

Abstract
The space between the t-tubule invagination and the sarcoplasmic reticulum (SR) membrane, the dyad, in ventricular myocytes has been predicted to experience very high [Ca2+] for short periods of time during a Ca2+ transient. The dyadic space accommodates many protein kinases responsible for the regulation of Ca2+ handling proteins of the cell. We show in vitro that cAMP-dependent protein kinase (PKA) is inhibited by high [Ca2+] through a shift in the ratio of CaATP/MgATP toward CaATP. We further generate a three-dimensional mathematical model of Ca2+ and ATP diffusion within dyad. We use this model to predict the extent to which PKA would be inhibited by an increased CaATP/MgATP ratio during a Ca2+ transient in the dyad in vivo. Our results suggest that under normal physiological conditions a myocyte paced at 1 Hz would experience up to 55% inhibition of PKA within the cardiac dyad, with inhibition averaging 5% throughout the transient, an effect which becomes more pronounced as the myocyte contractile frequency increases (at 7 Hz, PKA inhibition averages 28% across the dyad throughout the duration of a Ca2+ transient).
INTRODUCTION
The calcium concentration ([Ca2+]) in cardiac myocytes varies significantly both spatially and temporally during a contraction. Cytosolic-free calcium ([Ca2+]free) typically averages ∼150 nM at rest but increases to ∼1 μM during a contraction, with half-maximal contraction occurring at ∼600 nM (1). Subsequent relaxation occurs when [Ca2+]free falls to ∼200 nM (2). In organelles within the myocyte such as the sarcoplasmic reticulum (SR), the [Ca2+] is much more difficult to measure accurately. Shannon and Bers (3) have estimated the total SR [Ca2+] in quiescent myocytes to be as high as 14 mM although the free [Ca2+] in the SR lumen is likely to be lower than this.
During contraction, Ca2+ enters the myocyte through the voltage-gated L-type calcium channels (dihydropyridine receptors, DHPR) in the t-tubular invagination of the sarcolemma. This triggers an additional, greater release of Ca2+ from the SR through the ryanodine receptors (RYR), a process termed calcium-induced calcium release (CICR, (4)). The cell architecture is such that both Ca2+ entry into the cell and Ca2+ release from the SR deposit Ca2+ into the cardiac dyad, the physical space formed between the t-tubule and the opposing SR membrane. The dyadic cleft between the t-tubule and SR membrane is typically a gap of ∼10 nm and covers an area with a radius of 200 nm (5). These dimensions are sufficiently small to result in restricted diffusion of soluble components, the diffusion of which is further restricted by the protrusion of protein domains, particularly RYR domains, into the dyadic space. The movement of Ca2+ in this space has not been observed experimentally, as the kinetics of movement and the small size of the dyad impose technical barriers that are difficult to overcome. Instead, a number of groups has used computational methods to predict dyadic free calcium [Ca2+]dyad, which range from ∼10 μM to ∼7 mM (5–9).
Many proteins resident in the dyadic region are controlled by both Ca2+ and site-specific phosphorylation (10). It has been long established that phosphorylation of DHPR increases Ca2+ current (ICa), probably by increasing the open probability (Po) of the channel (11). Ca2+ also regulates RYR. Submicromolar Ca2+ is capable of activating RYR, but the maximal Po is reached at ∼100 μM, with higher (>5 mM) Ca2+ leading to inactivation of the channel (12,13). Although it is well established that both cAMP-dependent protein kinase (PKA) and calcium-calmodulin-dependent kinase II (CaMKII) phosphorylate RYR (14–16), the role of phosphorylation of RYR remains a contentious issue. It has been shown that RYR can be phosphorylated by both PKA and CaMKII in vitro at Ser-2809 (or Ser-2808, in humans) (14,15,17). Changes occurring upon in vitro phosphorylation at Ser-2809 are significant, including an increased Po (15,17), the abrogation of the inhibitory effects of calmodulin (CaM) (15) and Mg2+ (18), an increased Ca2+ sensitivity of Po (19), dissociation of regulatory factors (e.g., FKBP12.6), expression of subconductance states, and the expression of channel activity at diastolic [Ca2+]free (17). Clinically, hyperphosphorylation of RYR at Ser-2808 has been described in situations such as heart failure, suggesting abnormal control of the phosphorylation status may contribute to abnormal Ca2+ handling (20), although more recent studies in vivo have noted no increase in Ser-2808 phosphorylation in failing canine, rat, or human hearts (21). Ser-2030 has also been described as a target for PKA phosphorylation, with a recent study suggesting that it is the major RYR phosphorylation target in response to β-adrenergic stimulation (21). To date no effect on Ca2+ handling has been reported relating to RYR Ser-2030 phosphorylation.
The RYR channels may play a more important role in β-adrenergic signaling as scaffold proteins, as they form a complex that localizes PKA and phosphatases 1 and 2A in the dyad (17). This is thought to ensure efficient local control of the phosphorylation status of RYR and other neighboring proteins.
A number of protein kinases have been reported to be affected by high [Ca2+] because of the effect of Ca2+ on the level of MgATP. An increase in [Ca2+] shifts the MgATP⇔CaATP equilibrium toward CaATP, thus reducing MgATP. Additionally, specific ATP hydrolyzing proteins such as Juvenile Hormone Diol kinase (22) and the H+,K+-ATPase (23) will bind CaATP but not hydrolyze it efficiently. In these enzymes CaATP blocks the access of MgATP to the catalytic sites of the kinases and so is effectively a competitive inhibitor. Bhatnagar et al. (24) have reported that PKA will bind CaATP with a similar apparent affinity as MgATP but cannot hydrolyze CaATP to CaADP. These data suggest that PKA will be competitively inhibited by Ca2+ (through an increase in CaATP). If this occurs, it poses an interesting situation for the control of dyadic proteins by phosphorylation, where the normal extremes of [Ca2+]dyad may affect kinase activity.
Here we show that PKA activity is inhibited by high [Ca2+] as a result of an increase in [CaATP]. We have also developed a computer model of a ventricular myocyte incorporating the dyad that allows us to predict the extent to which PKA inhibition would occur in vivo.
METHODS
Materials
PKA was purchased from Upstate (Charlottesville, VA), PL919Y (synthetic peptide substrate, RSAIRRASTIEY-amide) was synthesized by Neosystem (Strasbourg, France), [γ-32P]ATP was obtained from MP Biomedicals (Irvine, CA), and P81 paper was purchased from Whatman (Brentford, UK). All other chemicals were purchased from Sigma-Aldrich (Poole, UK).
Phosphorylation assays
Phosphorylation reactions were conducted at 37°C in 100 μl buffer containing 400 U (1 U is 1 pmol phosphate incorporation into PL919Y at 37°C per min) PKA, 0.1 mM PL919Y, and 50 mM histidine (pH 7.0), 5 or 25 mM MgSO4, 6.25 mM NaF, 1 mM EGTA, and CaCl2 to achieve [Ca2+]free of 3 μM–10 mM (calculated using Bound and Determined (BAD) 4.42 (25)). After 2 min of equilibration the phosphorylation reaction was initiated by the addition of 0.1 mM [γ-32P]ATP (0.1 μCi/nmol, final concentration to 10 μM). After 1 min of incubation the reaction was terminated by the addition of 100 μl 1% (v/v) H3PO4 to the sample and transferring 180 μl of sample to P81 paper. P81 paper was then washed 4× 5 min in 1% (v/v) H3PO4 before drying. Incorporated [γ-32P] was determined by scintillation counting in Emulsifier-safe using a Packard TriCarb 1900TR scintillation counter, counting for 2 min per sample (both from Canberra-Packard Ltd., Pangbourne, UK).
Three-dimensional model of a sarcomere
The three-dimensional model of the half cardiac sarcomere is shown diagrammatically in Fig. 1. The sarcomere was cylindrical in shape (radius 500 nm; length 1000 nm). Ca2+ diffusion and regulatory processes were described by the following partial differential equation, which was solved using the implicit Euler's method of solution
 |
(1) |
where r, θ, and z are the radial, angular, and length dimensions in a cylindrical coordinate system, D is the diffusion coefficient, and F(r,θ,z,[Ca],t) is a function describing the position, concentration, and time-dependent Ca2+ regulatory processes and buffering included in the model. Similar equations were used to describe the diffusion of Mg2+ and the Ca2+ and Mg2+-bound forms of ATP and ADP in the model. The modeled cell segment was divided into 50 radial elements (each one extending 10 nm in the radial direction), 50 length elements (10-nm long within the dyad, 20.6-nm long outside of the dyad), and 20 (18°) angular elements. The SR was represented by a disk located 10 nm from the plasma membrane with a radius of 200 nm, and a thickness of 20 nm in the z-direction (see Fig. 1). These dimensions are in keeping with those reported in the literature (5). At the start of the simulations, extracellular [Ca2+] was assumed to be 1 mM, SR luminal [Ca2+] was assumed to be 0.7 mM, and resting [Ca2+]free was assumed to be 100 nM (26). Euler's method of solution of Eq. 1 requires that diffusion be computed into one element at each position beyond the boundaries of the model. This was achieved by setting all of the components in each element beyond the boundary equal to the value at the boundary as described previously (27).
FIGURE 1.

Cartoon of the computer model representing half of a sarcomere (Z-line to M-line). The position in the sarcomere is expressed in cylindrical coordinates (r,z,θ). The model is a cylinder with radius, r = 500 nm, length, z = 1000 nm. The junctional SR is modeled as an impermeable disk with radius, r1 = 200 nm, depth = 20 nm, situated 10 nm from the Z-line surface. Longitudinal SR is represented as a cylinder with radius = 90 nm and length = 970 nm. Free diffusion was allowed to occur throughout the longitudinal SR; however, SERCA uptake also occurred in this region. In r, the model is divided into 10-nm elements; in z the element size is 10 nm above and including the SR and 20.6 nm below, radially, the model is divided into 20 even segments. Calcium enters the model through the 20 radial segments at r = 1. Free [Ca2+], CaATP, MgATP, CaADP, and MgATP are obtained for each element at 1-ms intervals.
Ca2+ influx and release into the dyadic space was modeled by increasing the [Ca2+] in all the radial elements where r and z = 1 (i.e., the center 20 elements of the dyadic space). Ca2+ entry into each of the 20 elements was modeled using the following exponential equation:
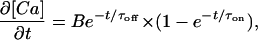 |
(2) |
where B is a current term and τoff and τon are time constants of 3 and 40 ms, respectively (10), for the rising and falling phases of the Ca2+ transient. The current term was chosen so that the Ca2+ increase in the whole myocyte (20 pl volume) was 16 μM and is consistent with Ca2+ increases in the literature (5,28,29). An exponential increase and decay in [Ca2+] was chosen over a square pulse to better represent the cooperative opening and closing of both DHPR and RYR. Ca2+ removal from the cytosol in the model was assumed to occur through uptake into the SR and extrusion across the plasma membrane. Uptake into the SR occurred on the cytosolic surface of the SR and throughout a cylinder (representing longitudinal SR), radius 90 nm, extending from the dyad opposing the SR surface to the distal end of the model (Fig. 1). Uptake into the SR mediated by SERCA was described by the Hill equation,
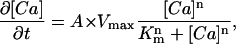 |
(3) |
where A is the number pumps per element, Km is the [Ca2+] required for half-maximal uptake velocity, Vmax is the theoretical maximum uptake velocity, and n is the Hill coefficient for uptake (see Table 1 for values).
TABLE 1.
Values used for mathematical model
| Ion or molecule | Concentration (mM) | Diffusion (μm2s−1) |
|---|---|---|
| Free Ca2+ | 0.0001* | 100 dyad*, 300 cytosol* |
| Free Mg2+ | 1† | 63 dyad*‡, 190 cytosol‡ |
| Total ATP | 7† | 60 dyad*§, 180 cytosol*§ |
| Total ADP | 0.005† | 60 dyad*§, 180 cytosol*§ |
| H+ | 0.0001 |
| Buffer | kon (μM−1s−1) | koff (s−1) | Concentration (μM) |
|---|---|---|---|
| Troponin C*† | 32.7 | 19.6 | 70 |
| Troponin C Ca/Mg†‡ | 2.37 | 0.032 | 140 |
| Myosin† | 13.8 | 0.46 | 140 |
| Calmodulin§ | 34 | 238 | 24 |
| Sarcolemma¶ | 100 | 1300 | 42 |
| Membrane‖ | 100 | 30 | 15 |
| ATP Ca** | 225 | 45,000 | 7000 |
| Mg** | 125 | 10,875 | |
| ADP Ca** | 125 | 193500 | 5 |
| Mg** | 125 | 84500 | |
| PKA MgATP‡‡ | 1.8 | 68 | |
| CaATP§§ | 1.8 ± 25% | 68 ± 25% |
| Calcium removal pathway | Km (μM) | Vmax (μMs−1) | Hill coefficient |
|---|---|---|---|
| SR uptake* | 0.3 | 210 | 2 |
| Plasma membrane extrusion† | 4 | 70 | 1 |
Peskoff and Langer (5).
Michailova and McCulloch (7).
Bernengo et al. (35).
Baylor and Hollingworth (30).
Metal ATP or ADP complex diffusion was assumed to be the same as free ATP or ADP.
Gao et al. (36).
Robertson et al. (37).
Pan and Solaro (38).
Haiech et al. (39).
Post and Langer (40).
Bers (41).
Michailova and McCulloch (7).and
Lew et al. (31).
No data available; rates based on those of MgATP (see text).
Bers (10).
Blaustein and Lederer (42).
Extrusion across the plasma membrane occurred at the Z-line, at all voxels outside of the dyad (z = 1, r > dyad radius). Extrusion was also described by Eq. 3, with n = 1 (see Table 1 for a complete list of values).
As described previously (27), passive leak from the SR and extracellular spaces was described by equations of the form
 |
(4) |
where [Ca]x is the free Ca2+ within the SR or in the extracellular space (set as 0.7 and 1 mM, respectively), and leak constants (Kleak) were adjusted so that under resting conditions ([Ca2+]free = 100 nM) leak from the SR was equal to SERCA uptake and leak into the cell was equal to plasma membrane extrusion.
Buffering within the model was the same at all locations and was based upon buffering components described by Bers (10) for a rabbit ventricle, with the addition of ATP and ADP, which are capable of binding Ca2+ or Mg2+ (see Table 1 for binding constants and concentrations). The action of these buffers was described by the equation
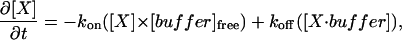 |
(5) |
where [X] is Ca2+ or Mg2+, kon and koff are the on and off rates for Ca2+ or Mg2+ binding to the buffer and [X · buffer] is the Ca2+ or Mg2+ bound form of the buffer.
The apparent metal-ATP occupancy of PKA was calculated at microsecond intervals using the above equation (with PKA acting as the buffer, see Table 1 for binding constants and concentrations). The rate constants for the binding of CaATP to PKA were assumed to be roughly the same as MgATP. To account for possible differences in the rate constants, the on and off rates for CaATP binding to PKA were varied by ±25% of MgATP values in simulations. This assumption was based upon evidence that the apparent affinities of the two species for PKA are similar (24).
RESULTS
Ca2+ (CaATP) sensitivity of PKA
The effect of [Ca2+] on PKA activity was examined by measuring the phosphorylation of a peptide substrate at a variety of [Ca2+]free. Fig. 2 A shows that high [Ca2+]free inhibits PKA activity and that this inhibitory profile is influenced by the concentration of Mg2+. Raising Mg2+ offered some protection to the enzyme from the inhibitory potential of Ca2+, consistent with the notion of competition between Ca2+ and Mg2+. As PKA does not possess an identified Ca2+-binding site, we explored the possibility that inhibition was achieved indirectly through competition of Ca2+ and Mg2+ for ATP. As [Ca2+]free increases (as in Fig. 2 A) it will shift the equilibrium of MgATP⇔CaATP toward CaATP, as shown in Fig. 2 B, resulting in PKA inhibition through both a reduction in MgATP concentration, and as a consequence of CaATP-binding. Others have shown that CaATP is not a substrate for the kinase (24). Fig. 2 C shows the activity of PKA from Fig. 2 A replotted against the CaATP/MgATP ratio. The data generated with both 5 mM and 25 mM Mg2+ are described by the same double exponential relationship, strengthening our assumption that the inhibition of PKA is caused by the increase in the CaATP/MgATP ratio.
FIGURE 2.

Effect of [Ca2+]free on PKA activity. (A) PKA (400 U) was incubated at a range of [Ca2+]free (3 μM–10 mM) for 2 min in 50 mM histidine (pH 7.0) containing 5 mM (•) or 25 mM (▴) MgSO4, 6.25 mM NaF, 1 mM EGTA, and 0.1 mM PL919Y. PL919Y was phosphorylated for 1 min by PKA at 37°C after the addition of 0.1 mM ATP-γ-32P (final concentration to 10 μM). The reaction was terminated by the addition of 100 μl 1% (v/v) H3PO4. Phosphorylated PL919Y was transferred to P81 paper, and [γ-32P] incorporation was measured by scintillation counting. The background-corrected levels of incorporation are shown and represent the mean ± SE (n = 3). (B) Effect of increasing [Ca2+]free on the CaATP/MgATP at total magnesium concentrations of 5 (•) and 25 mM (▴). The concentrations of CaATP and MgATP were calculated using BAD 4.42 (25). (C) Effect of CaATP/MgATP on PKA activity. Data from A were replotted using the ratio generated in B.
With the determination of the relationship between [Ca2+] and PKA activity, we sought to investigate whether the conditions required for the inhibition of PKA occur physiologically. The vast majority of cellular locations can be discounted because the [Ca2+]free required to sufficiently alter the CaATP/MgATP balance would never occur. The cardiac dyad, however, may be a site where this could occur. The dyadic cleft is a highly restricted space where high levels of Ca2+ occur transiently during excitation contraction coupling. Predicted transient [Ca2+]dyad in this region range between 10 μM and 7 mM (5–9). [Ca2+] in this range could significantly alter the CaATP/MgATP ratio, resulting in the inhibition of PKA (Fig. 2).
Computer simulation of half a sarcomere (model validation)
Many models of the cardiac sarcomere have been developed (see above), all of which in some way simplify the structure to focus on a specific area of interest. In this study, we have generated a model of a half sarcomere containing a single dyadic cleft using morphological structures of realistic dimensions (SR volume, dyadic area, plasma membrane area, sarcomeric volume (5); Fig. 1) to focus on Ca2+ and CaATP movement and concentration in and around the dyad. The spatial elements of the model are divided into voxels using the cylindrical coordinate system (r,z,θ; see Methods), and the Ca2+ flux rates, Ca2+ diffusion rates, and Ca2+-buffering characteristics are taken from the relevant literature (Table 1). The model allows for the entry of Ca2+ at a single site within the dyad, its free diffusion from one voxel to another, its uptake into the SR, and its efflux from the cell, thereby generating a dynamic model of Ca2+ homeostasis in the myocyte. To assess the predictive power of this model, and the kinetic parameters therein, we examined its ability to simulate a realistic cytosolic Ca2+ transient. Furthermore, we assessed the stability of the model by simulating a train of Ca2+ transients (9th and 10th shown in Fig. 3 A), the 10th of which is displayed in Fig. 3 B. A stable average cytosolic Ca2+ transient was predicted by the simulation, which rose to a peak of 680 nM at 95 ms and declined following a monoexponential function (τ = −0.67 s) to a diastolic [Ca2+]free of 220 nM. These values are similar to those reported for ventricular myocytes, paced at 1 Hz (8) and by other mathematical models (7,10).
FIGURE 3.

[Ca2+]free transients generated by the mathematical model. [Ca2+]free was averaged throughout the cytosolic elements of the model. (A) Transients 9 and 10 from a train of 20. (B) Transient generated on the 10th stimulation, expanded timescale from A.
[Ca2+]dyad during a Ca2+ transient
The establishment of a model that accurately simulates the average cytosolic Ca2+ transient allowed us to predict the [Ca2+]dyad. The [Ca2+] in the model was calculated at 1-ms intervals at each segment of the model (using the radial coordinate system r,z,θ). The three-dimensional plot in Fig. 4 A shows the [Ca2+] at z = 1 (the Z-line) plotted as a function of radial position and time. The shaded region represents the space within the dyadic cleft, where [Ca2+]dyad rises to 300 μM (average concentration in the cleft; Fig. 4 C), or 2.1 mM close to the channel opening (Fig. 4 B) for a short period of time after the arrival of an action potential. These concentrations are at least 1000-fold higher than those experienced in the bulk cytosol, and these excessive concentrations are experienced almost exclusively in the dyad as a steep gradient occurs at the junction between dyad and free cytosol (Fig. 4 A). The inclusion of ATP as a diffusible Ca2+ buffer has dramatic effects on [Ca2+]dyad within the cleft. Without ATP diffusion the peak [Ca2+]dyad is almost triple that observed with ATP diffusion (data not shown). This agrees with Michailova and McCulloch (7) in illustrating the importance of ATP as a mobile Ca2+ buffer.
FIGURE 4.

[Ca2+] generated within the dyad. (A) Three-dimensional plots of [Ca2+] at the Z-line (z = 1) as a function of time. The shaded region represents the dyadic space. (B) [Ca2+] generated at r = 1, akin to the 10-nm space immediately surrounding the RYR and DHPR channels. (C) [Ca2+] averaged across the whole of the dyad.
Ratio of CaATP/MgATP within the cardiac dyad during a calcium transient
In addition to predicting [Ca2+]free, our model also calculates the diffusion and concentration of ATP/CaATP/MgATP during the Ca2+ transient, so it is possible to calculate the CaATP/MgATP ratio generated throughout the model cell. Again the three foci of interest are the site of Ca2+ entry (Fig. 5 A), the dyad averaged as a whole (Fig. 5 B), and the average in the bulk cytosol (Fig. 5 C).
FIGURE 5.

CaATP/MgATP generated within the model. (A and B) Ratios within the dyad; broken line represents raw CaATP/MgATP ratio, bold black line represents the PKA-CaATP/PKA-MgATP-bound occupancy ratio using the same kinetics for both CaATP and MgATP binding to PKA. The gray lines show the effect of varying CaATP-binding kinetics by ±25%. (A) The ratio at r = 1, (B) across the dyad as a whole, and (C) average ratio throughout the cytosol.
Although the ratio of CaATP/MgATP in the bulk cytosol undergoes a modest increase (threefold change at its maximum) as predicted by Michailova and McCulloch (7), the ratio changes greatly in the dyad throughout the calcium transient. At the point of Ca2+ entry the CaATP/MgATP ratio displayed a 2000-fold increase from an initial ratio of 9.4 × 10−5 to a final ratio of 0.18. This represents a change from an initial condition of 10,000 molecules of MgATP for every molecule of CaATP, to a situation where there are only six molecules of MgATP for every CaATP.
As would be expected, the CaATP/MgATP ratio observed across the whole dyad changed substantially during a single Ca2+ transient; however the magnitude of change was less than that observed at the channel mouth. The increase, however, is still substantial being 650-fold at its height. Using the prediction of the CaATP/MgATP generated within the model along with the kinetic rates of CaATP and MgATP binding to PKA, we next were able to estimate the occupancy of PKA for either of the two metal ATP complexes (PKA-CaATP, PKA-MgATP, and their ratio) (see Fig. 5, A and B). We were further able to predict the impact of these changes in metal ATP occupancy on PKA activity throughout the sarcomere during a calcium transient.
The activity of PKA is reduced in the dyad of a cardiac myocyte during contraction
Having previously defined the relationship between PKA activity and the CaATP/MgATP ratio in vitro, we were able to use the CaATP/MgATP occupancy ratio of PKA to calculate the activity of PKA at each time point, relative to theoretical maximum activity of PKA, throughout the sarcomere. Fig. 6 A shows the activity of PKA across the t-tubule end of a sarcomere (z = 1, Z-line) during a Ca2+ transient. The shaded region illustrates the activity of PKA within the dyad. There is a gradient of PKA activity across the cleft, with maximal inhibition occurring at the point of calcium entry (r = 1). There is little or no effect on PKA activity beyond the radius of the cleft. At the radial edge of the cleft PKA, activity would only experience modest inhibition (∼10%) due to the CaATP/MgATP at the maxima of the Ca2+ transient. Thus, the oscillation in PKA activity in neighboring regions (e.g., corbular SR, longitudinal SR) would be slight. Fig. 6 B replots the activity of PKA at the channel mouth showing the effect of varying the kinetics of CaATP binding to PKA by ±25%. It shows there is a significant reduction in PKA activity at the channel mouth and that a 25% variation in CaATP-binding kinetics only results in ±5% shift in the relative activity of PKA. The time course of PKA inhibition within the cleft shows a maximal inhibition of 55% occurring at the point of calcium entry (r = 1). Fig. 6 C shows the average reduction in PKA activity across the whole cleft, again with inclusion of a 25% variation in CaATP-binding kinetics, here the reduction in PKA activity is 40%. In both cases the peak inhibitory effect occurs at 37 ms with maximal activity being returned around 200 ms, if one assumes a 1-Hz stimulatory frequency. Integrating the data allows us to predict the level of inhibition during both the whole and a portion of the transient. At 1 Hz this equates to an 8% reduction in PKA activity over the entire transient at the channel mouth (5% across the whole dyad). If we were to assume an increased stimulation frequency of 3 Hz (around that of exercising humans), the contraction cycle would last 333 ms, throughout which PKA activity would remain suppressed by 24% at the channel mouth (14% across the whole dyad). Increasing the frequency, yet higher, to that of small mammals (7 Hz) shows a reduction in PKA activity of nearly 47% (channel mouth) for the duration of the transient (28% across the whole dyad). As the rate of diffusion within the dyad is essentially an informed estimate based upon the diffusion in the free cytosol (5), we varied the dyadic diffusion coefficient to understand the impact an error in the original estimate may have. Peskoff and Langer (5) assume that due to the physical constants of diffusion (as described in the introduction) within the dyad, the rate of diffusion will be a third of that of the cytosol. Fig. 6, D and E, shows the reduction in PKA activity predicted by our model if no such reduction in diffusion is assumed within the dyad. Fig. 6 D shows the effect at the channel mouth, with Fig. 6 E illustrating the reduction in PKA activity throughout the dyad. As could be expected an increase in the diffusion rate blunts the magnitude of the reduction in PKA activity at both locations, leading to peak reductions of 38% and 19% in PKA activity located near to the channel mouth and throughout the dyad, respectively. We also modeled the effect of underestimating the restriction of diffusion within the dyad by halving the rates proposed by Peskoff and Langer (5) (Fig. 6, F and G). Under these conditions the reduction in PKA activity is magnified, resulting in peak inhibitions of 65% and 50% for PKA near the channel mouth and throughout the dyad, respectively.
FIGURE 6.

Transient inhibition of PKA activity in the dyad. (A) Three-dimensional plot of PKA activity at the Z-line (z = 1) as a function of time. The shaded region represents the dyadic space. (B) PKA activity at r = 1, akin to the 10-nm space immediately surrounding the RYR and DHPR channels and (C) averaged across the whole of the dyad. (D and E) As B and C, respectively, with double the dyadic diffusion rate. (F and G) As B and C, respectively, with half the dyadic diffusion rate. Black line represents equal CaATP and MgATP kinetics; gray lines show the effect of varying CaATP-binding kinetics by ± 25%.
DISCUSSION
In this study, we sought to examine if the high [Ca2+] in the cardiac muscle dyadic space would influence the enzymatic activity of a protein kinase that phosphorylates protein targets resident in these spaces. PKA was shown to be inhibited at high [Ca2+] in a manner consistent with competitive inhibition of MgATP binding by CaATP. The physiological significance of these observations was explored using a mathematical model of a ventricular myocyte (half) sarcomere. The model was generated using structurally accurate subcellular compartments (dyadic volume) and kinetic parameters (fluxes, buffering, diffusion). The model produced physiological realistic cytosolic Ca2+ transients. [Ca2+]dyad rose to concentrations exceeding 2 mM at sites close to the channel mouth and peaked at 300 μM across the dyadic cleft. This elevated [Ca2+]dyad caused a pronounced shift in the CaATP/MgATP ratio, which in turn was predicted to inhibit PKA activity by 55% at the mouth of the channel and 40% across the cleft. The extent of inhibition varied temporally and was significant for the first 200 ms of a Ca2+ transient, with the average level of inhibition throughout the transient being 8% (calculated at a Ca2+ transient frequency of 1 Hz). PKA activity in the cytosol was unaffected by the modest elevation in CaATP/MgATP in that space, even regions close to the cleft, such as the corbular SR would be unlikely to experience inhibition of kinase activity, although it must be noted we do not include Ca2+ release from the corbular SR itself, which could alter this conclusion.
Our experimental data show that increasing [Ca2+] does inhibit PKA by increasing the level of CaATP, as there is a direct relationship between the activity of PKA and the CaATP/MgATP ratio, independent of total [Ca2+] or [Mg2+]. CaATP is likely acting as a competitive inhibitor of MgATP binding to PKA.
We developed a computer simulation to predict the changes in the [Ca2+] and CaATP/MgATP ratio during a Ca2+ transient, generated in a ventricular myocyte. The model was based upon Bazzazi et al. (27), with Ca2+ buffering as described by Bers (10), with the addition of diffusible ATP and ADP able to bind Ca2+ or Mg2+ (30) and kinetic binding of CaATP and MgATP to PKA (24,31). Our model was able to simulate Ca2+ transients that replicated experimentally derived transients (8), in terms of both concentration and duration. In the simulation, [Ca2+]dyad rose to over 2 mM at the Ca2+ release site and 300 μM averaged across the whole cleft. These [Ca2+]dyad are within the range predicted by some, but not all, models of cardiac muscle (5,7–9). The discrepancy in predicted [Ca2+]dyad may relate to the representation of Ca2+ influx as a “common pool” or “all-or-none” phenomenon in some models (5,7,8) (ours included) as opposed to a representation of entry as multiple stochastic events leading to more graded entries (9). We and others (5) used fixed Ca2+ entry derived experimentally, whereas other groups have used a change in membrane potential to estimate Ca2+ influx (7,8). Our [Ca2+]dyad is also higher than that determined empirically using Na2+/Ca2+ exchanger (NCX) currents to estimate near membrane [Ca2+] (32). The data presented in Fig. 4 suggest that a steep [Ca2+] gradient occurs at the edge of the dyad; this coupled with the more recent data that NCXs are not localized within the dyad (33) suggest that NCXs would underestimate [Ca2+]dyad. The effects of reducing the peak [Ca2+]dyad levels to those proposed by Michailova and McCulloch (30 μM) (7) are shown in Fig. 7, A and B. The 10-fold reduction in proposed [Ca2+]dyad reduces the inhibition of PKA by 50%, (from 55% to 27%) at the Ca2+ release site and by 70% (from 40% to 12%) throughout the dyad as a whole. Although the effect on PKA activity is reduced under these conditions of lower [Ca2+]dyad, a 27% reduction in PKA activity could still prove significant. Fig. 7, C–F, shows the effect of varying the diffusion rates within the dyad, at 30 μM [Ca2+]dyad, as previously discussed for 300 μM in the results.
FIGURE 7.

Transient inhibition of PKA activity in the dyad with reduced [Ca2+]dyad. PKA activity (A) at r = 1, akin to the 10-nm space immediately surrounding the RYR and DHPR channels and (B) averaged across the whole of the dyad, where [Ca2+]dyad peaks a 30 μM. (C and D) As A and B, respectively, with double the dyadic diffusion rate. (E and F) As A and B, respectively, with half the dyadic diffusion rate. Black line represents equal CaATP and MgATP kinetics; gray lines show the effect of varying CaATP-binding kinetics by ± 25%.
Our model shows that the high [Ca2+]dyad causes a substantial shift in the CaATP/MgATP ratio within the dyadic cleft during a Ca2+ transient. At diastole CaATP/MgATP is 9.4 × 10−5, which quickly rises to 0.18 during systole. The ratio at systole is comparable with those predicted previously (34) in smooth muscle and in agreement with those predicted in cardiac muscle (7). The rate at which the CaATP/MgATP ratio alters over the initial 200 ms is faster than can be accommodated by kinetic binding of CaATP or MgATP to PKA. This leads to a delay between maximum of the CaATP/MgATP ratio and the PKA-CaATP/PKA-MgATP-bound ratio as is illustrated in Fig. 5, with the peak PKA-CaATP/PKA-MgATP bound ratio being 0.14.
Combining the experimental data describing the relationship of PKA activity and the CaATP/MgATP ratio with the simulated data predicting the bound PKA-CaATP/PKA-MgATP ratio transient within the dyadic cleft allowed us to predict the extent to which PKA activity would be reduced. The reduction in PKA activity peaked at ∼55% and was maintained ∼40% for 100 ms at a stimulatory frequency of 1 Hz.
To understand the full implication of a 40%–55% transient reduction in PKA activity on the phosphorylation status of dyadic proteins, it is necessary to have estimates of the level of activity (relative to theoretical maximum in vitro) required to maintain or increase the phosphorylation of the various proteins and channels residing in the dyadic cleft; however the empirical information regarding this region is not currently available. Our model does not attempt to predict the effect increasing CaATP may have on other dyadic enzymes, such as the phosphatases opposing PKA activity. Phosphatase activity is unlikely to be modulated by an increase in CaATP, as ATP is not a substrate, but the high [Ca2+]dyad alone could affect Ca2+-dependent phosphatases, although the effect on PKA described would be in addition to any other factors modulating dyad phosphorylation status. Recent models (17) have proposed that PKA is targeted to the dyad through muscle A kinase anchoring protein (mAKAP) as part of the RYR macromolecular complex. If this is the case it would lead to a high ratio of enzyme/substrate. This may counter some of the effect of CaATP on PKA and reflect a requirement for robust phosphorylation level of local proteins, although in this PKA-targeted model, PKA would be localized near the channel mouth and therefore experience the greatest level of inhibition predicted by our simulation.
There are perhaps greater implications for the effect of CaATP at higher stimulation frequencies. As there are no major Ca2+ removal mechanisms described as being located within the dyadic cleft, the major factors influencing [Ca2+]dyad are Ca2+ entry and the diffusion rate through and out of the dyad. This would suggest that at higher stimulation frequencies (i.e., during exercise or those found in small mammals), where diffusion rates and Ca2+ entry would remain the same, [Ca2+]dyad would remain at elevated levels for much more of the transient. At a contraction frequency of ∼3 Hz (human during exercise) our current model would suggest [Ca2+]dyad remains ∼10 μM for half of the contraction cycle, this would equate to a 14%–24% reduction in activity even at diastole. In small mammals Peskoff et al. (6) have suggested that at a frequency of ∼7 Hz (a typical heart rate of rats), the [Ca2+]dyad remains above 100 μM for half of the contraction cycle. This would lead to a chronic reduction in PKA activity (∼30% reduction during diastole), agreeing well with the integration of our data, suggesting an average reduction in PKA activity throughout a Ca2+ transient at 7 Hz stimulation frequency of 28%. It would be interesting to examine what strategies are employed in these situations to achieve rapid and efficient phosphorylation of target proteins in an environment unfavorable to protein kinase action.
Acknowledgments
The authors thank Margaret Kargacin for critical reading of the manuscript and Clive Orchard for critical input.
This work was supported by research grants from the Heart and Stroke foundation of Alberta and Canadian Institutes of Health Research to G.J.K. and from the British Heart Foundation to J.C. P.P.J. was a recipient of a Biotechnology and Biological Sciences Research Council studentship.
References
- 1.Bers, D. M. 2002. Cardiac excitation-contraction coupling. Nature. 415:198–205. [DOI] [PubMed] [Google Scholar]
- 2.Berne, R. M., and M. N. Levy. 1993. Physiology. Mosby Year Book, St. Louis, MO.
- 3.Shannon, T. R., and D. M. Bers. 2004. Integrated Ca2+ management in cardiac myocytes. Ann. N. Y. Acad. Sci. 1015:28–38. [DOI] [PubMed] [Google Scholar]
- 4.Fabiato, A. 1983. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 245:C1–14. [DOI] [PubMed] [Google Scholar]
- 5.Peskoff, A., and G. A. Langer. 1998. Calcium concentration and movement in the ventricular cardiac cell during an excitation-contraction cycle. Biophys. J. 74:153–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peskoff, A., J. A. Post, and G. A. Langer. 1992. Sarcolemmal calcium binding sites in heart: II. Mathematical model for diffusion of calcium released from the sarcoplasmic reticulum into the diadic region. J. Membr. Biol. 129:59–69. [DOI] [PubMed] [Google Scholar]
- 7.Michailova, A., and A. McCulloch. 2001. Model study of ATP and ADP buffering, transport of Ca(2+) and Mg(2+), and regulation of ion pumps in ventricular myocyte. Biophys. J. 81:614–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Winslow, R. L., J. Rice, S. Jafri, E. Marban, and B. O'Rourke. 1999. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, II: model studies. Circ. Res. 84:571–586. [DOI] [PubMed] [Google Scholar]
- 9.Rice, J. J., M. S. Jafri, and R. L. Winslow. 1999. Modeling gain and gradedness of Ca2+ release in the functional unit of the cardiac diadic space. Biophys. J. 77:1871–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bers, D. M. 2001. Excitation-Contraction Coupling and Cardiac Contractile Force. Kluwer Academic Publishers, Dordrecht, The Netherlands.
- 11.Reuter, H. 1967. The dependence of slow inward current in Purkinje fibres on the extracellular calcium-concentration. J. Physiol. 192:479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rousseau, E., and G. Meissner. 1989. Single cardiac sarcoplasmic reticulum Ca2+-release channel: activation by caffeine. Am. J. Physiol. 256:H328–H333. [DOI] [PubMed] [Google Scholar]
- 13.Xu, L., A. Tripathy, D. A. Pasek, and G. Meissner. 1998. Potential for pharmacology of ryanodine receptor/calcium release channels. Ann. N. Y. Acad. Sci. 853:130–148. [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez, P., M. S. Bhogal, and J. Colyer. 2003. Stoichiometric phosphorylation of cardiac ryanodine receptor on serine 2809 by calmodulin-dependent kinase II and protein kinase A. J. Biol. Chem. 278:38593–38600. [DOI] [PubMed] [Google Scholar]
- 15.Witcher, D. R., R. J. Kovacs, H. Schulman, D. C. Cefali, and L. R. Jones. 1991. Unique phosphorylation site on the cardiac ryanodine receptor regulates calcium channel activity. J. Biol. Chem. 266:11144–11152. [PubMed] [Google Scholar]
- 16.Witcher, D. R., B. A. Strifler, and L. R. Jones. 1992. Cardiac-specific phosphorylation site for multifunctional Ca2+/calmodulin-dependent protein kinase is conserved in the brain ryanodine receptor. J. Biol. Chem. 267:4963–4967. [PubMed] [Google Scholar]
- 17.Marx, S. O., S. Reiken, Y. Hisamatsu, T. Jayaraman, D. Burkhoff, N. Rosemblit, and A. R. Marks. 2000. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 101:365–376. [DOI] [PubMed] [Google Scholar]
- 18.Hain, J., H. Onoue, M. Mayrleitner, S. Fleischer, and H. Schindler. 1995. Phosphorylation modulates the function of the calcium release channel of sarcoplasmic reticulum from cardiac muscle. J. Biol. Chem. 270:2074–2081. [DOI] [PubMed] [Google Scholar]
- 19.Valdivia, H. H., J. H. Kaplan, G. C. Ellis-Davies, and W. J. Lederer. 1995. Rapid adaptation of cardiac ryanodine receptors: modulation by Mg2+ and phosphorylation. Science. 267:1997–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reiken, S., M. Gaburjakova, S. Guatimosim, A. M. Gomez, J. D'Armiento, D. Burkhoff, J. Wang, G. Vassort, W. J. Lederer, and A. R. Marks. 2003. Protein kinase A phosphorylation of the cardiac calcium release channel (ryanodine receptor) in normal and failing hearts. Role of phosphatases and response to isoproterenol. J. Biol. Chem. 278:444–453. [DOI] [PubMed] [Google Scholar]
- 21.Xiao, B., M. T. Jiang, M. Zhao, D. Yang, C. Sutherland, F. A. Lai, M. P. Walsh, D. C. Warltier, H. Cheng, and S. R. Chen. 2005. Characterization of a novel PKA phosphorylation site, serine-2030, reveals no PKA hyperphosphorylation of the cardiac ryanodine receptor in canine heart failure. Circ. Res. 96:847–855. [DOI] [PubMed] [Google Scholar]
- 22.Maxwell, R. A., W. H. Welch, and D. A. Schooley. 2002. Juvenile hormone diol kinase. I. Purification, characterization, and substrate specificity of juvenile hormone-selective diol kinase from Manduca sexta. J. Biol. Chem. 277:21874–21881. [DOI] [PubMed] [Google Scholar]
- 23.Mendlein, J., and G. Sachs. 1989. The substitution of calcium for magnesium in H+,K+-ATPase catalytic cycle. Evidence for two actions of divalent cations. J. Biol. Chem. 264:18512–18519. [PubMed] [Google Scholar]
- 24.Bhatnagar, D., R. Roskoski Jr., M. S. Rosendahl, and N. J. Leonard. 1983. Adenosine cyclic 3′,5′-monophosphate dependent protein kinase: a new fluorescence displacement titration technique for characterizing the nucleotide binding site on the catalytic subunit. Biochemistry. 22:6310–6317. [DOI] [PubMed] [Google Scholar]
- 25.Brooks, S. P., and K. B. Storey. 1992. Bound and determined: a computer program for making buffers of defined ion concentrations. Anal. Biochem. 201:119–126. [DOI] [PubMed] [Google Scholar]
- 26.Hinch, R., J. L. Greenstein, A. J. Tanskanen, L. Xu, and R. L. Winslow. 2004. A simplified local control model of calcium-induced calcium release in cardiac ventricular myocytes. Biophys. J. 87:3723–3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bazzazi, H., M. E. Kargacin, and G. J. Kargacin. 2003. Ca2+ regulation in the near-membrane microenvironment in smooth muscle cells. Biophys. J. 85:1754–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuan, W., K. S. Ginsburg, and D. M. Bers. 1996. Comparison of sarcolemmal calcium channel current in rabbit and rat ventricular myocytes. J. Physiol. 493:733–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cleemann, L., W. Wang, and M. Morad. 1998. Two-dimensional confocal images of organization, density, and gating of focal Ca2+ release sites in rat cardiac myocytes. Proc. Natl. Acad. Sci. USA. 95:10984–10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baylor, S. M., and S. Hollingworth. 1998. Model of sarcomeric Ca2+ movements, including ATP Ca2+ binding and diffusion, during activation of frog skeletal muscle. J. Gen. Physiol. 112:297–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lew, J., S. S. Taylor, and J. A. Adams. 1997. Identification of a partially rate-determining step in the catalytic mechanism of cAMP-dependent protein kinase: a transient kinetic study using stopped-flow fluorescence spectroscopy. Biochemistry. 36:6717–6724. [DOI] [PubMed] [Google Scholar]
- 32.Weber, C. R., V. Piacentino 3rd, K. S. Ginsburg, S. R. Houser, and D. M. Bers. 2002. Na(+)-Ca(2+) exchange current and submembrane [Ca(2+)] during the cardiac action potential. Circ. Res. 90:182–189. [DOI] [PubMed] [Google Scholar]
- 33.Scriven, D. R., P. Dan, and E. D. Moore. 2000. Distribution of proteins implicated in excitation-contraction coupling in rat ventricular myocytes. Biophys. J. 79:2682–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kargacin, M. E., and G. J. Kargacin. 1997. Predicted changes in concentrations of free and bound ATP and ADP during intracellular Ca2+ signaling. Am. J. Physiol. 273:C1416–C1426. [DOI] [PubMed] [Google Scholar]
- 35.Bernengo, J. C., C. Collet, and V. Jacquemond. 2001. Intracellular Mg2+ diffusion within isolated rat skeletal muscle fibers. Biophys. Chem. 89:35–51. [DOI] [PubMed] [Google Scholar]
- 36.Gao, W. D., P. H. Backx, M. Azan-Backx, and E. Marban. 1994. Myofilament Ca2+ sensitivity in intact versus skinned rat ventricular muscle. Circ. Res. 74:408–415. [DOI] [PubMed] [Google Scholar]
- 37.Robertson, S. P., J. D. Johnson, and J. D. Potter. 1981. The time-course of Ca2+ exchange with calmodulin, troponin, parvalbumin, and myosin in response to transient increases in Ca2+. Biophys. J. 34:559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pan, B. S., and R. J. Solaro. 1987. Calcium-binding properties of troponin C in detergent-skinned heart muscle fibers. J. Biol. Chem. 262:7839–7849. [PubMed] [Google Scholar]
- 39.Haiech, J., C. B. Klee, and J. G. Demaille. 1981. Effects of cations on affinity of calmodulin for calcium: ordered binding of calcium ions allows the specific activation of calmodulin-stimulated enzymes. Biochemistry. 20:3890–3897. [DOI] [PubMed] [Google Scholar]
- 40.Post, J. A., and G. A. Langer. 1992. Sarcolemmal calcium binding sites in heart: I. Molecular origin in “gas-dissected” sarcolemma. J. Membr. Biol. 129:49–57. [DOI] [PubMed] [Google Scholar]
- 41.Bers, D. M., L. V. Hryshko, S. M. Harrison, and D. D. Dawson. 1991. Citrate decreases contraction and Ca current in cardiac muscle independent of its buffering action. Am. J. Physiol. 260:C900–C909. [DOI] [PubMed] [Google Scholar]
- 42.Blaustein, M. P., and W. J. Lederer. 1999. Sodium/calcium exchange: its physiological implications. Physiol. Rev. 79:763–854. [DOI] [PubMed] [Google Scholar]