE gel contains acrylamide to which purified plasminogen and fibrinogen were substrates before polymerization. After polymerization, equal amounts of proteins in the samples were electrophoresed and the gel was washed and stained as described previously (Mohanam et al., 2002).
Immunohistochemical analysis
SNB19 cells (1×104 well−1) were seeded in 8-well chamber slides, incubated for 24 hours and transfected with EV/SV, puPAR, puPA, pMMP-9 and pU2M. After 72 hours, cells were fixed in 3.7% formaldehyde and incubated with 1% bovine serum albumin in PBS at room temperature for 1 hour for blocking. The slides were then incubated with PBS and 1% bovine serum albumin, with mouse IgG anti-uPAR (R&D Systems), mouse IgG anti-uPA (Calbiochem), or rabbit IgG anti-MMP-9 (Biomedia) at a concentration of 1:500 and incubated at room temperature for 1 hour as described previously (Lakka et al., 2004). The slides were subsequently washed three times with PBS to remove excess primary antibody and then incubated with either anti-mouse HRP conjugate or anti-rabbit HRP conjugate IgG (1:500 dilution) for 1 hour at room temperature. The slides were washed three times, DAB peroxidase substrate (Sigma #D-4168) was added, covered with glass cover slips, and observed in a bright-field microscope.
Matrigel invasion assay
Invasion of glioma cells in vitro was measured by the invasion of cells through Matrigel-coated (Collaborative Research Inc.) transwell inserts (Costar). Briefly, transwell inserts with 8 μm pores were coated with a final concentration of 1 mg ml−1 of Matrigel, SNB19 cells transfected with EV/SV, puPAR, puPA, pMMP-9 and pU2M or parental cells alone were trypsinized and 200 μl aliquots of cell suspension (1×106 cells ml−1) were added in triplicate wells. After a 24-hour incubation, cells that passed through the filter into the lower wells were quantified as described earlier (Mohan et al., 1999; Mohanam et al., 1997) and expressed as a percentage of the sum of cells in the upper and lower wells. Cells on the lower side of the membrane were fixed, stained with Hema-3 and photographed.
In vitro angiogenic assay
SNB19 cells (2×104 well−1) were seeded in 8-well chamber slides and transfected with EV/SV, puPA, puPAR, pMMP-9 and pU2M or with parental cells above. After a 24-hour incubation period, the conditioned media was removed and added to a monolayer of 4×104 human dermal endothelial cells in 8-well chamber slides, and the human dermal endothelial cells were allowed to grow for 72 hours. Cells were then fixed in 3.7% formaldehyde, blocked with 2% bovine serum albumin and the endothelial cells were incubated with factor VIII primary antibody (DAKO Corp.). The cells were then washed with PBS and incubated with a FITC-conjugated secondary antibody for 1 hour. The slides were washed and the formation of capillary-like structures was observed using a fluorescent microscope. Similar slides were H&E stained to visualize network formation. Image Pro software was used to quantify angiogenesis. The degree of angiogenesis was measured by the following method: number of branch points and the total number of branches per point were counted, with the product indicating the degree of angiogenesis.
In vitro spheroid invasion assay
Multicellular SNB19 spheroids were cultured in 6-well ultra low attachment plates. Briefly, 3×106 cells were suspended in 10 ml of medium, seeded onto the plates and cultured until spheroids formed. Spheroids of 100–200 μm diameter were selected and transfected with EV/SV, puPAR, puPA, pMMP-9 and pU2M or with parental spheroids alone. Three days after infection, tumor spheroids were stained with the fluorescent dye DiI and confronted with fetal rat brain aggregates stained with DiO. The progressive destruction of fetal rat brain aggregates and invasion of SNB19 spheroids were observed by confocal laser scanning microscopy and photographed as described previously (Lakka et al., 2003; Lakka et al., 2004). The remaining volume of the rat brain aggregates at 24, 48 and 72 hours by these glioma spheroids with these constructs were quantified using image analysis software as described previously (Gondi et al., 2003; Gondi et al., 2004; Lakka et al., 2003).
Intracranial tumor growth inhibition
For the intracerebral tumor model, 2×106 SNB19 GFP cells were injected intracerebrally into nude mice. Tumors were allowed to grow for 10 days. At this time, animals were randomized into seven groups and EV/SV, puPAR, puPA, pMMP-9 and pU2M (150 μg of each construct were injected into the brain using Alzet mini pumps at the rate of 0.25 μl h−1 (six mice in each group). Five weeks after tumor inoculation, mice were sacrificed by cardiac perfusion with 3.5% formaldehyde in PBS. Their brains were removed and placed in 4% paraformaldehyde for 24 hours, paraffin embedded and sectioned. The sections were screened for GFP fluorescence to examine tumor growth under a fluorescent microscope. The sections were reviewed blindly and scored semiquantitatively for tumor size. The average tumor area in each section was used to calculate tumor volume and compared between controls and treated groups.
RESULTS
Effect siRNA constructs on uPAR protein, and uPA and MMP-9 enzymatic activity in SNB19 glioblastoma cells
To simultaneously inhibit three endogenous genes with hairpin siRNA, we constructed a vector (pU2M) expressing siRNA for uPAR (77–98 bases of human uPAR in RNA), uPA (346–367 bases of human uPA in RNA) and MMP-9 (360–381 bases of human MMP-9 in RNA) under the control of the CMV promoter (Fig. 1). Western blot analysis was performed to examine the effect of empty vector/scrambled vector (EV/SV), puPAR, puPA, MMP-9 and pU2M transfection on uPAR protein concentrations in SNB19 cells. The uPAR protein band was present in SNB19 cells transfected with EV/SV, puPA and pMMP-9, whereas it was reduced significantly in puPAR- and pU2M-treated cells (Fig. 2A). The effect of the tricistronic construct (pU2M) was greater than the puPAR (Fig. 2A). The levels of GAPDH determined that equal quantities of protein were loaded in the gel (Fig. 2A). Fibrin zymography was performed to examine the effect of EV/SV, puPAR, puPA, pMMP-9 and pU2M-treated SNB 19 cells on uPA enzymatic activity. Gelatin zymography was performed to determine the effect of these constructs on the levels of MMP-9 in SNB19 cells. MMP-9 levels were significantly reduced in SNB19 cells treated with puPAR, pMMP-9 and pU2M compared to parental, EV/SV- and puPA-treated cells (Fig. 2B). Interestingly, MMP-2 levels were also downregulated in pU2M-treated cells. Care was taken to load equal quantity of proteins. (Fig. 2B). The uPA enzymatic activity (MR 55 000) was reduced significantly in puPA- and pU2M-treated cells compared with the parental, EV/SV-, puPAR-and pMMP-9-treated groups (Fig. 2C)
Fig. 2. Western blotting, fibrin/gelatin zymography and immunohistochemical analysis of uPA and MMP-9.
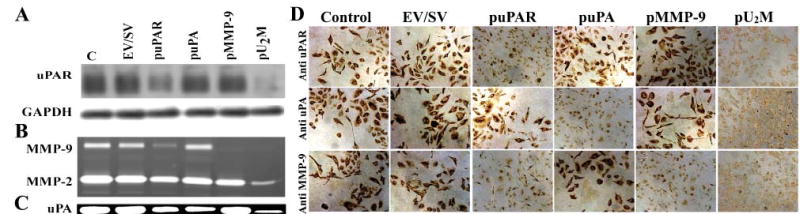
SNB19 cells were either mock transfected or transfected with an empty vector/scrambled vector (EV/SV) and vectors encoding siRNA uPAR (puPAR), uPA (puPA), MMP-9 (pMMP-9) and a combination of the three together (pU2M). After a 3-day-incubation period, total cell lysates were prepared in extraction buffer and 50 μg of protein from these samples were separated by 12% non-reducing SDS-PAGE and immunoblotted with anti-uPAR antibody (A). GAPDH was immunoprobed simultaneously as a loading control. Conditioned medium was collected from these samples (20 μg) and gelatin and fibrin zymography performed to detect MMP-9 (B) and uPA activity (C). (D) SNB19 cells (1×104) were seeded onto Lab-Tek II chamber slides and either mock transfected or transfected with EV/SV and vectors encoding siRNA puPA, puPAR and pMMP-9 either singly or together (pU2M). After 72 hours, cells were fixed, washed for 1 hour with blocking buffer and stained for uPAR, uPA and MMP-9 expression using specific antibodies for uPA, uPAR and MMP-9.
The effect of the tricistronic construct was more pronounced than that of the single siRNA constructs for these molecules. Determined by immunohistochemical analysis, puPAR, puPA, pMMP-9 and pU2M transfection decreased uPAR, uPA and MMP-9 concentrations in SNB19 cells. Figure 2D shows the protein levels of uPAR, uPA and MMP-9 in parental, EV/SV-, puPAR-, puPA-, pMMP-9- and pU2M-transfected cells using specific antibodies for uPAR, uPA and MMP-9. The respective intensities of uPAR, uPA and MMP-9 were very high in parental cells and in cells transfected with EV/SV. By contrast, uPAR intensity decreased in SNB19 cells transfected with puPAR and pU2M. puPA and pU2M transfection significantly decreased the intensity of uPA protein compared with parental, EV/SV-, puPAR- and pMMP-9-transfected cells. Further, MMP-9 protein concentration decreased significantly in pMMP-9- and pU2M-transfected cells compared with parental, EV/SV, puPA- and puPAR-transfected cells. These results demonstrate that the effect of the single constructs is molecule-specific and that the effect of the tricistronic construct is much more pronounced than that the single constructs alone.
puPAR, puPA, pMMP-9 and pU2M inhibit tumor-induced capillary network formation
The growth of a glial tumor depends on the induction of new capillary blood vessels that are necessary to support the developing tumor mass. We used a co-culture system in which microvascular endothelial cells were induced by conditioned media from glial cells to form capillary-like structures to examine the effect of RNAi-mediated suppression of uPAR, uPA and MMP-9. We performed immunohistochemical analysis using factor VIII antigen to evaluate tumor-induced vessel formation in an in vitro co-culture system and performed H&E staining. Endothelial cells form capillary-like structures in the presence of conditioned media from SNB19 parental and EV/SV-transfected cells (Fig. 3A). By contrast, transfection of SNB19 cells with vectors expressing siRNA for uPA, uPAR and MMP-9 either individually or in combination partially or completely inhibited tumor-induced microvessel formation (Fig. 3A). New branch points and/or an increase in the number of branches were not detected in pU2M-transfected cells compared with EV/SV-treated cells (Fig. 3A). Furthermore, compared with EV/SV-treated cells, the formation of capillary-like structures was inhibited by ~55% in puPAR-treated cultures, ~36% in puPA-treated cultures and ~60% in pMMP-9-treated cultures (Fig. 3B).
Fig. 3. Inhibition of glioma angiogenesis and invasion by siRNA constructs.
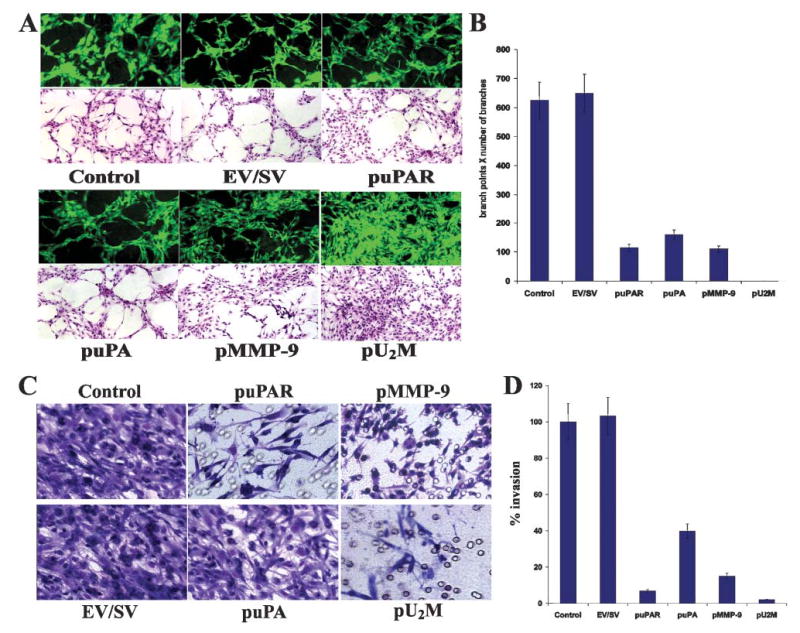
SNB19 cells (2×104) were seeded onto 8-well-chamber slides and transfected with EV/SV and vectors encoding siRNA uPAR (puPAR), uPA (puPA), MMP-9 (pMMP-9) and a combination of three together (pU2M). After a 24-hour-incubation period, the medium was removed, cells were co-cultured with either 4×104 human endothelial cells or 4×104 endothelial cells alone and were grown in the presence of conditioned media. After 72 hours endothelial cells were stained for factor VIII antigen in the co-cultures (green florescence). Cells grown in the preserved conditioned media were H&E stained and examined under either a florescent microscope or a bright field microscope (A). (B) Quantification of angiogenesis in co-cultures infected with EV/SV, puPAR, puPA, pMMP-9 and pU2M vectors. Values are mean±SD of four experiments. SNB19 cells were trypsinized 72 hours after transfection with EV/SV, puPAR, puPA, pMMP-9 and pU2M, washed with PBS and resuspended in serum-free medium. (C) Invasion assays were carried out in a 12-well transwell unit on polycarbonate filters with 8-μm pores coated with matrigel (0.7 mg ml−1). After a 24-hour-incubation period, the cells that had passed through the filter into the lower wells were stained, counted and photographed under a bright-field microscope. (D) The invasion was quantified as described Methods (D). Values are mean±SD of four experiments.
puPAR, puPA, pMMP-9 and pU2M inhibit invasion in SNB19 cells
Proteolytic degradation of ECM components is crucial for tumor-cell invasion. To evaluate the impact of siRNA-mediated inhibition of uPAR, uPA and MMP-9 on glioma invasiveness, we utilized two models. In the first model, we compared the invasive ability of SNB19 cells transfected with puPAR, puPA, pMMP-9 and pU2M to those infected with the EV/SV vector. SNB19 cells transfected with EV/SV and parental cells demonstrated extensive invasion through Matrigel-coated transwell inserts, as indicated by the intense staining of cells. By contrast, puPAR-, puPA-, pMMP-9- and pU2M-transfected cultures were markedly less invasive through the reconstituted basement membrane, as indicated by the staining intensity compared with the controls (Fig. 3C). Quantification confirmed that transfection with puPAR, puPA, pMMP-9 and pU2M vectors reduced invasion by SNB19 cells to 9%, 40%, 15% and 2%, respectively, of that of parental and EV/SV transfected controls (Fig. 3D). Inhibition of invasion was higher in cells transfected with the tricistronic construct compared to single constructs alone.
We further examined the effect of puPAR, puPA, pMMP-9 and pU2M vectors using a spheroid invasion assay. A significant, potential advantage of using glioma spheroids is that tumor cells grown in three-dimensional cultures exhibit properties that more closely resemble those of tumors in vivo. In the spheroid co-culture system, control spheroids and spheroids transfected with the EV/SV vector progressively invaded fetal rat-brain aggregates whereas spheroids transfected with puPAR, puPA, pMMP-9 and pU2M demonstrated partial to almost complete inhibition of invasion (Fig. 4A). Quantification revealed that glioma spheroids invaded the fetal rat brain aggregates by 30% within 1 day, 55% within 2 days and 95% by 3 days, at which time the tumor spheroid and brain aggregates had combined into single entity (Fig. 4B). A similar trend was observed with glioma spheroids transfected with the EV/SV vector. By contrast, tumor spheroids transfected with the pU2M vector did not invade fetal rat brain aggregates. By 3 days, the rat brain aggregates were invaded by approximately 96%, 95%, 45%, 25% and 15% in the parental, EV/SV-, puPA-, pMMP-9- and puPAR-transfected co-cultures, and by 1% in the pU2M-transfected co-cultures (Fig. 4B). These results provide strong evidence that pU2M strongly inhibits glioma invasion in both in vitro models.
Fig. 4. Inhibition and regression of invasiveness and tumor growth by siRNA in spheroid and intracranial assays.
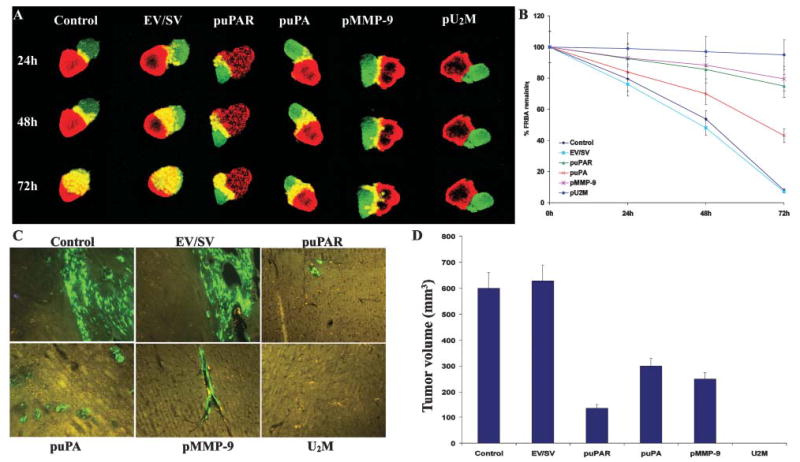
(A) Invasiveness of glioma spheroids was measured by co-culturing glioma spheroids with fetal rat brain aggregates. Spheroids of SNB19 cells were transfected with EV/SV, puPA, puPAR, pMMP-9 and pU2M and stained with DiI and co-cultured with DiO-stained fetal rat brain aggregates. Serial, 1-μm-thick sections were obtained from the surface through the center of the co-cultures with a confocal laser scanning microscope at the indicated time points. (B) The remaining volume of the rat brain aggregate transfected with EV/SV, puPA, puPAR, pMMP-9 and pU2M was measured as described in Methods. The values are mean ±SD of three experiments. (C,D) RNA-mediated regression of pre-established tumor growth. SNB19 GFP cells in suspension (2×106 in 10 μl serum-free medium) were injected intracranially. One week later, the mice were injected with either EV/SV or siRNA-expressing vectors (puPAR, puPA, pMMP-9 and pU2M) using an Alzet mini osmotic pump (constructs diluted to 1.5 μg ml−1 in PBS and injected at 0.25 μg hour−1, with six mice in each group). After a 5-week follow-up period, mice were sacrificed, their brains removed, paraffin embedded and sectioned. Sections were observed under fluorescence microscopy for GFP-expressing cells. (D) Semi-quantification of tumor volume in control, EV/SV, puPAR, puPA, pMMP-9 and pU2M-treated groups was assessed after 5 weeks. Data are shown mean±SD of six animals from each group.
pU2M completely regresses intracranial tumor growth
Here, we determined whether the downregulation of uPAR, uPA and MMP-9 levels using either single or tricistronic constructs causes regression of pre-established intracranial tumor growth in nude mice. All animals in the control and EV/SV-treated groups had intact cerebral tumors that were characterized by strong GFP fluorescence (Fig. 4C) whereas brain sections of mice treated with puPAR, puPA and pMMP-9 had small tumors, illustrated by minimal GFP fluorescence. Notably, GFP fluorescence was not detected in brain sections of mice treated with pU2M (Fig. 4C). Further quantification of these sections (scored by a neuropathologist blinded to treatment conditions) revealed no difference in tumor size between the parental and EV/SV treated groups and significant regression of pre-established intracranial tumor growth in the groups treated with puPAR, puPA and pMMP-9 (80%, 55% and 68% respectively) compared to control groups (Fig. 4D). However, complete regression of pre-established intracranial tumor growth was revealed in the pU2M treated group. These results demonstrate that RNAi-mediated suppression of uPAR, uPA and MMP-9 using a tricistronic construct completely eradicated malignant glioma growth in nude mice.
Inhibition of ERK1/2 phosphorylation
To better understand the effect of siRNA-mediated down-regulation of uPAR, uPA and MMP-9 on signaling pathways, we assayed for total and phosphorylated levels of ERK1/2, which are involved directly in tumor-cell survival, migration and proliferation. Western blots showed that there was no significant difference in total ERK1/2 concentrations in control and EV/SV-transfected cells compared with puPAR-, puPA-, pMMP-9- and pU2M-transfected cells (Fig. 5). However, the concentration of phospho-ERK1/2 was reduced significantly in SNB19 cells transfected with the pU2M vector compared with the control, EV/SV-, puPAR-, puPA- and pMMP-9-transfected SNB19 cells. Notably, there was no effect on the levels phospho-ERK in SNB19 cells transfected with any of the single constructs. GAPDH levels indicated that equal quantities of protein were loaded in the gel (Fig. 5).
Fig. 5. RNAi-mediated downregulation of uPAR, uPA and MMP-9 reduces phosphorylation of ERK.
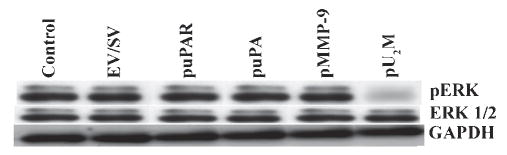
Western blot analysis of total and phosphorylated ERK (pERK) protein after transfection of glioblastoma cells with EV/SV, puPAR, puPA, pMMP-9 and pU2M constructs. GAPDH levels served as loading control.
CONCLUSIONS
Plasmid-based, CMV promoter-driven hpRNA targeting uPAR, uPA and MMP-9, either singly or simultaneously, induces RNAi in the SNB19 human glioma cell line.
The simultaneous, RNAi-mediated downregulation of uPAR, uPA and MMP-9 in SNB19 human glioma cells caused:
Inhibition of invasion and angiogenesis in vitro.
Regression of pre-established intracranial tumors in nude mice in vivo.
Reduction in the phosphorylation of ERK 1 and 2 signaling molecules.
DISCUSSION
In mammals, dsRNA larger than 30 nt activates an antiviral defense mechanism that includes the production of interferons and results in the non-specific degradation of RNA and a general shutdown of the host protein-synthesis machinery (Williams, 1997). To circumvent this major defense response, small, 21 bp dsRNA molecules were used, so that the dsRNAs were sufficiently large to induce gene-specific suppression but small enough to evade the antiviral defense mechanism (Caplen et al., 2001; Elbashir et al., 2001). The potential application for RNAi in mammals is the simultaneous inhibition of multiple genes in somatic cells. Earlier observations have demonstrated that inhibition of a synthetic reporter by a transfected hairpin siRNA vector is constant, even in the presence of another hairpin siRNA vector. This demonstrates that there is no significant competition between co-transfected hairpin siRNA vectors. In the present study, we demonstrated that plasmid-based RNA interference has the potential to inhibit uPA, uPAR and MMP-9 target molecules. Figure 2 showed that there are significantly decreased levels of uPAR protein, and uPA and MMP-9 enzymatic activity in glioma cells transfected with pU2M vector compared to mock and empty vector. We also demonstrated the significant reduction in the levels of these molecules by immunohistochemistry using specific antibodies for these molecules in puPAR, puPA, pMMP-9 and pU2M-transfected cells. It has been reported that treatment of a human T98G glioblastoma cell line with uPA antisense phosphorothioate oligonucleotides resulted in greater decreases of uPA mRNA expression than sense vectors (Engelhard et al., 1996). Our own results demonstrate that uPA and uPAR protein and mRNA levels were decreased significantly by antisense uPA and antisense uPAR clones (Mohanam et al., 1997; Mohanam et al., 2002). In a rat sarcoma model, MMP-9 expression was decreased with ribozyme directed against MMP-9 mRNAi. (Hua and Muschel, 1996). The reduced levels of MMP-9 in stable MMP-9 antisense clones and SNB19 cells transfected with antisense MMP-9 Ad-constructs were also determined (Kondraganti et al., 2000; Lakka et al., 2002b). Earlier studies showed that RNAi can be transmitted from cell to cell, and to the next generation, and might also be amplified by an RNA-dependent RNA polymerase (Carmell et al., 2003; Shuey et al., 2002).
Our results show that conditioned medium from a glioblastoma cell line transfected with pU2M inhibited the capillary-like structures compared with mock or empty vector (Fig. 3A,B). This indicates that the angiogenic signal necessary for the induction of angiogenesis was not present in pU2M-transfected cells. As demonstrated by the absence of angiogenic induction in pU2M-transfected SNB19 glioma cells, downregulation of uPA, uPAR and MMP-9 by hpRNA caused the downregulation of angiogenic factors. This accords with our previous work in which antisense-mediated downregulation of uPAR and MMP-9 retarded angiogenesis (Lakka et al., 2003). Other studies have shown that VEGF induces sustained cellular permeability in capillary endothelial cells that is mediated by activation of the uPA/uPAR system, which substantiates the involvement of uPAR in angiogenesis (Behzadian et al., 2003). This also indicates possible cross-talk between uPA/uPAR and VEGF. It was shown that the absence of uPA or tissue type plasminogen activator (tPA) significantly decreased the development of experimental choroidal neovascularization compared with either wild-type or uPAR-deficient mice (uPA-/-) (Li et al., 1998). It has been reported that primary tumor growth is reduced significantly in uPA-/- and plasmino-gen activator inhibitor-1-deficient (PAI-1-/-) mice relative to wild-type mice. Moreover, tumors in uPA-/- and PAI-/- mice have lower proliferative and higher apoptotic indices, and different neovascularmorphology compared with wild-type mice (Gutierrez et al., 2000).
Previous studies have demonstrated that several peptides that have been shown to inhibit uPA binding by bacteriophage display inhibit angiogenesis and primary tumor growth in syngenic mice (Gutierrez et al., 2000).
MMPs also regulate tumor angiogenesis and might be required for the ‘angiogenic switch’ that occurs during tumor neovascularization (Bergers et al., 2000). Two transgenic models of tumor progression, the K14-HPV16 [G] skin cancer model (Coussens et al., 2000) and the RIP1-Tag2 [G] model (Bergers et al., 2000), demonstrate that MMP-9 plays a significant role in angiogenesis. We used a co-culture system in which glioma and endothelial cells were cultured together to evaluate endothelial-tube formation. In this model, we showed that capillary-like structure formation was decreased in the presence of anti-MMP-9 antibodies, TIMP-1, a synthetic inhibitor batimastat (BB-94) and the protein kinase C inhibitor calphostin because of reduced activity of MMP-9 (Chandrasekar et al., 2000). Other studies have demonstrated that the inhibition of the ERK1/ERK 2 pathway with PD98059, a specific inhibitor of MEK1, abrogated basic fibroblast growth factor-mediated tube formation by mouse endothelial cells (Tanaka et al., 1999).
Diffuse single-cell invasion, which occurs in all glial tumors regardless of histological grade, is defined as a translocation of neoplastic cells through host cellular and ECM barriers. Malignant gliomas express higher levels of uPA, uPAR and MMP-9 compared with low grade and normal brain tissue (Forsyth et al., 1999; Raithatha et al., 2000; Rao et al., 1993; Rao et al., 1996). Inhibitors and antibodies of uPA inhibit the invasiveness of tumor cells into ECM, and amniotic and chick chorioallantoic membranes (Alonso et al., 1998; Holst-Hansen et al., 1996; Jarrard et al., 1995; Min et al., 1996). Moreover, uPA antibodies blocked metastasis of Hep3 human carcinoma cells in chick embryos and inhibited the local invasiveness of subcutaneous Hep3 tumors in nude mice (Ossowski et al., 1991). Our previous results showed that antisense, stable clones of uPA, uPAR and MMP-9 were much less invasive in in vitro models (Go et al., 1997; Kondraganti et al., 2000; Mohanam et al., 1997; Mohanam et al., 2001). We also showed that clones stably expressing an aminoterminal fragment of uPA (ATF-uPA) were markedly less invasive in in vitro models compared to controls (Mohanam et al., 2002). CD44 has been demonstrated to serve as a dokins molecule to retain MMP-9 proteolytic activity at the cell surface (Yu et al., 1996; Yu and Stamenkovic, 2000) and cleavage of MMP-9 from CD44 resulted in impairment of migration (Kajita et al., 2001). Localization of MMP-9 to the cell surface mediated the activation of latent transforming growth factor β and promotes tumor cell invasion and angiogenesis (Yu et al., 2000).
In the present study, almost complete inhibition of glioblastoma-cell invasion was observed in both Matrigel and spheroid-invasion assays (Fig. 3C,D and Fig. 4A,B). Thus, downregulation of uPA, uPAR and MMP-9 by RNAi significantly inhibit the ability of glioma cells to invade the surrounding ECM compared to approaches described earlier.
These results correlate well with the spheroid and Matrigel invasion assays, in that the pU2M-transfected cells do not invade, even in the presence of brain tissue in an in vitro model. This might be because the cells have lost their ability to anchor to the ECM and are unable to invade the confronted brain tissue. Earlier studies showed that binding of uPA to uPAR in MCF-7 cells activates ERK 1 and ERK 2, which are required for cell motility (Nguyen et al., 1998). Jo et al. (Jo et al., 2003) have also shown that an alternate pathway links uPAR to ERK in the absence of the epidermal growth factor receptor (EGFR). However, this pathway is silenced by EGFR expression, which indicates the involvement of uPAR in cell motility. Physical association between uPAR and the integrin αV chain of the vectronectin receptor leads to functional interaction of these receptors, indicating that uPAR directs cytoskeletal rearrangements and cell migration by altering αVβ3 signaling specificity (Carriero et al., 1999). Our findings confirm previous reports that both uPA and uPAR are required for cell migration in a Vitronectin environment (Busso et al., 1994). Other studies have reported that high-molecular-weight serum–protein complexes promote both uPAR-focal adhesion colocalization and cell migration of glioma cells (Hedberg et al., 2000). Studies utilizing native MMP inhibitors, such as the TIMPs, have demonstrated that downregulation of both TIMP-1 and TIMP-2 contributed significantly to the invasive potential of gliomas (Lampert et al., 1998; Mohanam et al., 1995; Nakano et al., 1995). Introduction of TIMP-1 cDNA into an invasive astrocytoma cell line reduced its invasive potential (Matsuzawa et al., 1996). In addition, the synthetic MMP inhibitors batimastat, marimastat and AG3340 reduced glioma invasion in vitro (Tonn et al., 1999). Stable transfection of phosphatase and tensin homologue (PTEN) reduced MMP-9 secretion caused by hyaluronic acid-induced phosphorylation of focal adhesion kinase and ERK1/ERK2 signaling (Park et al., 2002). Glioblastomas with EGFR VIII amplification demonstrated the highest levels of MMP-9 (Choe et al., 2002).
In earlier work (Lakka et al., 2000) we have shown that transient transfection of SNB19 cells with either mt-ERK or mt-JNK repressed MMP-9 promoter activity, which indicated that interfering with either pathway might inhibit MMP-9 expression. Inhibition of ERK by MEK-specific inhibitors blocked MMP-9 expression in breast cancer cells (Yao et al., 2001), and decreased MMP-9 production and attenuated the invasiveness in vivo in head and neck squamous carcinoma cells (Simon et al., 1998). Our recent results showed that cells stably transfected with mt-ERK were less invasive and had significantly reduced levels of MMP-9 (Lakka et al., 2002a). It has been reported that these two signaling pathways (MAPK and ERK1/2) are activated when uPA binds to uPAR (Konakova et al., 1998; Tang et al., 1998). Figure 5 shows that the pU2M construct inhibited the phosphorylation of ERK1/2.
In the present study, we completely suppressed pre-established intracranial tumor growth during the 5-week follow-up period in nude mice injected with pU2M vector compared to mock and empty vector/scrambled vector (Fig. 4C,D). These results in vivo correlate well with the spheroid-invasion assay. In contrast to our earlier experiments, in which uPAR-antisense cells were raised ex vivo before intracranial implantation (Go et al., 1997), the pU2M plasmid was delivered after tumor implantation.
Several studies using selective inhibitors of uPA and small, synthetic, cyclic, competitive uPA antagonists derived from the binding site of uPAR resulted in a significant reduction of tumor burden (Evans et al., 1998; Sato et al., 2002; Sturzebecher et al., 1999). Recent studies have also shown that fusion of diphtheria toxin with ATF-uPA caused a significant regression of glial tumors and was highly potent and selective at killing uPAR-expressing glioblastoma cell lines (Vallera et al., 2002). Adenovirus-mediated downregulation of uPA and uPAR (Gondi et al., 2003), and cathepsin B and uPAR (Gondi et al., 2004) constructs inhibited the migration and invasion of glioma cells, and inhibited ex vivo tumor growth compared with vector-treatment. Intracranial injections of SNB19 glioblastoma cells infected with an adenovirus expressing antisense MMP-9 did not produce tumors in nude mice (Lakka et al., 2002b). A bicistronic Ad-construct with antisense uPAR and MMP-9 was more effective at inhibiting invasion, angiogenesis and tumor growth in vivo than a single construct (ad-uPAR) (Lakka et al., 2003). Our recent studies demonstrated that a siRNA bicistronic construct for cathepsin B and MMP-9 completely repressed pre-established intracranial tumors (Lakka et al., 2004). From our results, it can be concluded that downregulation of uPA, uPAR and MMP-9 is achieved via a triple hpRNA that targets the corresponding mRNA molecules. Because no viral delivery methods were used, the question of viral toxicity does not arise (Alemany et al., 1999). Thus, it can be concluded that RNAi is a powerful alternative to genetic tools such as antisense oligonucleotides and ribozyme technologies to reduce target-gene expression. Our results prove that downregulation of uPA, uPAR and MMP-9 by hpRNA decreases migration, invasion and angiogenesis in the SNB19 human glioma cell line. As such, the use of RNAi-based gene therapy has great potential for the treatment of gliomas and other metastatic tumors.
Acknowledgments
We would like to thank Noorjehan Ali for technical assistance, Tina Wilson for preparing the manuscript and Sushma Jasti and Diana Meister for reviewing the manuscript. This work was supported by NIH grants CA092393, CA85216, CA75557, CA95058 and CA76350 (to J.S.R.).
References
- Alemany R, Gomez-Manzano C, Balague C, Yung WK, Curiel DT, Kyritsis AP, Fueyo J. Gene therapy for gliomas: molecular targets, adenoviral vectors, and oncolytic adenoviruses. Experimental Cell Research. 1999;252:1–12. doi: 10.1006/excr.1999.4623. [DOI] [PubMed] [Google Scholar]
- Alonso DF, Farias EF, Ladeda V, Davel L, Puricelli L, Bal de Kier JE. Effects of synthetic urokinase inhibitors on local invasion and metastasis in a murine mammary tumor model. Breast Cancer Research Treatment. 1996;40:209–223. doi: 10.1007/BF01806809. [DOI] [PubMed] [Google Scholar]
- Alonso DF, Tejera AM, Farias EF, Bal de Kier JE, Gomez DE. Inhibition of mammary tumor cell adhesion, migration, and invasion by the selective synthetic urokinase inhibitor B428. Anticancer Research. 1998;18:4499–4504. [PubMed] [Google Scholar]
- Andreasen PA, Kjoller L, Christensen L, Duffy MJ. The urokinase-type plasminogen activator system in cancer metastasis: a review. International Journal of Cancer. 1997;72:1–22. doi: 10.1002/(sici)1097-0215(19970703)72:1<1::aid-ijc1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Behzadian MA, Windsor LJ, Ghaly N, Liou G, Tsai NT, Caldwell RB. VEGF-induced paracellular permeability in cultured endothelial cells involves urokinase and its receptor. FASEB Journal. 2003;17:752–754. doi: 10.1096/fj.02-0484fje. [DOI] [PubMed] [Google Scholar]
- Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nature Cell Biology. 2000;2:737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand JR, Pottier M, Vekris A, Opolon P, Maksimenko A, Malvy C. Comparison of antisense oligonucleotides and siRNAs in cell culture and in vivo. Biochemical Biophysical Research Communications. 2002;296:1000–1004. doi: 10.1016/s0006-291x(02)02013-2. [DOI] [PubMed] [Google Scholar]
- Blasi F. The urokinase receptor. A cell surface, regulated chemokine. APMIS. 1999;107:96–101. doi: 10.1111/j.1699-0463.1999.tb01531.x. [DOI] [PubMed] [Google Scholar]
- Bugge TH, Kombrinck KW, Xiao Q, Holmback K, Daugherty CC, Witte DP, Degen JL. Growth and dissemination of Lewis lung carcinoma in plasminogen-deficient mice. Blood. 1997;90:4522–4531. [PubMed] [Google Scholar]
- Busso N, Masur SK, Lazega D, Waxman S, Ossowski L. Induction of cell migration by pro-urokinase binding to its receptor: possible mechanism for signal transduction in human epithelial cells. Journal of Cell Biology. 1994;126:259–270. doi: 10.1083/jcb.126.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplen NJ, Parrish S, Imani F, Fire A, Morgan RA. Specific inhibition of gene expression by small double-stranded RNAs in invertebrate and vertebrate systems. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:9742–9747. doi: 10.1073/pnas.171251798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmell MA, Zhang L, Conklin DS, Hannon GJ, Rosenquist TA. Germline transmission of RNAi in mice. Nature Structural Biology. 2003;10:91–92. doi: 10.1038/nsb896. [DOI] [PubMed] [Google Scholar]
- Carriero MV, Del Vecchio S, Capozzoli M, Franco P, Fontana L, Zannetti A, Botti G, D’Aiuto G, et al. Urokinase receptor interacts with alpha(v)beta5 vitronectin receptor, promoting urokinase-dependent cell migration in breast cancer. Cancer Research. 1999;59:5307–5314. [PubMed] [Google Scholar]
- Carroll VA, Binder BR. The role of the plasminogen activation system in cancer. Seminars in Thrombosis and Hemostasis. 1999;25:183–197. doi: 10.1055/s-2007-994920. [DOI] [PubMed] [Google Scholar]
- Chandrasekar N, Jasti S, Alfred-Yung WK, Ali-Osman F, Dinh DH, Olivero WC, Gujrati M, Kyritsis AP, et al. Modulation of endothelial cell morphogenesis in vitro by MMP-9 during glial–endothelial cell interactions. Clinical Experimental Metastasis. 2000;18:337–342. doi: 10.1023/a:1010833730407. [DOI] [PubMed] [Google Scholar]
- Choe G, Park JK, Jouben-Steele L, Kremen TJ, Liau LM, Vinters HV, Cloughesy TF, Mischel PS. Active matrix metalloproteinase 9 expression is associated with primary glioblastoma subtype. Clinical Cancer Research. 2002;8:2894–2901. [PubMed] [Google Scholar]
- Coussens LM, Tinkle CL, Hanahan D, Werb Z. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell. 2000;103:481–490. doi: 10.1016/s0092-8674(00)00139-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley CW, Cohen RL, Lucas BK, Liu G, Shuman MA, Levinson AD. Prevention of metastasis by inhibition of the urokinase receptor. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:5021–5025. doi: 10.1073/pnas.90.11.5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewerchin M, Nuffelen AV, Wallays G, Bouche A, Moons L, Carmeliet P, Mulligan RC, Collen D. Generation and characterization of urokinase receptor-deficient mice. Journal of Clinical Investigation. 1996;97:870–878. doi: 10.1172/JCI118489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nature Reviews Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Ellis V, Behrendt N, Dano K. Plasminogen activation by receptor-bound urokinase A kinetic study with both cell-associated and isolated receptor. Journal of Biological Chemistry. 1991;266:12752–12758. [PubMed] [Google Scholar]
- Ellis V, Pyke C, Eriksen J, Solberg H, Dano K. The urokinase receptor: involvement in cell surface proteolysis and cancer invasion. Annals of the New York Academy of Sciences. 1992;667:13–31. doi: 10.1111/j.1749-6632.1992.tb51591.x. [DOI] [PubMed] [Google Scholar]
- Engelhard H, Narang C, Homer R, Duncan H. Urokinase antisense oligodeoxynucleotides as a novel therapeutic agent for malignant glioma: in vitro and in vivo studies of uptake, effects and toxicity. Biochemical Biophysical Research Communication. 1996;227:400–405. doi: 10.1006/bbrc.1996.1519. [DOI] [PubMed] [Google Scholar]
- Evans DM, Sloan-Stakleff K, Arvan M, Guyton DP. Time and dose dependency of the suppression of pulmonary metastases of rat mammary cancer by amiloride. Clinical Experimental Metastasis. 1998;16:353–357. doi: 10.1023/a:1006517614491. [DOI] [PubMed] [Google Scholar]
- Forsyth PA, Wong H, Laing TD, Rewcastle NB, Morris DG, Muzik H, Leco KJ, Johnston RN, et al. Gelatinase-A (MMP-2), gelatinase-B (MMP-9) and membrane type matrix metalloproteinase-1 (MT1-MMP) are involved in different aspects of the pathophysiology of malignant gliomas. British Journal of Cancer. 1999;79:1828–1835. doi: 10.1038/sj.bjc.6990291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese A, Westphal M. Glioma invasion in the central nervous system. Neurosurgery. 1996;39:235–250. doi: 10.1097/00006123-199608000-00001. [DOI] [PubMed] [Google Scholar]
- Gladson CL, Pijuan-Thompson V, Olman MA, Gillespie GY, Yacoub IZ. Up-regulation of urokinase and urokinase receptor genes in malignant astrocytoma. American Journal of Pathology. 1995;146:1150–1160. [PMC free article] [PubMed] [Google Scholar]
- Go Y, Chintala SK, Mohanam S, Gokaslan Z, Venkaiah B, Bjerkvig R, Oka K, Nicolson GL, et al. Inhibition of in vivo tumorigenicity and invasiveness of a human glioblastoma cell line transfected with antisense uPAR vectors. Clinical Experimental Metastasis. 1997;15:440–446. doi: 10.1023/a:1018410523635. [DOI] [PubMed] [Google Scholar]
- Gondi CS, Lakka SS, Yanamandra N, Olivero WC, Dinh DH, Gujrati M, Tung CH, Weissleder R, et al. Adenovirus-mediated expression of antisense urokinase plasminogen activator receptor and antisense cathepsin B inhibits tumor growth, invasion, and angiogenesis in gliomas. Cancer Research. 2004;64:4069–4077. doi: 10.1158/0008-5472.CAN-04-1243. [DOI] [PubMed] [Google Scholar]
- Gondi CS, Lakka SS, Yanamandra N, Siddique K, Dinh DH, Olivero WC, Gujrati M, Rao JS. Expression of antisense uPAR and antisense uPA from a bicistronic adenoviral construct inhibits glioma cell invasion, tumor growth, and angiogenesis. Oncogene. 2003;22:5967–5975. doi: 10.1038/sj.onc.1206535. [DOI] [PubMed] [Google Scholar]
- Gutierrez LS, Schulman A, Brito-Robinson T, Noria F, Ploplis VA, Castellino FJ. Tumor development is retarded in mice lacking the gene for urokinase-type plasminogen activator or its inhibitor, plasminogen activator inhibitor-1. Cancer Research. 2000;60:5839–5847. [PubMed] [Google Scholar]
- Hedberg KK, Stauff C, Hoyer-Hansen G, Ronne E, Griffith OH. High-molecular-weight serum protein complexes differentially promote cell migration and the focal adhesion localization of the urokinase receptor in human glioma cells. Experimental Cell Research. 2000;257:67–81. doi: 10.1006/excr.2000.4873. [DOI] [PubMed] [Google Scholar]
- Holst-Hansen C, Johannessen B, Hoyer-Hansen G, Romer J, Ellis V, Brunner N. Urokinase-type plasminogen activation in three human breast cancer cell lines correlates with their in vitro invasiveness. Clinical Experimental Metastasis. 1996;14:297–307. doi: 10.1007/BF00053903. [DOI] [PubMed] [Google Scholar]
- Hua J, Muschel RJ. Inhibition of matrix metalloproteinase 9 expression by a ribozyme blocks metastasis in a rat sarcoma model system. Cancer Research. 1996;56:5279–5284. [PubMed] [Google Scholar]
- Jarrard DF, Hansen NM, Patai B, Rukstalis DB. Urokinase plasminogen activator is necessary but not sufficient for prostate cancer cell invasion. Invasion Metastasis. 1995;15:34–45. [PubMed] [Google Scholar]
- Jo M, Thomas KS, O’Donnell DM, Gonias SL. Epidermal growth factor receptor-dependent and -independent cell-signaling pathways originating from the urokinase receptor. Journal of Biological Chemistry. 2003;278:1642–1646. doi: 10.1074/jbc.M210877200. [DOI] [PubMed] [Google Scholar]
- Kajita M, Itoh Y, Chiba T, Mori H, Okada A, Kinoh H, Seiki M. Membrane-type 1 matrix metalloproteinase cleaves CD44 and promotes cell migration. Journal of Cell Biology. 2001;153:893–904. doi: 10.1083/jcb.153.5.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konakova M, Hucho F, Schleuning WD. Downstream targets of urokinase-type plasminogen-activator-mediated signal transduction. European Journal of Biochemistry. 1998;253:421–429. doi: 10.1046/j.1432-1327.1998.2530421.x. [DOI] [PubMed] [Google Scholar]
- Kondraganti S, Mohanam S, Chintala SK, Kin Y, Jasti SL, Nirmala C, Lakka SS, Adachi Y, et al. Selective suppression of matrix metalloproteinase-9 in human glioblastoma cells by antisense gene transfer impairs glioblastoma cell invasion. Cancer Research. 2000;60:6851–6855. [PubMed] [Google Scholar]
- Lakka SS, Gondi CS, Yanamandra N, Dinh DH, Olivero WC, Gujrati M, Rao JS. Synergistic down-regulation of urokinase plasminogen activator receptor and matrix metalloproteinase-9 in SNB19 glioblastoma cells efficiently inhibits glioma cell invasion, angiogenesis, and tumor growth. Cancer Research. 2003;63:2454–2461. [PubMed] [Google Scholar]
- Lakka SS, Gondi CS, Yanamandra N, Olivero WC, Dinh DH, Gujrati M, Rao JS. Inhibition of cathepsin B and MMP-9 gene expression in glioblastoma cell line via RNA interference reduces tumor cell invasion, tumor growth and angiogenesis. Oncogene. 2004;23:4681–4689. doi: 10.1038/sj.onc.1207616. [DOI] [PubMed] [Google Scholar]
- Lakka SS, Jasti SL, Gondi C, Boyd D, Chandrasekar N, Dinh DH, Olivero WC, Gujrati M, et al. Downregulation of MMP-9 in ERK-mutated stable transfectants inhibits glioma invasion in vitro. Oncogene. 2002a;21:5601–5608. doi: 10.1038/sj.onc.1205646. [DOI] [PubMed] [Google Scholar]
- Lakka SS, Jasti SL, Kyritsis AP, Yung WK, Ali-Osman F, Nicolson GL, Rao JS. Regulation of MMP-9 (type IV collagenase) production and invasiveness in gliomas by the extracellular signal-regulated kinase and jun amino-terminal kinase signaling cascades. Clinical Experimental Metastasis. 2000;18:245–252. doi: 10.1023/a:1006724826083. [DOI] [PubMed] [Google Scholar]
- Lakka SS, Rajan M, Gondi C, Yanamandra N, Chandrasekar N, Jasti SL, Adachi Y, Siddique K, et al. Adenovirus-mediated expression of antisense MMP-9 in glioma cells inhibits tumor growth and invasion. Oncogene. 2002b;21:8011–8019. doi: 10.1038/sj.onc.1205894. [DOI] [PubMed] [Google Scholar]
- Lampert K, Machein U, Machein MR, Conca W, Peter HH, Volk B. Expression of matrix metalloproteinases and their tissue inhibitors in human brain tumors. American Journal of Pathology. 1998;153:429–437. doi: 10.1016/S0002-9440(10)65586-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Lu H, Griscelli F, Opolon P, Sun LQ, Ragot T, Legrand Y, Belin D, et al. Adenovirus-mediated delivery of a uPA/uPAR antagonist suppresses angiogenesis-dependent tumor growth and dissemination in mice. Gene Therapy. 1998;5:1105–1113. doi: 10.1038/sj.gt.3300742. [DOI] [PubMed] [Google Scholar]
- Matsuzawa K, Fukuyama K, Hubbard SL, Dirks PB, Rutka JT. Transfection of an invasive human astrocytoma cell line with a TIMP-1 cDNA: modulation of astrocytoma invasive potential. Journal of Neuropathology and Experimental Neurology. 1996;55:88–96. doi: 10.1097/00005072-199601000-00009. [DOI] [PubMed] [Google Scholar]
- Min HY, Doyle LV, Vitt CR, Zandonella CL, Stratton-Thomas JR, Shuman MA, Rosenberg S. Urokinase receptor antagonists inhibit angiogenesis and primary tumor growth in syngeneic mice. Cancer Research. 1996;56:2428–2433. [PubMed] [Google Scholar]
- Miyagishi M, Taira K. U6 promoter-driven siRNAs with four uridine 3’ overhangs efficiently suppress targeted gene expression in mammalian cells. Nature Biotechnology. 2002;20:497–500. doi: 10.1038/nbt0502-497. [DOI] [PubMed] [Google Scholar]
- Mohan PM, Chintala SK, Mohanam S, Gladson CL, Kim ES, Gokaslan ZL, Lakka SS, Roth JA, et al. Adenovirus-mediated delivery of antisense gene to urokinase-type plasminogen activator receptor suppresses glioma invasion and tumor growth. Cancer Research. 1999;59:3369–3373. [PubMed] [Google Scholar]
- Mohanam S, Chandrasekar N, Yanamandra N, Khawar S, Mirza F, Dinh DH, Olivero WC, Rao JS. Modulation of invasive properties of human glioblastoma cells stably expressing aminoterminal fragment of urokinase-type plasminogen activator. Oncogene. 2002;21:7824–7830. doi: 10.1038/sj.onc.1205893. [DOI] [PubMed] [Google Scholar]
- Mohanam S, Chintala SK, Go Y, Bhattacharya A, Venkaiah B, Boyd D, Gokaslan ZL, Sawaya R, Rao JS. In vitro inhibition of human glioblastoma cell line invasiveness by antisense uPA receptor. Oncogene. 1997;14:1351–1359. doi: 10.1038/sj.onc.1200963. [DOI] [PubMed] [Google Scholar]
- Mohanam S, Jasti SL, Kondraganti SR, Chandrasekar N, Kin Y, Fuller GN, Lakka SS, Kyritsis AP, et al. Stable transfection of urokinase-type plasminogen activator antisense construct modulates invasion of human glioblastoma cells. Clinical Cancer Research. 2001;7:2519–2526. [PubMed] [Google Scholar]
- Mohanam S, Wang SW, Rayford A, Yamamoto M, Sawaya R, Nakajima M, Liotta LA, Nicolson GL, et al. Expression of tissue inhibitors of metalloproteinases: negative regulators of human glioblastoma invasion in vivo. Clinical and Experimental Metastasis. 1995;13:57–62. doi: 10.1007/BF00144019. [DOI] [PubMed] [Google Scholar]
- Nakano A, Tani E, Miyazaki K, Yamamoto Y, Furuyama J. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in human gliomas. Journal of Neurosurgery. 1995;83:298–307. doi: 10.3171/jns.1995.83.2.0298. [DOI] [PubMed] [Google Scholar]
- Naldini L, Vigna E, Bardelli A, Follenzi A, Galimi F, Comoglio PM. Biological activation of pro-HGF (hepatocyte growth factor) by urokinase is controlled by a stoichiometric reaction. Journal of Biological Chemistry. 1995;270:603–611. doi: 10.1074/jbc.270.2.603. [DOI] [PubMed] [Google Scholar]
- Nguyen DH, Hussaini IM, Gonias SL. Binding of urokinase-type plasminogen activator to its receptor in MCF-7 cells activates extracellular signal-regulated kinase 1 and 2 which is required for increased cellular motility. Journal of Biological Chemistry. 1998;273:8502–8507. doi: 10.1074/jbc.273.14.8502. [DOI] [PubMed] [Google Scholar]
- Ossowski L, Russo-Payne H, Wilson EL. Inhibition of urokinase-type plasminogen activator by antibodies: the effect on dissemination of a human tumor in the nude mouse. Cancer Research. 1991;51:274–281. [PubMed] [Google Scholar]
- Park MJ, Kim MS, Park IC, Kang HS, Yoo H, Park SH, Rhee CH, Hong SI, et al. PTEN suppresses hyaluronic acid-induced matrix metalloproteinase-9 expression in U87MG glioblastoma cells through focal adhesion kinase dephosphorylation. Cancer Research. 2002;62:6318–6322. [PubMed] [Google Scholar]
- Pedersen PH, Edvardsen K, Garcia-Cabrera I, Mahesparan R, Thorsen J, Mathisen B, Rosenblum ML, Bjerkvig R. Migratory patterns of lac-z transfected human glioma cells in the rat brain. International Journal of Cancer. 1995;62:767–771. doi: 10.1002/ijc.2910620620. [DOI] [PubMed] [Google Scholar]
- Raithatha SA, Muzik H, Muzik H, Rewcastle NB, Johnston RN, Edwards DR, Forsyth PA. Localization of gelatinase-A and gelatinase-B mRNA and protein in human gliomas. Neuro-oncology. 2000;2:145–150. doi: 10.1093/neuonc/2.3.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao JS, Steck PA, Mohanam S, Stetler-Stevenson WG, Liotta LA, Sawaya R. Elevated levels of M(r) 92,000 type IV collagenase in human brain tumors. Cancer Research. 1993;53:2208–2211. [PubMed] [Google Scholar]
- Rao JS, Yamamoto M, Mohaman S, Gokaslan ZL, Fuller GN, Stetler-Stevenson WG, Rao VH, Liotta LA, et al. Expression and localization of 92 kDa type IV collagenase/gelatinase B (MMP-9) in human gliomas. Clinical and Experimental Metastasis. 1996;14:12–18. doi: 10.1007/BF00157681. [DOI] [PubMed] [Google Scholar]
- Sato S, Kopitz C, Schmalix WA, Muehlenweg B, Kessler H, Schmitt M, Kruger A, Magdolen V. High-affinity urokinase-derived cyclic peptides inhibiting urokinase/urokinase receptor-interaction: effects on tumor growth and spread. FEBS Letters. 2002;528:212–216. doi: 10.1016/s0014-5793(02)03311-2. [DOI] [PubMed] [Google Scholar]
- Shapiro RL, Duquette JG, Roses DF, Nunes I, Harris MN, Kamino H, Wilson EL, Rifkin DB. Induction of primary cutaneous melanocytic neoplasms in urokinase-type plasminogen activator (uPA)-deficient and wild-type mice: cellular blue nevi invade but do not progress to malignant melanoma in uPA-deficient animals. Cancer Research. 1996;56:3597–3604. [PubMed] [Google Scholar]
- Shuey DJ, McCallus DE, Giordano T. RNAi: gene-silencing in therapeutic intervention. Drug Discovery Today. 2002;7:1040–1046. doi: 10.1016/s1359-6446(02)02474-1. [DOI] [PubMed] [Google Scholar]
- Simon C, Goepfert H, Boyd D. Inhibition of the p38 mitogen-activated protein kinase by SB 203580 blocks PMA-induced Mr 92,000 type IV collagenase secretion and in vitro invasion. Cancer Research. 1998;58:1135–1139. [PubMed] [Google Scholar]
- Sturzebecher J, Vieweg H, Steinmetzer T, Schweinitz A, Stubbs MT, Renatus M, Wikstrom P. 3-Amidinophenylalanine-based inhibitors of urokinase. Bioorganic Medical Chemistry Letter. 1999;9:3147–3152. doi: 10.1016/s0960-894x(99)00541-7. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Abe M, Sato Y. Roles of extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase in the signal transduction of basic fibroblast growth factor in endothelial cells during angiogenesis. Japan Journal of Cancer Research. 1999;90:647–654. doi: 10.1111/j.1349-7006.1999.tb00796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Kerins DM, Hao Q, Inagami T, Vaughan DE. The urokinase-type plasminogen activator receptor mediates tyrosine phosphorylation of focal adhesion proteins and activation of mitogen-activated protein kinase in cultured endothelial cells. Journal of Biological Chemistry. 1998;273:18268–18272. doi: 10.1074/jbc.273.29.18268. [DOI] [PubMed] [Google Scholar]
- Tonn JC, Kerkau S, Hanke A, Bouterfa H, Mueller JG, Wagner S, Vince GH, Roosen K. Effect of synthetic matrix-metalloproteinase inhibitors on invasive capacity and proliferation of human malignant gliomas in vitro. International Journal of Cancer. 1999;80:764–772. doi: 10.1002/(sici)1097-0215(19990301)80:5<764::aid-ijc22>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Vallera DA, Li C, Jin N, Panoskaltsis-Mortari A, Hall WA. Targeting urokinase-type plasminogen activator receptor on human glioblastoma tumors with diphtheria toxin fusion protein DTAT. Journal of the National Cancer Institute. 2002;94:597–606. doi: 10.1093/jnci/94.8.597. [DOI] [PubMed] [Google Scholar]
- Werb Z, Ashkenas J, MacAuley A, Wiesen JF. Extracellular matrix remodeling as a regulator of stromal-epithelial interactions during mammary gland development, involution and carcinogenesis. Brazil Journal of Medical Biology Research. 1996;29:1087–1097. [PubMed] [Google Scholar]
- Williams BR. Role of the double-stranded RNA-activated protein kinase (PKR) in cell regulation. Biochemical Society Transactions. 1997;25:509–513. doi: 10.1042/bst0250509. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sawaya R, Mohanam S, Bindal AK, Bruner JM, Oka K, Rao VH, Tomonaga M, et al. Expression and localization of urokinase-type plasminogen activator in human astrocytomas in vivo. Cancer Research. 1994a;54:3656–3661. [PubMed] [Google Scholar]
- Yamamoto M, Sawaya R, Mohanam S, Rao VH, Bruner JM, Nicolson GL, Rao JS. Expression and localization of urokinase-type plasminogen activator receptor in human gliomas. Cancer Research. 1994b;54:5016–5020. [PubMed] [Google Scholar]
- Yao J, Xiong S, Klos K, Nguyen N, Grijalva R, Li P, Yu D. Multiple signaling pathways involved in activation of matrix metalloproteinase-9 (MMP-9) by heregulin-beta1 in human breast cancer cells. Oncogene. 2001;20:8066–8074. doi: 10.1038/sj.onc.1204944. [DOI] [PubMed] [Google Scholar]
- Yu Q, Grammatikakis N, Toole BP. Expression of multiple CD44 isoforms in the apical ectodermal ridge of the embryonic mouse limb. Developmental Dynamics. 1996;207:204–214. doi: 10.1002/(SICI)1097-0177(199610)207:2<204::AID-AJA8>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Development. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- Yu W, Kim J, Ossowski L. Reduction in surface urokinase receptor forces malignant cells into a protracted state of dormancy. Journal of Cell Biology. 1997;137:767–777. doi: 10.1083/jcb.137.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamore PD. RNA interference: listening to the sound of silence. Nature Structural Biology. 2001;8:746–750. doi: 10.1038/nsb0901-746. [DOI] [PubMed] [Google Scholar]