

Abstract
Using a variety of biochemical and cell-based approaches, we show that estrogen receptor a (ERα) is acetylated by the p300 acetylase in a ligand- and steroid receptor coactivator (SRC)-dependent manner. Using mutagenesis and mass spectrometry, we identified two conserved lysine residues in ERα (Lys266 and Lys268) that are the primary targets of p300-mediated acetylation. These residues are acetylated in cells, as determined by immunoprecipitation-Western blotting experiments using an antibody that specifically recognizes ERα acetylated at Lys266 and Lys268. The acetylation of ERα by p300 is reversed by native cellular deacetylases, including TSA-sensitive enzymes (i.e., Class I and II deacetylases) and NAD+-dependent/nicotinamide-sensitive enzymes (i.e., Class III deacetylases, such as SIRT1). Acetylation at Lys266 and Lys268, or substitution of the same residues with glutamine (i.e., K266/268Q), a residue that mimics acetylated lysine, enhances the DNA binding activity of ERα in gel electrophoretic mobility shift assays. Likewise, substitution of Lys266 and Lys268 with glutamine enhances the ligand-dependent activity of ERα in a cell-based reporter gene assay. Collectively, our results implicate acetylation as modulator of the ligand-dependent gene regulatory activity of ERα. Such regulation is likely to play a role in estrogen-dependent signaling outcomes in a variety of estrogen target tissues in both normal and pathological states.
Keywords: acetylation, estrogen receptor alpha, deacetylation, DNA binding, p300, SIRT1
Introduction
The gene-regulatory actions of estrogens are mediated through two nuclear estrogen receptor (ER) proteins, ERα and ERβ, which belong to the nuclear receptor (NR) superfamily (1-3). ERs bind estrogens with high affinity and function as ligand-regulated transcription factors to control global patterns of gene expression. ERα and ERβ have unique, but overlapping, patterns of expression in a variety of estrogen target tissues, including mammary glands, uterus, and bone (4-6). In addition, the two ERs may exhibit distinct gene-regulatory activities under certain promoter and cell contexts (7-15).
ERα and ERβ share a conserved structural and functional organization, including: (1) an amino-terminal A/B region containing a transcriptional activation function (AF-1), (2) a DNA-binding domain (DBD), (3) and a carboxyl-terminal ligand-binding domain (LBD) containing a second transcriptional activation function (AF-2) (see Fig. 2B) (1, 3). In additional to these canonical nuclear receptor domains, recent studies have also begun to characterize the ″C-terminal extension″ (CTE) of the ERα and ERβ DBDs (amino acids 251-288 and 170-207, respectively), which plays a role in regulating the DNA-binding activities of the receptors (16, 17). The coordinated actions of the aforementioned ER functional domains allow for precisely controlled signal-regulated transcription in response to both natural and synthetic ER ligands.
Fig. 2.

The ERα DNA binding domain is the major target for E2- and SRC-dependent acetylation by p300. (A) The twenty nine lysine residues in ERα are listed by the ERα domain in which they appear. (B) Schematic diagram of the ERα deletion and point mutants used in the ERα acetylation assays. The wild type (Wt), K302/303R, ΔAB, and ΔDBD ERα are FLAG-tagged, whereas the LBD polypeptides (i.e., 282-595 and 282-420) are GST-tagged. The locations of Lys302 and Lys303 are indicated by white dots. (C) Acetylation assays with the point and deletion mutants shown in (B). Acetylation assays were performed as described for Fig. 1B. GST-SRC2(RID/PID) was added as indicated. The reactions were analyzed by polyacrylamide-SDS gel electrophoresis with subsequent fluorography. To allow direct comparisons of acetylation levels, equal molar (not equal mass) amounts of each protein were used, as verified by staining with Coomassie brilliant blue R-250.
The binding of agonistic ligands by ERα and ERβ promotes a conformational change in the receptor LBDs that allows the receptor to interact directly or indirectly with a diverse set of coregulatory proteins (1, 3). These include members of the steroid receptor coactivator (SRC) family (i.e., SRC1, 2, and 3), which function primarily as bridging factors to recruit other coregulators (18-20), including a diverse set of protein-modifying enzymes (e.g., acetylases, methyltransferases, kinases) (21, 22). p300 and its paralog CREB-binding protein (CBP) are the two best characterized mammalian acetylases. They function as coregulators for a variety of transcription factors, including ERs and other NRs (23-25). p300 and CBP are recruited to ERα and ERβ in a ligand-dependent manner via interactions with SRC proteins. During an estrogen-dependent transcriptional response, p300 and CBP can acetylate nucleosomal histones to alter chromatin structure and function (19, 22), as well as components of the transcription complex to alter transcriptional activity (see below). A variety of deacetylases, including trichostatin A (TSA)-sensitive enzymes (i.e., Class I and II deacetylases, such as histone deacetylase 1 or HDAC1) and NAD+-dependent/nicotinamide-sensitive enzymes (i.e., Class III deacetylases, such as SIRT1), also function as coregulators and can reverse the protein acetylation reactions catalyzed by acetylases (26-29).
A number of recent studies have shown that acetylation is an important covalent post-translational modification for regulating the activity of transcription-related factors, including p53, SRC3 (a.k.a. ACTR), NF-κB p65, and poly(ADP-ribose) polymerase-1 (30-39). Interestingly, different acetylases (e.g., p300/CBP versus PCAF) have different substrate preferences as demonstrated by the fact that some factors can be acetylated by one but not the other (35, 36, 38, 40). With regard to modulating the activity of transcription-related factors, the consequence of acetylation may be either transcriptional activation or inhibition. Acetylation of transcription-related factors can increase their transcriptional activity by: (1) enhancing DNA binding activity, (2) stimulating interactions with positive transcriptional regulators, such as chromatin remodeling factors or coactivators, (3) inhibiting interactions with negative regulators, resulting in a loss of transcription repression, (4) increasing the stability of the factors, and (5) altering subcellular localization (35, 36, 38, 40). Likewise, acetylation may inhibit the activity of transcription-related factors by reducing binding to DNA or chromatin, as well as reducing protein-protein interactions required for transcriptional activation (35, 36, 38, 40). Thus, the specific biochemical effects of acetylation are varied and differ with each target protein.
In the studies described herein, we show that ERα, but not ERβ, is a target for acetylation by p300. Using a variety of biochemical and cell-based assays, we have identified the sites of acetylation, and explored the mechanisms and functional consequences of p300-mediated acetylation on ERα activity. Collectively, our results implicate acetylation as modulator of the ligand-dependent gene regulatory activity of ERα.
Results
Human ERα is acetylated by p300 and deacetylated by TSA- and nicotinamide-sensitive deacetylases
To examine the acetylation of human ERα by p300, we used an in vitro acetylation assay with [3H]-acetyl coenzyme A (acetyl CoA) and the following purified recombinant proteins: ERα, p300, and GST-fused SRC2(RID/PID), which contains the receptor interaction domain (RID) and p300/CBP interaction domain (PID) of SRC2 (Fig. 1A). With this assay, we showed previously that SRCs play a key role in the targeted acetylation of nucleosomal core histones by p300, functioning as a bridging factor between p300 and E2-bound ERα (ref. 41 and Fig. 1B, bottom). Interestingly, these same interactions also promote the acetylation of ERα (Fig.1B, top), indicating that interactions among agonist-bound ERα, SRC and p300 are required for efficient acetylation of ERα by p300 (see also Supplemental Fig. 1). SRC2(RID/PID), which lacks the putative SRC acetylase domain (18, 21), was unable to promote the acetylation of ERα in the absence of p300 (data not shown).
Fig. 1.

ERα is acetylated by p300 and deacetylated by TSA- and nicotinamide-sensitive deacetylases. (A) Polyacrylamide-SDS gel analyses of purified recombinant ERα, GST-SRC2(RID/PID), p300, and SIRT1 stained with Coomassie brilliant blue R-250. Size markers in kDa are shown. The asterisk for the GST-SRC2(RID/PID) sample indicates a major breakdown product with minor breakdown products below. (B) p300 acetylates ERα and nucleosomal core histones in an E2- and SRC-dependent manner. ERα and salt-dialyzed chromatin were incubated with p300 in the presence of GST-SRC2(RID/PID), E2, and [3H]-acetyl CoA. The reactions were analyzed by polyacrylamide-SDS gel electrophoresis with subsequent fluorography. (C) ERα is deacetylated by TSA- and nicotinamide-sensitive deacetylases. [3H]-ERα was incubated with HeLa cell nuclear extract (HeLa NE) in the presence or absence of NAD+, TSA, or nicotinamide as indicated. The reactions were analyzed by polyacrylamide-SDS gel electrophoresis with subsequent fluorography. (D) ERα is deacetylated by the NAD+-dependent deacetylase SIRT1. The assays were set up as in (C) except that purified recombinant SIRT1 was used in place of the HeLa cell NE.
Next, we examined whether the p300-dependent acetylation of ERα can be reversed by native deacetylases present in a HeLa cell nuclear extract (HNE). Immobilized [3H]-acetylated ERα was incubated with HNE, which contains a variety of deacetylases including TSA-sensitive enzymes (i.e., Class I and II deacetylases) and NAD+-dependent/nicotinamide-sensitive enzymes (i.e., Class III deacetylases, such as SIRT1) (26-29). As shown in Fig. 1C, incubation with HNE dramatically reduced the acetylation of ERα (compare lanes 1 and 2). The addition of TSA inhibited deacetylation of ERα by the HNE (lane 3), indicating that one or more Class I/II deacetylases present in the HNE can deacetylate ERα. The addition of NAD+ in the presence of TSA (i.e., under conditions where Class I/II deacetylases were inhibited) also resulted in the deacetylation of ERα (compare lanes 3 and lane 4). The effect of NAD+ was blocked by the addition of nicotinamide (lane 5), indicating that one or more Class III deacetylases present in the HNE, such as SIRT1, can deacetylate ERα. To explore this last result further, we performed a similar set of experiments using purified recombinant SIRT1 (Fig. 1A) in place of the HNE. As shown in Fig. 1D, SIRT1 without added NAD+ had no effect on the acetylation of ERα (compare lanes 1 and 2), whereas SIRT1 in the presence of NAD+ was a potent deacetylase of ERα (lane 3). The deacetylation of ERα by SIRT1 + NAD+ was inhibited by nicotinamide (lane 4), but not by TSA (lane 5). Taken together, these results indicate that the acetylation of ERα by p300 can be reversed by native TSA- and nicotinamide-sensitive deacetylases, including SIRT1.
Initial identification of Lys268 and Lys266 as sites of acetylation in ERα
Full length ERα contains 29 lysine residues that are potential sites of acetylation by p300: 4 in the A/B region, 10 in the DBD, and 15 in the LBD (for the purposes of this study, the domains have the boundaries defined in Fig. 2A). In order to identify the lysine residues in ERα that are acetylated by p300, we used deletion mutants (Fig. 2, B and C), point mutants (Fig. 3), and an unbiased quantitative mass spectrometry approach (Fig. 4). ERα deletion mutants lacking either the A/B region (i.e., ERαΔAB), DBD (i.e., ERαΔDBD), or both the A/B region and the DBD [i.e., GST-LBD(282-595)] (Fig. 2B) were assayed for E2- and SRC-dependent acetylation by p300 in the presence of [3H]-acetyl CoA. Importantly, all three of these receptor deletions contain intact LBDs and SRC interaction domains, since efficient acetylation of ERα requires the binding of E2 and SRC (Figs. 1B and Supplemental Fig. 1). Note that for these assays, we used equal molar amounts of the purified receptor proteins, as opposed to equal mass amounts (Fig. 2C, bottom), so that the relative number of [3H]-acetyl groups added per mole of protein could be assessed. The ERα deletion mutant lacking the A/B region (i.e., ERαΔAB) showed a modest reduction in E2- and SRC-dependent acetylation by p300, whereas the ERα deletion mutants lacking the DNA binding domain [i.e., ERαΔDBD and GST-LBD(282-595)] showed a dramatic reduction in acetylation (Fig. 2C, top). These results suggested that the DBD is the major target for acetylation by p300. This result was confirmed by the independent approaches described below.
Fig. 3.

Mutation of Lys266 and Lys268 in ERα reduces acetylation by p300. (A) Acetylation assays with ERα lysine mutants. Purified FLAG-tagged wild type and Lys to Arg single-point mutant ERαs were assayed for acetylation by p300 in the presence of GST-SRC2(RID/PID) and E2 as described for Fig. 1B. top, Polyacrylamide-SDS gel analysis of the purified ERα proteins with subsequent staining using Coomassie brilliant blue R-250 to confirm equal protein amounts. Size markers in kDa are shown. bottom, Summary of the results from the acetylation assays. The ERα bands were excised from the gels after fluorography and quantified by liquid scintillation counting. The acetylation level of each lysine point mutant was expressed relative to wild type. Each bar represents the mean plus the SEM from at least three different experiments. (B) Acetylation assays with Lys to Arg 266/268 double-point mutant ERαs. Wild type and mutant ERαs were assayed for acetylation by p300 in the presence of GST-SRC2(RID/PID) and E2 as described for Fig. 1B. top, Polyacrylamide-SDS gel analysis of the purified ERα proteins with subsequent fluorography. bottom, The same gel stained using Coomassie brilliant blue R-250 to confirm equal protein amounts.
Fig. 4.

Quantitative mass spectrometric analysis of ERα acetylation by p300. (A) Schematic diagram of the quantitative mass spectrometric procedure for determining site-specific ERα acetylation levels. (B) Quantitative mass spectrometric determination of SRC- and E2-dependent acetylation by p300 of peptide 264-269 from trypsin-digested full-length wild type or K268Q mutant ERα. The ERαs were assayed for acetylation by p300 in the presence of GST-SRC2(RID/PID) and E2 as described for Fig. 1B and then subjected to quantitative MALDI-QqTOF spectrometry as described in (A). The data are expressed as amount of acetylated 264-269 peptide (i.e., AcK) relative to the total amount of 264-269 peptide in the reaction (i.e., AcK + DAcK). As indicated in Table 1, K268 is the major site (>95%) of single acetylation in the 264-269 peptide. In the K268Q mutant, all of the acetylation of the 264-269 peptide is at K266.
To determine the sites of acetylation more precisely, we individually changed the 10 lysine residues in the DBD and the 4 lysine residues in the A/B region to arginine, an amino acid that has a positively charged side chain like lysine, but cannot be acetylated. The 14 lysine to arginine (K⇒R) point mutant ERαs were expressed and purified as recombinant proteins, and then assayed for E2- and SRC-dependent acetylation by p300 versus wild type ERα. Of the 14 K⇒R point mutants tested, only two showed a reduction in acetylation: K268R (∼50% reduction) and K266R (∼20% reduction) (Fig. 3A and data not shown). When both Lys266 and Lys268 were mutated, there was a dramatic (>90%) reduction in acetylation (Fig. 3B). These results provided a first indication that Lys268 and Lys266, both of which are in the CTE of the DBD (17), are major and minor targets, respectively, for E2- and SRC-dependent acetylation by p300.
Confirmation by mass spectrometry of Lys268 and Lys266 as sites of acetylation in ERα
To confirm the sites of acetylation in ERα using an independent and unbiased assay, we used the quantitative mass spectrometry approach outlined in Fig. 4A (42). In vitro acetylation reactions with full length ERα plus SRC, p300, and E2 were performed in the absence or presence of cold acetyl CoA. Following enzymatic acetylation of the lysine residues in ERα specifically targeted by p300, all of the remaining unacetylated lysine residues were chemically acetylated using deuterated-acetic anhydride. The modified ERα protein was then digested with trypsin. The resulting peptides contained light acetyl groups (AcK) on lysines acetylated by p300 and heavy (deuterated) acetyl groups (DAcK) on all other lysines. The mass difference of 3 Da between AcK and DAcK on the peptides was visualized by matrix-assisted laser desorption/ionization (MALDI) quadrupole-quadrupole-time-of-flight (QqTOF) mass spectrometry (43). The percent acetylation of the chemically identical, isotopically distinct peptides (i.e., heavy versus light) was calculated directly from ratios of the intensities of the monoisotopic peaks. (42).
The mass spectrometry approach provided good coverage of the lysine residues in the A/B region, DBD, and hinge region spanning amino acids 1 through 298 (12 of the 14 lysine residues in this region were detected and quantified; Lys48 and Lys257 were in peptides that were not observed by the mass spectrometry approach) (Supplemental Table 1). Coverage in the LBD spanning amino acids 299 through 595 was less complete (only 8 of the 15 lysine residues in this region were detected and quantified) (Supplemental Table 1). However, the results in Fig. 2C indicate that the LBD is not a major target of acetylation by p300. Furthermore, when taken in combination, the three analytical approaches that we used (i.e., deletion mutants, point mutants, and mass spectrometry) yielded complete coverage of all 29 lysine residues in ERα (Supplemental Table 1).
Three of the detectable tryptic peptides from the mass spectrometry analysis had quantifiable levels of acetyl CoA-dependent acetylation (expressed as a percentage of the total amount of each peptide present in the reaction) (Table 1). The acetylated peptides spanned the following residues in ERα: (1) 244-256, containing Lys244 and Lys252, (2) 264-269, containing Lys266 and Lys268, and (3) 288-300, containing Lys299 (Table 2). The 264-269 peptide had approximately three- and six-fold more total p300-dependent acetylation than the 244-256 and 288-300 peptides, respectively (Table 1; 31.5 ± 0.9 % versus 11.1 ± 0.7 % and 4.5 ± 0.6 %, respectively), indicating that the 264-269 peptide contains the major target for E2- and SRC-dependent acetylation by p300. These results fit well with our mutagenesis experiments which identified Lys268 and Lys266 as major and minor sites of acetylation, respectively. Based on these results, we focused on Lys266 and Lys268 and explored the E2- and SRC-dependent acetylation of these residues by p300 in more detail.
Table 1.
Quantitative mass spectrometric analysis of ERα reveals Lys268 as a major site of acetylation by p300.
| Percentage of the total amount of ERα Tryptic Peptide Acetylated by p300 in an Acetyl CoA-dependent Manner1 | ||||
|---|---|---|---|---|
| ERα Peptide (Lysines) | Total Acetylation | Single Acetylation2 | Double Acetylation2 | Major Site of Single Acetylation2 |
| 244-256 (K244, K252) | 11.1 ± 0.7 % | 10.4 ± 0.9 % (∼10.1% from K252, ∼0.3% from K244) | 0.7 ± 0.4 % | K252 (∼97%) |
| 264-269 (K266, K268) | 31.5 ± 0.9 % | 24.7 ± 0.9 % (∼23.5% from K268, ∼1.2% from K266) | 6.8 ± 0.9 % | K268 (∼95%) |
| 288-300 (K299) | 4.5 ± 0.6 % | 4.5 ± 0.6 % | N/A | K299 (100%) |
Full-length ERα was acetylated by p300 in the presence of SRC(RID/PID) and E2. The acetylated ERα was then digested with trypsin and subjected to quantitative mass spectrometry as described in Materials and Methods. The percentage of the indicated peptide that was acetylated in an acetyl CoA-dependent manner was determined by comparing the results from acetylation reactions run with and without added acetyl CoA. Note that only the three peptides listed showed appreciable acetyl CoA-dependent acetylation by p300 in the mass spectrometry analysis. The values represent the mean ± SEM for three separate determinations.
As determined by MS/MS analysis using MALDI-ion trap mass spectrometry. The numbers in parentheses indicate the percentage of the singly acetylated peptide that was acetylated at the site listed.
MS/MS analysis using MALDI-ion trap mass spectrometry was used to quantify the acetylation of the two individual lysine residues in the 264-269 peptide (i.e., Lys266 and Lys268). For the singly acetylated species of the 264-269 peptide, >95% of the acetylation was on Lys268 (Table 1 and data not shown). In addition, the singly acetylated (i.e., primarily Ac-Lys268) species of the 264-269 peptide was present at approximately three- to four-fold more than the doubly acetylated (i.e., Ac-Lys266 + Ac-Lys268) species (Fig. 4B and Table 1). Furthermore, mutation of Lys268 to an unacetylatable residue (i.e., glutamine, Q) reduced the amount of both single and double acetylation of the 264-269 peptide (Fig. 4B), even though a dramatic (∼10-fold) compensatory increase in the acetylation of Lys266 was observed, from ∼1% of the total peptide for wild type ERα (Table 1, see ″Single Acetylation″ column for K266) to ∼10% of the total peptide for the K268Q mutant (Fig. 4B). Further MS/MS analyses demonstrated that both single and double acetylation of the 264-269 peptide occurred efficiently only when acetylation of the full length ERα was carried out in the presence of E2 and GST-SRC2(RID/PID) (Fig. 4B). Collectively, the mass spectrometry data support the conclusions that (1) Lys268 is the major site of E2- and SRC-dependent acetylation by p300 and (2) Lys266 is a minor site whose acetylation can be increased by mutation or prior acetylation of Lys268. These conclusions fit well with the conclusions from the mutagenesis studies described above.
Lys302 and Lys303 are not major sites of acetylation by p300 in full length ERα
Results from a recent study by Wang et al. (44) showed that Lys302 and Lys303 in an isolated fragment of ERα (i.e., amino acids 282-420) can be acetylated by p300 in the absence of E2 and SRC. We examined whether these same sites might be potential sites of acetylation in full length ERα by mutating both sites to arginine, a residue that cannot be acetylated. We found no difference in the acetylation of wild type or K302/303R ERα by p300 with or without E2 or GST-SRC2(RID/PID) (Figs. 2C and 3A, data not shown), indicating that Lys302 and Lys303 are not targets for acetylation by p300 in full length of ERα. These results we confirmed in our mass spectrometry analysis, which showed no detectable acetylation on Lys302 and Lys303 (Tables 1, Supplemental Table 1, and data not shown). Interestingly, a fragment of ERα containing the entire LBD and including Lys302 and Lys303 (i.e., amino acids 282-595) was not acetylated by p300 (Fig. 2C, lanes 9 and 10), yet the smaller fragment used by Wang et al. (44) (i.e., amino acids 282-420) was acetylated (Fig. 2C, lanes 11 and 12). These results suggest that Lys302 and Lys303 are cryptic residues which are not normally accessible to p300 in full length ERα. Truncation of the LBD (as in the 282-420 fragment) presumably disrupts the secondary and tertiary structure of the LBD, which could cause Lys302 and Lys303 to become exposed and accessible to acetylation by p300. Taken together, our results indicate that Lys302 and Lys303 are not major sites of acetylation by p300 in full length ERα.
Lys268 and Lys266 are conserved across species
Alignment of human ERα with the ERαs from a variety of other species shows that Lys266 and Lys268 have been conserved throughout evolution, at least from amphibians through mammals (Fig. 5A). Furthermore, three of the four fish species examined have at least one lysine in the same position as Lys266 and Lys268, or within one residue (Fig. 5A). Although alignment of human ERα with other human nuclear receptors is difficult for the CTE due to low sequence conservation, the alignment can be fixed at the most carboxyl-terminal conserved cysteine residue in the second zinc finger of the DBD (Cys240 for ERα; Fig. 5B). Such an analysis reveals that almost all non-orphan nuclear receptors have at least one, but typically two or more, and as many as six, lysine residues located in the amino-terminal portion of the CTE (+11 to +32 relative to the aforementioned conserved cysteine residue). This region includes the three lysine residues in the androgen receptor (AR) that are acetylated by p300/CBP (Lys630, Lys632, and Lys633) (45-47). These lysine-rich regions may be targeted for acetylation in other receptors as well. Interestingly, ERβ lacks lysine residues homologous to Lys266 and Lys268 in ERα (Fig. 5B), fitting well with our observation that ERβ is not acetylated by p300 under the same conditions that lead to acetylation of ERα (data not shown).
Fig. 5.

Alignment of the amino acid sequences surrounding Lys266 and Lys268 in human ERα with corresponding regions from other ERαs and other nuclear receptors. Sequence alignment of the C-terminal extension (CTE) of the human ERα DBD with the corresponding region of ERs from other species and other nuclear receptors. The alignments were anchored at the last conserved cysteine residue in the second zinc finger of the DBD (underlined C). The boxed region demarcates the amino-terminal portion of the CTE (i.e., those residues located within +11 to +32 of the aforementioned cysteine residue; note that the full CTE, as defined by Melvin et al. (17), extends to +47). Asterisks indicate residues that are conserved in all the receptors shown. (A) Alignment of ERs. The lysine residues (K) shown in bold correspond to Lys266 and Lys268 in human ERα. (B) Alignment of human nuclear receptors. All of the lysine residues (K) in the boxed region are shown in bold. K266 and K268 of ERα, and K630, K632, and K633 of AR are underlined.
Lys268 and Lys266 of ERα are acetylated in vivo
Next, we determined whether Lys266 and Lys268 are bona fide acetylation sites in vivo. For these studies, we generated an antiserum that specifically recognizes acetylated Lys266/Lys268 ERα (AcK266/268-ERα) using a doubly acetylated peptide antigen spanning amino acids 263 through 273 of human ERα. Due to sequence conservation among mammalian ERαs (Fig. 5A), this antiserum should recognize similarly acetylated ERα from most, if not all, mammalian species. As shown in the Western blots in Fig. 6A, this antiserum specifically recognizes purified wild type ERα that has been acetylated by p300 (middle panels), but not unacetylated wild type ERα (left panels). Furthermore, this antiserum does not recognize K266/268Q ERα (right panels) or K268Q ERα (data not shown) that has been incubated with p300 under conditions that lead to the acetylation of wild type ERα (i.e., +Acetyl CoA, +E2, +SRC) (right panels). These results, as well as the assays described below, demonstrate that this antiserum specifically recognizes acetylated K266/268 ERα.
Fig. 6.

Lys266 and Lys268 of ERα are acetylated in vivo. (A) Characterization of the AcK266/268-ERα antibody. Increasing amounts (10 ng to 300 ng) of purified acetylated or unacetylated wild type or K266/268Q mutant ERαs were subjected to polyacrylamide-SDS gel electrophoresis, followed by Western blotting with either anti-ERα antibody or anti-AcK266/268-ERα antibody. (B) Schematic diagram of the in vivo ERα acetylation assay. NA = nicotinamide. (C) and (D) Analysis of ERα acetylation in 231/ERα (C) and 293T cells transiently transfected with an ERα expression vector (D). Assays were performed as outlined in (B). The immunoprecipitated material was deacetylated by the addition of recombinant SIRT1 and NAD+ as indicated to demonstrate specificity of the AcK266/268-ERα antibody for the acetylated form of ERα.
Once we verified the specificity of our antiserum, we carried out in vivo acetylation assays by using the experimental scheme illustrated in Fig. 6B. Briefly, FLAG-tagged ERα was immunoprecipitated from one of two cell lines (231/ERα cells, which stably express FLAG-tagged ERα, or transfected 293T cells, which transiently express FLAG-tagged ERα). Note that the cells were treated with TSA and nicotinamide prior to collection to block the actions of deacetylases during the experiment. In the absence of these treatments, the acetylation of ERα was dramatically reduced (data not shown), suggesting that deacetylation of the receptor occurs rapidly in vivo. The immunoprecipitates were then analyzed for acetylated ERα using the acetylated Lys266/Lys268 ERα antiserum described above. To demonstrate acetylation dependence for the observed signal, the immunoprecipitated material was deacetylated by the addition of recombinant SIRT1 + NAD+ in some cases prior to Western blotting.
In 231/ERα cells, we observed basal acetylation of Lys266/Lys268 that was increased about two- to three-fold in the presence of E2 (Fig. 6C, compare lanes 1 and 2). As expected, the signal for acetylated Lys266/Lys268 ERα in the immunoprecipitated material was lost upon incubation with SIRT1 + NAD+ prior to Western blotting (compare lanes 3 and 4). These results indicate that ERα from a well-characterized estrogen-responsive cell line that stably expresses the receptor (see refs. 48, 49, for example) is acetylated at Lys266 and Lys268, and that the level of acetylation can be modulated by E2. Next, we used transient expression of FLAG-tagged ERα in 293T cells, which are ERα-negative, so that we could compare the acetylation of wild type ERα with an unacetylatable Lys266/Lys268 mutant (i.e., K266/268R) in vivo. Wild type ERα immunoprecipitated from the transfected 293T cells was acetylated at Lys266/Lys268 (Fig. 6D, lane 1). Again, as expected, the signal for acetylated Lys266/Lys268 ERα was lost upon incubation of the immunoprecipitated material with SIRT1 + NAD+ prior to Western blotting (lane 2). In contrast to wild type ERα, no acetylation of the K266/268R mutant was observed (Fig. 6D, lanes 3 and 4). Together, these cell-based studies using an antiserum that specifically recognizes acetylated Lys266/Lys268 ERα clearly demonstrate that ERα is acetylated at Lys266 and Lys268 in vivo.
Interestingly, similar experiments using an antiserum that specifically recognizes ERα singly acetylated at Lys266 (AcK266-ERα) or Lys268 (AcK268-ERα) failed to detect acetylated ERα in the immunoprecipitated material from 231/ERα cells and transfected 293T cells (data not shown). These results suggest that although the AcK268-ERα species may predominate in the in vitro reaction (Table 1), the AcK266/268-ERα species predominates in cells.
Acetylation or mutation of ERα at Lys266/Lys268 does not affect E2 binding, interaction with SRC2, or subcellular localization of ERα
Our initial studies using (1) a transcriptionally inactive ERα mutant defective in SRC binding (i.e., L540Q) and (2) a set of previously characterized polypeptide inhibitors that block ERα-SRC and SRC-p300/CBP interactions showed that acetylation of ERα by p300 correlates with E2-dependent transcriptional activation (Supplemental Fig. 1). In order to explore the possible effects of acetylation on the activities of ERα in more detail, we performed a number of functional assays to examine ligand binding (Supplemental Fig. 2A), ERα-SRC2 interactions (Supplemental Fig. 2B), subcellular localization (Supplemental Fig. 2C), DNA binding (Fig 7), and transactivation (Fig. 8). For these assays, we used a set of Lys266/Lys268 double mutants in which the lysine residues were changed to arginine (R, which has a positively charged side chain and mimics unacetylated lysine) or glutamine (Q, which has a neutral side chain and mimics acetylated lysine). As shown in Fig. 3B, ERα mutants harboring these changes (i.e., K266/268R and K266/268Q) showed a dramatic reduction in acetylation by p300 in vitro compared to wild type ERα (see also Figs. 6A and 6D). Our results indicate that acetylation or mutation of ERα at Lys266/Lys268 does not appreciably affect E2 binding, interaction with SRC2, or subcellular localization of ERα (Supplemental Fig. 2)
Fig. 7.

Acetylated ERα and K266/268Q ERα exhibit increased DNA binding activity. (A) Acetylation of ERα by p300 in vitro increases ERα DNA binding activity in EMSAs. Aliquots of purified ERα were incubated with p300, GST-SRC2(RID/PID), and E2 in the absence (i.e., no acetylation) or presence (i.e., acetylation) of unlabeled acetyl CoA as indicated. Aliquots of the reactions were subjected to Western blotting using an acetyl lysine antibody (top) or an ERα antibody (middle), or analyzed by EMSA (bottom). The fold increase of ERα:ERE complex formation upon acetylation is indicated. (B) Changing the p300 acetylation sites in ERα from Lys to Arg (which mimics unacetylated Lys) inhibits acetylation-dependent increases in the DNA binding activity of ERα in EMSAs. The assays were set up as described for (A) using wild type, K180R, and K266/268R mutant ERαs, followed by an EMSA. The fold increase of ERα:ERE complex formation upon acetylation is indicated. (C) Changing Lys266 and Lys268 in ERα to Gln (Q) increases the DNA binding activity of ERα in EMSAs. The EMSA assays were set up using increasing amounts of unacetylated wild type, K180R, and K266/K268Q ERαs. The DNA binding activities of the mutant ERαs were calculated relative to wild type ERα. Each bar represents the mean plus the SEM from at least three separate determinations.
Fig. 8.
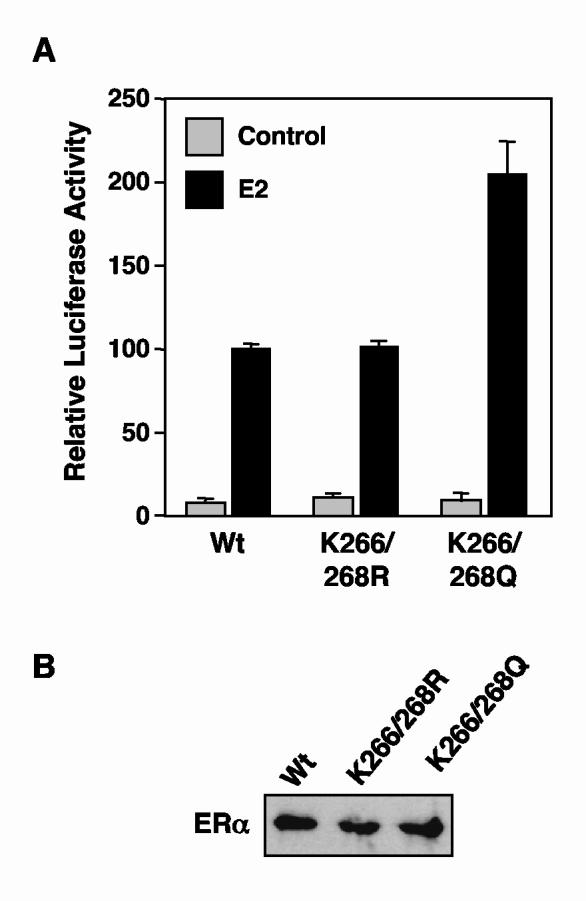
K266/268Q ERα, but not K266/268R ERα, exhibits increased transactivation activity in cells. (A) The transactivation activities of wild type and K266/268 mutant ERαs were assessed by transient transfection reporter gene assays. HeLa cells were transfected with expression vectors for wild type or K266/268 mutant ERαs, pGL3-2ERE-pS2-Luc (an E2-responsive luciferase reporter vector), and a β-galactosidase expression vector (used as an internal control). Transfected cells were treated with 10 nM E2 for 18 hrs prior to collection for determination of luciferase and β-galactosidase activities. The luciferase activity in each sample was normalized to β-galactosidase activity. Each bar represents the mean plus the SEM from at least three separate determinations. (B) Expression levels of wild type and K266/268 mutant ERαs in the transfected HeLa cells. Extracts from the transfected HeLa cells described in (A) were analyzed by Western blotting for ERα. The samples were normalized for β-galactosidase activity and total protein amount.
Acetylated ERα and K266/268Q ERα exhibit increased DNA binding activity
Next, we examined the effects of acetylation on ERα DNA binding activity by using electrophoretic mobility shift assays (EMSAs). In our initial assays, purified wild type ERα was incubated with p300 in the presence of E2 and GST-SRC(RID/PID) with or without unlabeled acetyl CoA to generate acetylated or unacetylated ERα, respectively. Aliquots of each reaction were subjected to Western blotting using: (1) an anti-acetyl-lysine antibody to confirm the acetylation of ERα, which occurred only in the presence of acetyl CoA, as expected (Fig. 7A, top) and (2) an anti-ERα antibody, which detected total ERα (Fig. 7A, middle). Another aliquot from each reaction was used in EMSAs with a [32P]-labeled double-stranded DNA probe containing an ERE sequence (Fig. 7A, bottom). Interestingly, acetylated ERα showed an approximate 4-fold increase in DNA binding activity compared to mock acetylated ERα (i.e., reaction without acetyl CoA). This effect was blocked by mutation of Lys266/Lys268 to arginine, but not Lys180, which was used as a control (Fig. 7B). Interestingly, mutation of Lys266/Lys268 to glutamine (i.e., K266/268Q), but not arginine (i.e., K266/268R), increased DNA binding activity of unacetylated ERα by about 5-fold (Fig. 7C). Thus, ERα DNA binding activity increases as the side chain charge at residues 266 and 268 are neutralized (from +1 at lysine to neutral at glutamine). Together, these results indicate that acetylation at Lys266/Lys268 can play a role in regulating the DNA binding activity of ERα.
K266/268Q ERα, but not K266/268R ERα, exhibits increased transactivation activity in cells
Finally, to determine if the increased DNA binding activity observed in the EMSAs might lead to enhanced transactivation, we tested the activity of the K266/268Q and K266/268R mutant ERαs using a cell-based reporter gene assay. Briefly, HeLa cells grown in estrogen-free medium were transfected with a vector for the expression of wild type, K266/268Q, or K266/268R ERα and a luciferase reporter construct containing two EREs upstream of the estrogen-regulated pS2 promoter. After treatment with vehicle or E2 for 18 hr, the cells were collected, extracts were prepared, luciferase activity was measured (Fig. 8A), and relative ERα levels were determined by Western blotting (Fig. 8B; no differences were observed). The K266/268Q mutant, which mimics acetylated ERα, gave a two-fold increase in reporter gene activity relative to wild type ERα (Fig. 8A), suggesting that acetylation at Lys266/Lys268 can play a role in regulating the transcriptional activity of ERα. In contrast, the K266/268R mutant, which mimics unacetylated ERα, gave reporter gene activity similar to wild type ERα (Fig. 8A). The lack of an inhibitory effect with the K266/268R mutant may be a consequence of the limited sensitivity of our reporter gene assays, as well as the extent of acetylation required for an observable effect. With regard to this latter point, note that with wild type ERα, perhaps as few as 5% of the receptor molecules in the cells are acetylated (data not shown), whereas with the K266/268Q mutant, 100% of the receptor molecules are ″acetylated″. Collectively, our results demonstrate a good correlation between the increased DNA binding and transactivation activities of the K266/268Q mutant ERα, when compared to wild type ERα.
Discussion
Acetylation modulates the activity of ERα, androgen receptor, and possibly other nuclear receptors
In the studies described herein, we have shown that ERα is acetylated by p300 at Lys266 and Lys268 in an SRC-dependent manner (Figs. 1B, 3, and 4), with Lys268 being the major site of acetylation in full-length ERα (Fig. 4 and Table 1). The extent of ERα acetylation is regulated by E2 both in vitro and in vivo (Figs. 1B and 6C). In addition, the acetylation of ERα is reversed by native cellular deacetylases, including TSA-sensitive enzymes (i.e., Class I and II deacetylases) and NAD+-dependent/nicotinamide-sensitive enzymes (i.e., Class III deacetylases, such as SIRT1) (Figs. 1C and 1D). Furthermore, our results indicate that acetylation at Lys266 and Lys268 regulates the DNA binding and transcriptional activities of ERα (Figs. 7 and 8). Collectively, our results implicate acetylation as modulator of the ligand-dependent gene regulatory activity of ERα. Ultimately, such regulation is likely to play a role in E2-dependent signaling outcomes in a variety estrogen target tissues in both normal and pathological states.
To date, two other nuclear receptors have been shown to be targets for acetylation: androgen receptor (AR) (46) and thyroid hormone receptor (TR) (50). Acetylation of AR by p300/CBP and PCAF decreases corepressor binding, increases coactivator binding, and increases ligand-dependent transactivation (45, 46). In addition, acetylation of AR may also regulate MEKK1-induced apoptosis (51). Mutation of the AR acetylation sites also inhibits proper trafficking of the receptor, although it is not clear whether this is directly related to impaired acetylation of AR (47). Interestingly, HDAC1 has been shown to interact with AR in the absence of androgen and dissociate in the presence of androgen, suggesting that reversible acetylation might play a role in regulating activity of AR (51). In contrast to AR, little is known about the functional consequences of TR acetylation (50).
Alignment of human ERα with the ERαs from a variety of other species shows that Lys266 and Lys268 are highly conserved from amphibians through mammals (Fig. 5A). In addition, most non-orphan nuclear receptors typically have two or more, and as many as six, lysine residues located in the amino-terminal portion of the CTE, corresponding to the region where Lys266 and Lys268 are located in ERα (Fig. 5B). It is interesting to speculate that this region of nuclear receptors might represent a common target for acetylation. In this regard, note that this region includes the three lysine residues in AR that are acetylated by p300/CBP (Lys630, Lys632, and Lys633) (45-47) (Fig. 5B). Interestingly, human ERβ lacks lysine residues homologous to Lys266 and Lys268 in ERα (Fig. 5B), fitting well with our observation that ERβ is not acetylated by p300 under the same conditions that lead to acetylation of ERα (data not shown). These differences in acetylation by p300 may account for some of the functional differences that have been noted for ERα and ERβ (7-15).
Acetylation of Lys266 and 268 enhances the DNA binding and transactivation activities of ERα
Our results demonstrate that acetylation of ERα at Lys266 and Lys268 by p300 enhances the DNA binding activity of ERα (Fig. 7), an effect that appears to be dependent on the neutralization of positive charges at those residues. In fact, substitution of Lys266 and Lys268 for glutamate (i.e., K266/268E), which has a negatively charged side chain, enhances ERα DNA binding activity to an even greater extent (∼10-fold more than wild type ERα; data not shown) than substitution for glutamine (i.e., K266/268Q; ∼5-fold; Fig. 7). Since we lack structural information about this region, however, it is not clear whether Lys266 and Lys268 directly contact the DNA or are involved in a critical intramolecular interactions that regulate the DNA binding activity of ERα. The former possibility seems less likely since one might expect the loss of positive charge at Lys266 and Lys268 upon acetylation to reduce, not enhance, interactions with negatively charged DNA. Furthermore, Lys266 and Lys268 are located outside of the core DBD in the CTE (Fig. 5A). Results from a recent study suggests that the CTE is required for the binding of ERα to imperfect EREs or half ERE sites and may be a target during the enhancement of ERα DNA binding by HMGB-1/-2 proteins (17). Acetylation of Lys266 and Lys268 may play a role in modulating the structure of the CTE to enhance the DNA binding activity of ERα. Whether the increase in ERα DNA binding activity observed upon acetylation Lys266 and Lys268 is solely responsible for the concomitant increase in transcriptional activity has not yet been determined.
A possible role for regulated post-translational modification in determining the activity of ERα
The enzymes that regulate the acetylation state of ERα (i.e., the acetylases and deacetylases) are likely to play a key role in modulating the activity of ERα. p300 and its paralog CREB binding protein are potent acetylases that play multiple roles in ERα-dependent gene regulation (18, 22). Likewise, deacetylases have also been shown to play important roles in ERα-dependent gene regulation (52-56). Distinguishing the direct effects of these enzymes on ERα acetylation status from their effects on other transcription-related targets will require further investigation.
Interestingly, resveratrol, an activator of the NAD+-dependent deacetylase SIRT1 (57), has been shown to modulate estrogen-dependent signaling pathways and inhibit estrogen-dependent cell proliferation (58). Although direct effects of resveratrol on the activity of ERα are impossible to rule out (59-62), it is interesting to speculate that perhaps some of the antagonistic actions of resveratrol on estrogen signaling relate to its ability to stimulate SIRT1 activity (i.e., resveratrol might enhance the deacetylation of ERα by SIRT1, thus reducing ERα DNA binding and transcriptional activities). The ability of SIRT1 to deacetylate ERα suggests a possible link between nuclear NAD+ metabolism, the regulation of ERα activity by acetylation, and cell proliferation. If this is the case, the acetylation status of ERα, as measured by acetylation-specific antibodies such as the ones described herein, might be a useful additional prognostic indicator for breast cancers.
Also of note with regard to the regulatory aspects of ERα post-translational modification is that Lys266 and Lys268 in ERα, which are sites of acetylation, have recently been shown to be targets for SUMOylation as well (63). This suggests an intriguing interplay between these two post-translational modifications in the regulation of ERα activity. As shown previously, multiple covalent post-translational modifications of a single protein can interact functionally to add additional levels of regulatory control (40, 64-68). This possibility, and the others noted above, will be examined in future studies.
Materials and Methods
Chemicals
Acetyl coenzyme A (acetyl CoA), 17β-estradiol (E2), nicotinamide, nicotinamide adenine dinucleotide (NAD+), and trichostatin A (TSA) were from Sigma (St. Louis, MO). [3H]-acetyl CoA was from PerkinElmer Life and Analytical Sciences (Boston, MA).
Synthesis and purification of recombinant proteins
FLAG-tagged wild type human ERα and his6-tagged human p300 were expressed in Sf9 cells by using recombinant baculoviruses and purified as described previously (69, 70). Mutant human ERα cDNAs, including hERα ΔAB (180-595), hERα ΔDBD (1-180/269-595), hERα L540Q, and various hER lysine point mutants, were generated either by PCR or site-directed mutagenesis. The corresponding FLAG-tagged mutant ERα proteins were expressed in Sf9 cells by using recombinant baculoviruses and purified using FLAG M2 affinity chromatography as described for wild type ERα. The his6-tagged mouse SIRT1 (a.k.a. Sir2a) expression construct was provided by Shin-ichiro Imai, Washington University, St. Louis. The corresponding protein was expressed in E. coli and purified by standard nickel-NTA affinity chromatography. The GST-LBD(282-595) and GST-LBD(282-420) expression plasmids were provided by Benita Katzenellenbogen, University of Illinois, Urbana-Champaign and Richard Pestell, Georgetown University, respectively. The corresponding GST-fusion proteins were expressed in E. coli and purified by standard glutathione-agarose affinity chromatography. GST-fused SRC2(RID/PID) was expressed in E. coli and purified by glutathione-agarose affinity chromatography as described previously (41). All purified proteins were frozen in aliquots in liquid N2 and stored at -80°C. Aliquots were analyzed by polyacrylamide-SDS gel electrophoresis with Coomassie brilliant blue R-250 staining relative to BSA mass standards.
In vitro ERα and nucleosomal core histone acetylation assays
ERα and nucleosomal core histone acetylation reactions with [3H]-acetyl-CoA were carried out essentially as described previously (41). Briefly, ERα was incubated in the presence (Fig. 1B) or absence (all other figures) of salt-dialyzed chromatin, with or without p300, GST-SRC2(RID/PID), E2, and [3H]-acetyl-CoA as indicated in a final volume of 35 mL under reaction conditions described previously (71, 72). The chromatin was prepared by salt dialysis using a plasmid DNA template with four tandem EREs and was purified on sucrose gradients to remove free histones (41, 73). The reactions were incubated at 27°C for 30 min and aliquots were analyzed by both 10% and 15% polyacrylamide-SDS gel electrophoresis to resolve ERα and core histones, respectively. The proteins in the gels were detected by staining using Coomassie brilliant blue R-250, followed by fluorography. The [3H]-labeled ERα and core histone bands were excised from the gel and quantified by liquid scintillation counting. Acetylation reactions with unlabeled acetyl-CoA were carried out under similar reaction conditions, however the acetylated target proteins were detected by Western blotting with antibodies to acetylated lysine (New England Biolabs, Ipswich, MA) or acetylated ERα (see description below). Mock acetylation reactions lacked acetyl CoA or GST-SRC2(RID/PID), as indicated.
In vitro ERα deacetylation assays
Purified FLAG-tagged ERα was immobilized on FLAG M2-agarose resin and acetylated by p300 in the presence of [3H]-acetyl-CoA as described above to generate [3H]-acetylated ERα. After extensive washing to remove the p300 and [3H]-acetyl-CoA, the ERα was eluted by using FLAG peptide, aliquoted, frozen in liquid N2, and stored at -80°C until use. For deacetylation reactions, [3H]-acetylated ERα was incubated with HeLa cell nuclear extract or purified SIRT1 in deacetylation buffer (50 mM Tris-HCl pH 7.5, 50 mM NaCl, 4 mM MgCl2) for 40 min at 27°C in the presence or absence of TSA (10 μM), NAD+ (400 μM), and nicotinamide (4 mM) in a final volume of 100 μL as indicated. After the reactions were complete, the samples were incubated with FLAG M2-agarose resin for 2 hrs at 4°C to concentrate the ERα protein, followed by extensive washing. The resin was boiled in SDS loading solution and the samples were resolved by polyacrylamide-SDS gel electrophoresis with subsequent fluorography.
Mass spectrometric analysis of acetylated ERα
In vitro acetylation of ERα was analyzed by quantitative mass spectrometry (42). ERα was acetylated by p300 in the presence of unlabeled acetyl CoA under the conditions described above. The ERα was separated from the other proteins in the reaction by polyacrylamide-SDS gel electrophoresis with subsequent staining using Coomassie brilliant blue R-250. Gel slices containing the ERα were excised, treated with iodoacetamide to block oxidation of cysteines, washed, and dehydrated with acetonitrile. A mixture of 30% deuterium-acetic anhydride (D6-acetic anhydride) in 100 mM ammonium bicarbonate was added to the gel slices to acetylate all unmodified lysine residues in ERα with deuterated acetyl groups. Following these chemical modifications, the ERα protein was digested with trypsin for 7 hours at 37°C in the gel slices. Because of the complete acetylation of all lysine residues, trypsin cut only after arginine in these reactions. The resulting peptides contained light acetylation on lysines acetylated by p300 and heavy (deuterated) acetylation on all other lysines. This translated into a mass difference of minus 3 Da for acetylation visualized by matrix-assisted laser desorption/ionization (MALDI) quadrupole-quadrupole-time-of-flight (QqTOF) mass spectrometry (43). The percent acetylation of the chemically identical, isotopically distinct peptides (i.e., heavy versus light) was calculated directly from ratios of the intensities of the monoisotopic peaks. The specific sites of acetylation in the peptides showing acetyl CoA-dependent acetylation were confirmed by MS/MS analysis using a MALDI-ion trap mass spectrometer (42).
Generation of acetylated ERα antibodies
Rabbit anti-acetylated Lys266/268 human ERα antiserum (AcK266/268-ERα) was generated by Covance Research Products, Inc. (Denver, PA) using standard techniques for peptide immunogens. Briefly, a double acetylated peptide spanning amino acids 263 through 273 of human ERα (Arg-Met-Leu-acetyl-Lys-His-acetyl-Lys-Arg-Gln-Arg-Asp-Asp) was conjugated to KLH and used as an immunogen in rabbits. Non-acetylation-specific antibodies were depleted from the antiserum by adsorption to a non-acetylated peptide affinity matrix with subsequent collection of the unbound material. The antiserum was screened by ELISA and Western blotting using acetylated and unacetylated purified recombinant human ERα.
In vivo ERα acetylation assays
MDA-MB-231 human breast cancer cells stably expressing FLAG-tagged human ERα (231/ERα cells) (49) were grown in DME/F12 containing 10% charcoal-dextran stripped calf serum (CDCS). 293T human kidney epithelial cells were grown in DMEM containing 10% FBS and were transfected with a vector (pCMV5) for the expression of FLAG-tagged wild type or K266/268R human ERα using Fugene 6 transfection reagent (Roche Diagnostics Corp., Indianapolis, IN) 18 hr prior to subsequent treatments. Both the 231/ ERα cells and the transfected 293T cells were treated with 2 μM TSA and 5 mM nicotinamide in the presence or absence of 100 nM E2 for two hours prior to collection. The cells were lysed in lysis buffer (10 mM Tris-HCl pH 7.5, 0.5 M NaCl, 0.1% NP-40, 0.5 mM EDTA, 10% glycerol) supplemented with a complete protease inhibitor cocktail (Roche Diagnostics Corp., Indianapolis, IN), TSA, and nicotinamide. The whole cell lysates were diluted with an equal volume of incubation buffer (25 mM Tris-HCl pH 7.9, 0.5% Triton X-100, 0.05% SDS, and 3 mM EDTA) and incubated with FLAG M2-agarose for 2 hr at 4°C with gentle mixing. Following the incubation, the resin was washed three times with wash buffer (20 mM Tris-HCl pH 7.5, 300 mM NaCl, 10% glycerol, 1.5 mM MgCl2, and 0.2% NP-40). After the final wash, the immunoprecipitated ERα was subjected to deacetylation or mock deacetylation in reactions containing 50 mM Tris-HCl pH 7.5, 50 mM NaCl, 4 mM MgCl2 with or without purified SIRT1 and NAD+ as indicated. The reactions were incubated for 1 hr at room temperature with gentle mixing, followed by extensive washing of the resin. After the final wash, the resin was boiled in SDS loading solution and the samples were resolved by polyacrylamide-SDS gel electrophoresis with subsequent Western blotting using an antibody to ERα or Ac-K266/268-ERα.
Electrophoretic mobility shift assays (EMSAs)
Purified recombinant wild type or lysine point mutant ERαs, which were either unmodified, acetylated by p300, or mock acetylated as described above, were used for the EMSAs. EMSAs were performed essentially as described previously (74). Briefly, 20 nM of ERα was incubated with [32P]-end-labeled double-stranded oligonucleotide containing a consensus ERE sequence on ice for 20 min in the presence or absence of E2 (100 nM). The samples were analyzed on nondenaturing 4.8% polyacrylamide gels run in 1x TBE. Quantification of the shifted ERα:ERE complexes was done by phosphorimager analysis with ImageQuant v1.2 software (Molecular Dynamics).
Transient transfection reporter gene assays
HeLa cells were grown in DME/F12 containing 10% charcoal-dextran stripped FBS (CDFBS). The cells were plated in 6-well plates 12 hr prior to transfection and reached ∼70% confluence prior to transfection using Fugene 6 transfection reagent (Roche Diagnostics Corp., Indianapolis, IN). Each well received the following combination of plasmid DNAs: (1) 5 ng of a pCMV5 vector for the expression of wild type or mutant ERα, or 5 ng of an empty pCMV5 control vector, (2) 250 ng of an estrogen-responsive luciferase reporter construct containing two EREs upstream of the human pS2 (a.k.a. TFF1) promoter (pGL3-2ERE-pS2-Luc), and (3) 100 ng of pCMVβ, a constitutive β-galactosidase expression vector used for normalization. Twelve hr after transfection, the cells were treated with vehicle or 10 nM of E2 for an additional 18 hr. Luciferase activity was measured in extracts from the transfected cells using a 96-well plate luminometer (Beckman Coulter LD400) and normalized to β-galactosidase activity measured in the same extracts using the plate reader. To ensure reproducibility, each assay was run in duplicate, and each experiment was performed at least three times.
Supplementary Material
Acknowledgements
We would like to thank the following people for their assistance with various aspects of this work: Tong Zhang, Miltos Kininis, Dave Wacker, Kristine Hope, Don Ruhl, Matt Gamble and Raga Krishnakumar for critical reading of the manuscript; Christian Fritze for the anti-acetylated ERα antibodies; Brian Chait for assistance with mass spectrometry; Richard Pestell, Shin-ichiro Imai, Benita Katzenellenbogen, and Michael Hottiger for plasmid DNA constructs; Mark Roberson for use of a luminometer; and Volker Vogt for advice and helpful discussions. This work was funded by grants from the American Cancer Society (RSG TBE-105130) and the NIH/NIDDK (R01 DK058110) to W.L.K.
Footnotes
Notice: This is an un-copyedited author manuscript copyrighted by The Endocrine Society. This may not be duplicated or reproduced, other than for personal use or within the rule of ″Fair Use of Copyrighted Materials″ (section 107, Title 17, U.S. Code) without permission of the copyright owner, The Endocrine Society. From the time of acceptance following peer review, the full text of this manuscript is made freely available by The Endocrine Society at http://www.endojournals.org/. The final copy edited article can be found at http://www.endojournals.org/. The Endocrine Society disclaims any responsibility or liability for errors or omissions in this version of the manuscript or in any version derived from it by the National Institutes of Health or other parties. The citation of this article must include the following information: author(s), article title, journal title, year of publication and DOI.
Disclosures: M.Y.K., E.M.W., Y.T.E.C., D.R.H., and W.L.K. have no potential conflicts of interests to declare with entities directly related to the material being published.
Grant Support: This work was funded by grants from the American Cancer Society (RSG TBE-105130) and the NIH/NIDDK (R01 DK058110) to W.L.K.
References
- 1.Enmark E, Gustafsson JA. Oestrogen receptors - an overview. J Intern Med. 1999;246:133–138. doi: 10.1046/j.1365-2796.1999.00545.x. [DOI] [PubMed] [Google Scholar]
- 2.Mangelsdorf DJ, Thummel C, Beato M, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Warner M, Nilsson S, Gustafsson JA. The estrogen receptor family. Curr Opin Obstet Gynecol. 1999;11:249–254. doi: 10.1097/00001703-199906000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Couse JF, Korach KS. Estrogen receptor null mice: what have we learned and where will they lead us? Endocr Rev. 1999;20:358–417. doi: 10.1210/edrv.20.3.0370. [DOI] [PubMed] [Google Scholar]
- 5.Nilsson S, Makela S, Treuter E, et al. Mechanisms of estrogen action. Physiol Rev. 2001;81:1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 6.Pettersson K, Gustafsson J. Role of estrogen receptor beta in estrogen action. Annu Rev Physiol. 2001;63:165–192. doi: 10.1146/annurev.physiol.63.1.165. [DOI] [PubMed] [Google Scholar]
- 7.Barkhem T, Carlsson B, Nilsson Y, Enmark E, Gustafsson J, Nilsson S. Differential response of estrogen receptor alpha and estrogen receptor beta to partial estrogen agonists/antagonists. Mol Pharmacol. 1998;54:105–112. doi: 10.1124/mol.54.1.105. [DOI] [PubMed] [Google Scholar]
- 8.Paech K, Webb P, Kuiper GG, et al. Differential ligand activation of estrogen receptors ERalpha and ERbeta at AP1 sites. Science. 1997;277:1508–1510. doi: 10.1126/science.277.5331.1508. [DOI] [PubMed] [Google Scholar]
- 9.Kuiper GG, Lemmen JG, Carlsson B, et al. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology. 1998;139:4252–4263. doi: 10.1210/endo.139.10.6216. [DOI] [PubMed] [Google Scholar]
- 10.McInerney EM, Weis KE, Sun J, Mosselman S, Katzenellenbogen BS. Transcription activation by the human estrogen receptor subtype beta (ER beta) studied with ER beta and ER alpha receptor chimeras. Endocrinology. 1998;139:4513–4522. doi: 10.1210/endo.139.11.6298. [DOI] [PubMed] [Google Scholar]
- 11.Cowley SM, Parker MG. A comparison of transcriptional activation by ER alpha and ER beta. J Steroid Biochem Mol Biol. 1999;69:165–175. doi: 10.1016/s0960-0760(99)00055-2. [DOI] [PubMed] [Google Scholar]
- 12.Delaunay F, Pettersson K, Tujague M, Gustafsson JA. Functional differences between the amino-terminal domains of estrogen receptors alpha and beta. Mol Pharmacol. 2000;58:584–590. doi: 10.1124/mol.58.3.584. [DOI] [PubMed] [Google Scholar]
- 13.Jones PS, Parrott E, White IN. Activation of transcription by estrogen receptor alpha and beta is cell type- and promoter-dependent. J Biol Chem. 1999;274:32008–32014. doi: 10.1074/jbc.274.45.32008. [DOI] [PubMed] [Google Scholar]
- 14.Meyers MJ, Sun J, Carlson KE, Marriner GA, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor-beta potency-selective ligands: structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J Med Chem. 2001;44:4230–4251. doi: 10.1021/jm010254a. [DOI] [PubMed] [Google Scholar]
- 15.Cheung E, Schwabish MA, Kraus WL. Chromatin exposes intrinsic differences in the transcriptional activities of estrogen receptors alpha and beta. Embo J. 2003;22:600–611. doi: 10.1093/emboj/cdg037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Melvin VS, Roemer SC, Churchill ME, Edwards DP. The C-terminal extension (CTE) of the nuclear hormone receptor DNA binding domain determines interactions and functional response to the HMGB-1/-2 co-regulatory proteins. J Biol Chem. 2002;277:25115–25124. doi: 10.1074/jbc.M110400200. [DOI] [PubMed] [Google Scholar]
- 17.Melvin VS, Harrell C, Adelman JS, Kraus WL, Churchill M, Edwards DP. The role of the C-terminal extension (CTE) of the estrogen receptor alpha and beta DNA binding domain in DNA binding and interaction with HMGB. J Biol Chem. 2004;279:14763–14771. doi: 10.1074/jbc.M313335200. [DOI] [PubMed] [Google Scholar]
- 18.McKenna NJ, Lanz RB, O'Malley BW. Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev. 1999;20:321–344. doi: 10.1210/edrv.20.3.0366. [DOI] [PubMed] [Google Scholar]
- 19.Lee KC, Kraus WL. Nuclear receptors, coactivators and chromatin: new approaches, new insights. Trends. Endrinol. Metab. 2001;12:191–197. doi: 10.1016/s1043-2760(01)00392-7. [DOI] [PubMed] [Google Scholar]
- 20.Leo C, Chen JD. The SRC family of nuclear receptor coactivators. Gene. 2000;245:1–11. doi: 10.1016/s0378-1119(00)00024-x. [DOI] [PubMed] [Google Scholar]
- 21.Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14:121–141. [PubMed] [Google Scholar]
- 22.Kraus WL, Wong J. Nuclear receptor-dependent transcription with chromatin. Is it all about enzymes? Eur J Biochem. 2002;269:2275–2283. doi: 10.1046/j.1432-1033.2002.02889.x. [DOI] [PubMed] [Google Scholar]
- 23.Blobel GA. CREB-binding protein and p300: molecular integrators of hematopoietic transcription. Blood. 2000;95:745–755. [PubMed] [Google Scholar]
- 24.Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- 25.Vo N, Goodman RH. CREB-binding Protein and p300 in Transcriptional Regulation. J Biol Chem. 2001;276:13505–13508. doi: 10.1074/jbc.R000025200. [DOI] [PubMed] [Google Scholar]
- 26.Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- 27.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marks PA, Miller T, Richon VM. Histone deacetylases. Curr Opin Pharmacol. 2003;3:344–351. doi: 10.1016/s1471-4892(03)00084-5. [DOI] [PubMed] [Google Scholar]
- 29.Gray SG, Ekstrom TJ. The human histone deacetylase family. Exp Cell Res. 2001;262:75–83. doi: 10.1006/excr.2000.5080. [DOI] [PubMed] [Google Scholar]
- 30.Luo J, Li M, Tang Y, Laszkowska M, Roeder RG, Gu W. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc Natl Acad Sci U S A. 2004;101:2259–2264. doi: 10.1073/pnas.0308762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 32.Chen H, Lin RJ, Xie W, Wilpitz D, Evans RM. Regulation of hormone-induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell. 1999;98:675–686. doi: 10.1016/s0092-8674(00)80054-9. [DOI] [PubMed] [Google Scholar]
- 33.Chen LF, Mu Y, Greene WC. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. Embo J. 2002;21:6539–6548. doi: 10.1093/emboj/cdf660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen L, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science. 2001;293:1653–1657. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- 35.Bannister AJ, Miska EA. Regulation of gene expression by transcription factor acetylation. Cell Mol Life Sci. 2000;57:1184–1192. doi: 10.1007/PL00000758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sterner DE, Berger SL. Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev. 2000;64:435–459. doi: 10.1128/mmbr.64.2.435-459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kiernan R, Bres V, Ng RW, et al. Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J Biol Chem. 2003;278:2758–2766. doi: 10.1074/jbc.M209572200. [DOI] [PubMed] [Google Scholar]
- 38.Kouzarides T. Acetylation: a regulatory modification to rival phosphorylation? Embo J. 2000;19:1176–1179. doi: 10.1093/emboj/19.6.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hassa PO, Haenni SS, Buerki C, et al. Acetylation of poly(ADP-ribose) polymerase-1 by p300/CREB-binding protein regulates coactivation of NF-kappaB-dependent transcription. J Biol Chem. 2005;280:40450–40464. doi: 10.1074/jbc.M507553200. [DOI] [PubMed] [Google Scholar]
- 40.Freiman RN, Tjian R. Regulating the regulators: lysine modifications make their mark. Cell. 2003;112:11–17. doi: 10.1016/s0092-8674(02)01278-3. [DOI] [PubMed] [Google Scholar]
- 41.Kim MY, Hsiao SJ, Kraus WL. A role for coactivators and histone acetylation in estrogen receptor alpha-mediated transcription initiation. Embo J. 2001;20:6084–6094. doi: 10.1093/emboj/20.21.6084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith CM, Gafken PR, Zhang Z, Gottschling DE, Smith JB, Smith DL. Mass spectrometric quantification of acetylation at specific lysines within the amino-terminal tail of histone H4. Anal Biochem. 2003;316:23–33. doi: 10.1016/s0003-2697(03)00032-0. [DOI] [PubMed] [Google Scholar]
- 43.Krutchinsky AN, Zhang W, Chait BT. Rapidly switchable matrix-assisted laser desorption/ionization and electrospray quadrupole-time-of-flight mass spectrometry for protein identification. J Am Soc Mass Spectrom. 2000;11:493–504. doi: 10.1016/S1044-0305(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 44.Wang C, Fu M, Angeletti RH, et al. Direct acetylation of the estrogen receptor alpha hinge region by p300 regulates transactivation and hormone sensitivity. J Biol Chem. 2001;276:18375–18383. doi: 10.1074/jbc.M100800200. [DOI] [PubMed] [Google Scholar]
- 45.Fu M, Rao M, Wang C, et al. Acetylation of androgen receptor enhances coactivator binding and promotes prostate cancer cell growth. Mol Cell Biol. 2003;23:8563–8575. doi: 10.1128/MCB.23.23.8563-8575.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fu M, Wang C, Reutens AT, et al. p300 and p300/cAMP-response element-binding protein-associated factor acetylate the androgen receptor at sites governing hormone-dependent transactivation. J Biol Chem. 2000;275:20853–20860. doi: 10.1074/jbc.M000660200. [DOI] [PubMed] [Google Scholar]
- 47.Thomas M, Dadgar N, Aphale A, et al. Androgen receptor acetylation site mutations cause trafficking defects, misfolding, and aggregation similar to expanded glutamine tracts. J Biol Chem. 2004;279:8389–8395. doi: 10.1074/jbc.M311761200. [DOI] [PubMed] [Google Scholar]
- 48.Acevedo ML, Lee KC, Stender JD, Katzenellenbogen BS, Kraus WL. Selective recognition of distinct classes of coactivators by a ligand-inducible activation domain. Mol Cell. 2004;13:725–738. doi: 10.1016/s1097-2765(04)00121-2. [DOI] [PubMed] [Google Scholar]
- 49.Ediger TR, Kraus WL, Weinman EJ, Katzenellenbogen BS. Estrogen receptor regulation of the Na+/H+ exchange regulatory factor. Endocrinology. 1999;140:2976–2982. doi: 10.1210/endo.140.7.6885. [DOI] [PubMed] [Google Scholar]
- 50.Lin HY, Hopkins R, Cao HJ, et al. Acetylation of nuclear hormone receptor superfamily members: thyroid hormone causes acetylation of its own receptor by a mitogen-activated protein kinase-dependent mechanism. Steroids. 2005;70:444–449. doi: 10.1016/j.steroids.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 51.Fu M, Wang C, Wang J, et al. Androgen receptor acetylation governs trans activation and MEKK1-induced apoptosis without affecting in vitro sumoylation and trans-repression function. Mol Cell Biol. 2002;22:3373–3388. doi: 10.1128/MCB.22.10.3373-3388.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Margueron R, Duong V, Bonnet S, et al. Histone deacetylase inhibition and estrogen receptor alpha levels modulate the transcriptional activity of partial antiestrogens. J Mol Endocrinol. 2004;32:583–594. doi: 10.1677/jme.0.0320583. [DOI] [PubMed] [Google Scholar]
- 53.Leong H, Sloan JR, Nash PD, Greene GL. Recruitment of histone deacetylase 4 to the N-terminal region of estrogen receptor alpha. Mol Endocrinol. 2005 doi: 10.1210/me.2005-0178. [DOI] [PubMed] [Google Scholar]
- 54.Kawai H, Li H, Avraham S, Jiang S, Avraham HK. Overexpression of histone deacetylase HDAC1 modulates breast cancer progression by negative regulation of estrogen receptor alpha. Int J Cancer. 2003;107:353–358. doi: 10.1002/ijc.11403. [DOI] [PubMed] [Google Scholar]
- 55.Liu XF, Bagchi MK. Recruitment of distinct chromatin-modifying complexes by tamoxifen-complexed estrogen receptor at natural target gene promoters in vivo. J Biol Chem. 2004;279:15050–15058. doi: 10.1074/jbc.M311932200. [DOI] [PubMed] [Google Scholar]
- 56.Kurtev V, Margueron R, Kroboth K, Ogris E, Cavailles V, Seiser C. Transcriptional regulation by the repressor of estrogen receptor activity via recruitment of histone deacetylases. J Biol Chem. 2004;279:24834–24843. doi: 10.1074/jbc.M312300200. [DOI] [PubMed] [Google Scholar]
- 57.Sinclair D. Sirtuins for healthy neurons. Nat Genet. 2005;37:339–340. doi: 10.1038/ng0405-339. [DOI] [PubMed] [Google Scholar]
- 58.Le Corre L, Chalabi N, Delort L, Bignon YJ, Bernard-Gallon DJ. Resveratrol and breast cancer chemoprevention: molecular mechanisms. Mol Nutr Food Res. 2005;49:462–471. doi: 10.1002/mnfr.200400094. [DOI] [PubMed] [Google Scholar]
- 59.Gehm BD, McAndrews JM, Chien PY, Jameson JL. Resveratrol, a polyphenolic compound found in grapes and wine, is an agonist for the estrogen receptor. Proc Natl Acad Sci U S A. 1997;94:14138–14143. doi: 10.1073/pnas.94.25.14138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gehm BD, Levenson AS, Liu H, et al. Estrogenic effects of resveratrol in breast cancer cells expressing mutant and wild-type estrogen receptors: role of AF-1 and AF-2. J Steroid Biochem Mol Biol. 2004;88:223–234. doi: 10.1016/j.jsbmb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 61.Bowers JL, Tyulmenkov VV, Jernigan SC, Klinge CM. Resveratrol acts as a mixed agonist/antagonist for estrogen receptors alpha and beta. Endocrinology. 2000;141:3657–3667. doi: 10.1210/endo.141.10.7721. [DOI] [PubMed] [Google Scholar]
- 62.Bouras T, Fu M, Sauve AA, et al. SIRT1 deacetylation and repression of p300 involves lysine residues 1020/1024 within the cell cycle regulatory domain 1. J Biol Chem. 2005;280:10264–10276. doi: 10.1074/jbc.M408748200. [DOI] [PubMed] [Google Scholar]
- 63.Sentis S, Le Romancer M, Bianchin C, Rostan MC, Corbo L. Sumoylation of the estrogen receptor alpha hinge region regulates its transcriptional activity. Mol Endocrinol. 2005;19:2671–2684. doi: 10.1210/me.2005-0042. [DOI] [PubMed] [Google Scholar]
- 64.Yang XJ. Multisite protein modification and intramolecular signaling. Oncogene. 2005;24:1653–1662. doi: 10.1038/sj.onc.1208173. [DOI] [PubMed] [Google Scholar]
- 65.Gronroos E, Hellman U, Heldin CH, Ericsson J. Control of Smad7 stability by competition between acetylation and ubiquitination. Mol Cell. 2002;10:483–493. doi: 10.1016/s1097-2765(02)00639-1. [DOI] [PubMed] [Google Scholar]
- 66.Desterro JM, Rodriguez MS, Hay RT. SUMO-1 modification of IkappaBalpha inhibits NF-kappaB activation. Mol Cell. 1998;2:233–239. doi: 10.1016/s1097-2765(00)80133-1. [DOI] [PubMed] [Google Scholar]
- 67.Fischle W, Wang Y, Allis CD. Histone and chromatin cross-talk. Curr Opin Cell Biol. 2003;15:172–183. doi: 10.1016/s0955-0674(03)00013-9. [DOI] [PubMed] [Google Scholar]
- 68.Berger SL. Histone modifications in transcriptional regulation. Curr Opin Genet Dev. 2002;12:142–148. doi: 10.1016/s0959-437x(02)00279-4. [DOI] [PubMed] [Google Scholar]
- 69.Kraus WL, Kadonaga JT. p300 and estrogen receptor cooperatively activate transcription via differential enhancement of initiation and reinitiation. Genes Dev. 1998;12:331–342. doi: 10.1101/gad.12.3.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kraus WL, Kadonaga JT. Ligand- and cofactor-regulated transcription with chromatin templates. In: Picard D, editor. Steroid/Nuclear Receptor Superfamily: A Practical Approach. Oxford University Press; Oxford/New York: 1999. pp. 167–189. [Google Scholar]
- 71.Kraus WL, Manning ET, Kadonaga JT. Biochemical analysis of distinct activation functions in p300 that enhance transcription initiation with chromatin templates. Mol Cell Biol. 1999;19:8123–8135. doi: 10.1128/mcb.19.12.8123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Manning ET, Ikehara T, Ito T, Kadonaga JT, Kraus WL. p300 forms a stable, template-committed complex with chromatin: role for the bromodomain. Mol Cell Biol. 2001;21:3876–3887. doi: 10.1128/MCB.21.12.3876-3887.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jeong SW, Lauderdale JD, Stein A. Chromatin assembly on plasmid DNA in vitro. Apparent spreading of nucleosome alignment from one region of pBR327 by histone H5. J Mol Biol. 1991;222:1131–1147. doi: 10.1016/0022-2836(91)90597-y. [DOI] [PubMed] [Google Scholar]
- 74.Kraus WL, Montano MM, Katzenellenbogen BS. Identification of multiple, widely spaced estrogen-responsive regions in the rat progesterone receptor gene. Mol Endocrinol. 1994;8:952–969. doi: 10.1210/mend.8.8.7997237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.