

Abstract
In this study, we investigate the extent to which techniques for homology modeling that were developed for water-soluble proteins are appropriate for membrane proteins as well. To this end we present an assessment of current strategies for homology modeling of membrane proteins and introduce a benchmark data set of homologous membrane protein structures, called HOMEP. First, we use HOMEP to reveal the relationship between sequence identity and structural similarity in membrane proteins. This analysis indicates that homology modeling is at least as applicable to membrane proteins as it is to water-soluble proteins and that acceptable models (with Cα-RMSD values to the native of 2 Å or less in the transmembrane regions) may be obtained for template sequence identities of 30% or higher if an accurate alignment of the sequences is used. Second, we show that secondary-structure prediction algorithms that were developed for water-soluble proteins perform approximately as well for membrane proteins. Third, we provide a comparison of a set of commonly used sequence alignment algorithms as applied to membrane proteins. We find that high-accuracy alignments of membrane protein sequences can be obtained using state-of-the-art profile-to-profile methods that were developed for water-soluble proteins. Improvements are observed when weights derived from the secondary structure of the query and the template are used in the scoring of the alignment, a result which relies on the accuracy of the secondary-structure prediction of the query sequence. The most accurate alignments were obtained using template profiles constructed with the aid of structural alignments. In contrast, a simple sequence-to-sequence alignment algorithm, using a membrane protein-specific substitution matrix, shows no improvement in alignment accuracy. We suggest that profile-to-profile alignment methods should be adopted to maximize the accuracy of homology models of membrane proteins.
INTRODUCTION
Membrane proteins are believed to comprise 20–30% of the proteins in a genome (1–3) and represent a significant proportion of therapeutic drug targets (4). However, as a result of difficulties in experimental structure determination, they constitute only ∼1% of the structures available in the protein data bank (PDB) (5). The absence of structural information severely limits our ability to understand membrane protein function. Based on previous experience with water-soluble proteins, it is likely that computational structure prediction will provide a useful approach to generating models for these proteins. Typically, the most accurate models of protein structures are achieved through homology modeling, where a known structure is used as a template for the construction of a model of a related protein (6). However, it remains unclear whether the methods and assumptions used in homology modeling of water-soluble proteins can be applied directly to membrane proteins without modification.
There are several features of membrane proteins that distinguish them from water-soluble proteins. The differences arise because the environment of the transmembrane regions of membrane proteins is different from that in aqueous solution: it is predominantly lipophilic, lacks hydrogen-bonding potential, and provides little screening of electrostatic interactions. At the primary sequence level, this results in significant differences in amino acid composition (7,8) and in the probabilities of amino acid substitutions during evolution (9,10), generally favoring residues with hydrophobic side chains, especially at the protein-lipid interface (11,12). In addition, amino acids have been shown to have different secondary-structure propensities in membrane environments and in aqueous solution (13–15).
The differences in the properties of the two types of protein might be expected to have consequences for the applicability of some homology modeling methods to membrane proteins. For example, differences in amino acid composition and evolutionary substitution probabilities imply that methods for the alignment of protein sequences may not be directly transferable. This possibility has led to the creation of novel amino acid substitution matrices (10,16), which are used to identify probable matches in sequences, and to the introduction of so-called bipartite alignment methods that utilize these matrices in transmembrane regions only (10,16,17).
A second aspect of modeling that may be affected by the differences between membrane proteins and water-soluble proteins is the prediction of secondary structure. We draw a distinction between the secondary structure of a residue and its location relative to the membrane, since every amino acid can be labeled as having both a specific secondary-structure type and a specific location. This distinction is useful because it allows for the unique description of secondary-structure elements peripheral to the membrane (18), as well as coil-like residues within the membrane, e.g., in reentrant loops or unwound helices (19). Thus, a method capable of accurately predicting the secondary structure of each residue in a membrane protein sequence would provide information that is supplementary to that obtained from the prediction of the location of a particular amino acid with respect to the bilayer. More generally, it is important to understand the extent to which secondary-structure prediction algorithms designed for soluble proteins are applicable to membrane proteins.
A third way the membrane environment may affect homology modeling studies involves the presence of unique topological constraints provided by the lipid bilayer (20). In principle, it is possible that the range of relative orientations of helices within the membrane is more restricted than in the aqueous phase, which may limit the structural diversity available to families of membrane proteins. It might also suggest that homology models of membrane proteins are more accurate than models of water-soluble proteins for the same level of sequence identity. It is therefore of interest to assess the relationship between sequence identity and structural similarity for membrane proteins.
In this work, we address the three issues raised above. We analyze the performance of state-of-the-art globular-protein homology modeling strategies using a set of 36 homologous membrane protein structures (HOMEP), comprising 11 families of topologically related proteins. Taking each protein in turn, we use all its family members as templates for the construction of homology models whose accuracy is then determined by comparison to the known structure. Although small on the scale of general sequence alignment benchmark sets such as BaliBase (21), the HOMEP set is carefully compiled and covers a wide range of sequence identities, varying from 80 to <10%.
METHODS
The HOMEP benchmark set
A data set of 36 HOMEP structures (see Supplementary Material Table 1; the data set is available at http://trantor.bioc.columbia.edu/∼lucy/homep) was selected from the PDB (5). All the proteins were solved using x-ray crystallography at a resolution of 3.5 Å or better. If two or more structures of the same protein were available, that with the highest resolution was selected. Polypeptide chains believed not to contact the membrane were omitted. Each family contains proteins with the same topology, defined here as the number and orientation of the transmembrane domains, excluding peripheral membrane-spanning domains that are not present in all members of the family. Taking each protein as a potential query sequence and all other members of its family as templates (for a homology model), the HOMEP data set contains 94 query-template pairs, from which 94 alignments and homology models can be constructed (Supplementary Material Table 2).
Two definitions of the transmembrane regions were adopted. The first, referred to as TM, was defined by hand to incorporate all residues in membrane-spanning secondary-structure elements according to DSSP (22) that were also superimposed in the structural alignment of all family members. Thus, the TM regions include residues located at the lipid-water interface as well as within the bilayer (Supplementary Material Table 3). The second definition, referred to as TMDET, comprises only residues in the hydrophobic core of the membrane, as defined by the TMDET algorithm (23) used by the PDB_TM database (24). Two short segments were incorrectly assigned by TMDET and thus excluded from the analysis: a strand (residues 128–133) in a loop region of 1osm and a helical region in the first two N-terminal residues of 1pw4.
Secondary-structure prediction accuracy
Since HOMEP is highly redundant by design, for the analysis of secondary-structure prediction algorithms we used the 40% nonredundant set of membrane proteins from the PDB_TM database from July 1, 2005. After excluding theoretical models, Cα-only structures, and proteins with missing residues, the set contained 106 chains from 71 membrane proteins, of which 92 chains were α-helical and 14 chains were β-barrels. Predictions were obtained with local installations of PSIPRED (25) v2.3, JNET (26), and PHDsec (27), and compared against assignments from DSSP. To obtain the multiple-sequence alignment input for each protein, we ran a PSI-BLAST search on the National Center for Biotechnology Information (NCBI) nonredundant database (nr); we ran three PSI-BLAST iterations including sequences below an E-value cutoff of 5 × 10−4 and reported sequences with an E-value cutoff of 1 × 10−3. No filtering of transmembrane regions was carried out.
We also assessed the composite prediction used by HMAP (28), which is a vector of probabilities for the three states (helix, strand, and coil) determined by direct averaging of the confidence scores from PSIPRED, JNET, and PHDsec. To enable comparison with the DSSP assignments, the prediction at each position was taken as the state with the highest probability.
Generation of sequence alignments
Sequence-to-sequence alignments
The dynamic programming algorithm in ClustalW v1.82 (29) was used to align each of the query-template sequence pairs. Gap-open penalties (po) of 9, 10, 11, 12, 15, and 20 were tested in combination with gap-extension penalties (pe) of 0.1 or 1. No clear difference was seen in the Q or AL0 scores (see below) of pairwise alignments using these different gap penalties (data not shown), so the default values (po = 10 and pe = 0.1) were used.
Sequence-to-profile alignments
We carried out PSI-BLAST (30) searches for each template sequence on the nr database, which was clustered at 65% sequence identity; five iterations of PSI-BLAST were carried out using E-value cutoffs as above. The sequence hits were compiled into a multiple-sequence alignment from which very remote homologs were removed according to the sequence threshold of Batalov and Abagyan as described by Tang et al. (28). This purged alignment was then used to create a sequence-based profile to which the query sequence was aligned with ClustalW, creating a sequence-to-profile alignment. A profile is an alternate representation of the primary sequence in which each amino acid position contains a set of probabilities.
Multiple-sequence alignments
These were generated by combining PSI-BLAST hits (as above) for both query and template into a single nonredundant set of sequences, which were then aligned using ClustalW, (T-Coffee (31), Muscle (32), and ProbCons (33)).
HMAP profile-to-profile alignments
HMAP is a program for the construction and alignment of structure-based profiles (28) that is similar in its algorithms to other profile-based approaches (34). For each template we generated two types of profile: HMAP [1,2] and HMAP [1,2,3], which combine sequence and secondary- and tertiary-structure information in different ways. The HMAP [1,2] template profiles combined sequence information from a PSI-BLAST search (as above) with a consensus secondary-structure assignment derived from all templates in the family, alongside position-specific weights reflecting the location of ungapped (i.e., core) positions in the alignment. The HMAP [1,2,3] template profiles differ in that the PSI-BLAST hits were taken from all available templates and merged using a structural alignment as a guide. For the query sequence we created a similar HMAP [1,2] profile, except that the secondary structure was obtained from a consensus prediction (see above) and the position-specific weights depended on the confidence levels of those predictions. Query and template profiles were then aligned using a score designed to favor matching of ungapped core regions and of secondary-structure types. Gap penalties were also assigned according to the location of core regions or secondary-structure elements. We used the local-global alignment method where unaligned terminal residues are only penalized in the query.
In the case of the reductase family of proteins, one member (PDB code: 1l0v) comprises two protein chains, whereas the homologous region in the other two reductase proteins is made up by a single chain. Alignment therefore required concatenation of the sequences or profiles of the two 1l0v chains; multiple sequence alignments were not possible.
Structure-based alignments
Structure-based sequence alignments were carried out with SKA (35,36). Residues that were matched in the structure alignment were used to define the correct alignment, which is the reference state in the calculation of the percentage of aligned positions that are correctly predicted, Q (see below). The sequence identity for each query-template pair was calculated using this alignment and was defined as the number of identical residues divided by the length of the shortest sequence.
Measures of accuracy
Models were built using Modeller 6v2 (37) and were assessed using several measures of structure similarity or model accuracy. In addition to the root mean squared deviation of the positions of the Cα atoms (Cα-RMSD), we compare the model with the native structure using two scores that are used to evaluate predictions in CASP (38). Both measures are based on the global distance test (GDT), which determines the number of model-template Cα-atom pairs, G(v) that are within a distance threshold, v Å (39). Using GDT results, the GDT_TS score (40) is then calculated as the average percentage of residues that fit within four different cutoff distances:
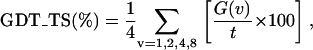 |
where t is the number of Cα-atoms in the template structure. A second measure, the AL0 score (37), is computed in a similar way but using a single threshold of 3.8 Å, that is
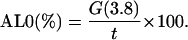 |
This threshold corresponds approximately to the distance between adjacent Cα atoms in a peptide chain, so that it tends to reflect structural differences corresponding to shifts in the sequence alignment.
Sequence alignment accuracy was also measured using the percentage of correctly aligned positions, Q:
 |
where Na is the number of nongapped positions in the structure-based SKA alignment and Nc is the number of correctly aligned positions in the test alignment compared to the SKA alignment.
For ease of comparison, the individual membrane protein models in our set (one for each query-template pair, M, have been ranked according to i) the fraction of the target structure that can be superimposed on the template within a cutoff distance of 5 Å, and ii) the sequence identity between the target and template. These two rankings, respectively denoted by 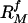 and
and 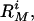 were combined into a relative difficulty score (41) for each model: Difficulty
were combined into a relative difficulty score (41) for each model: Difficulty
RESULTS
Benchmark of membrane protein homology model accuracy
For each of the 94 pairs of membrane proteins in the HOMEP data set, a homology model was built using the structure-based sequence alignment, which we take as the correct alignment. The Cα-RMSD and GDT_TS scores of these models, plotted against sequence identity (Fig. 1), provide a benchmark of the likely quality of a membrane protein homology model for a given level of sequence identity, assuming that the correct alignment can be achieved and that no refinement is carried out. Fig. 1 shows that the quality of a membrane protein homology model decreases exponentially with decreasing sequence identity.
FIGURE 1.

Structural relationship between membrane protein models and their templates. The sequence identity of the structure-based (correct) alignment is plotted against (A) the Cα-RMSD and (B) the GDT_TS scores of the corresponding model compared to the native structure. Data are shown for the whole protein (•) and for the transmembrane regions (□). Six models had RMSD values of between 10 and 40 Å; for clarity these points are plotted at RMSD = 10 Å.
Since the alignments used to generate these homology models are based on structural (i.e., optimal) alignments, Fig. 1 also contains information on the structural similarity between the target and query crystal structures. As such, the exponential relationship between sequence and structure for these membrane proteins appears to be very similar to that observed for pairs of homologous water-soluble proteins (42–44). The TM definition used here corresponds loosely to the common core defined by Chothia and Lesk (44); the Cα-RMSD values of the two data sets match reasonably well. The membrane protein whole-protein Cα-RMSDs are more similar to the values of Flores et al. (43), which also represent whole proteins, although this comparison is more difficult due to the large number of outliers in our data set. These outliers are caused by the absence of template regions for certain long (>10 residue) loops and termini, resulting in large local errors to which the RMSD measure is particularly sensitive. When AL0 and GDT_TS scores are used, however, it is clear that the scores for the whole models are indeed significantly lower than the scores for the transmembrane regions (Fig. 2). This suggests that there is a marked structural variability in the connecting regions between membrane-spanning segments of topologically related proteins (i.e., with the same number of transmembrane domains and the same N- to C-terminal orientations), as indicated by the variability in their length and sequences.
FIGURE 2.

Relationship between model quality and model-building difficulty. (Top) Alignment accuracy measured by AL0 for the whole protein (•) and transmembrane regions (□). (Bottom) Structural accuracy measured by GDT_TS for the whole protein (•) and transmembrane regions (□).
The AL0 scores of the transmembrane regions approach 100% in the majority of the models, whereas the GDT_TS scores for the same regions are often below 100%, suggesting that the errors in the easier models are local deviations that might be removed given an effective refinement protocol.
Secondary-structure prediction accuracy
We ran three different programs on a nonredundant set of membrane proteins of known structure and compared the results with assignments calculated using DSSP (Table 1). The per-residue three-state accuracy (helix, strand, or coil) of the three methods was found to be between 68 and 79%, which is comparable to the ∼76% found for globular proteins (25,26,45,46). Similar results were obtained for the composite prediction used by HMAP. Note that the standard deviations are large in all cases, especially for PHDsec and JNET, reflecting a variation in scores that is larger than the 7–10% deviation found for soluble proteins. When considering only the hydrophobic cores, as defined using TMDET, the accuracy improves further, especially for PSIPRED (87%). Comparing the different fold types, we found that α-helical residues in membrane proteins (particularly in the membrane regions) are on average more accurately predicted than β-strand residues, although the data set is smaller for the latter, making such comparisons tentative.
TABLE 1.
Secondary-structure prediction accuracy
| Residues* | PSIPRED | PHDsec | JNET | Composite† | |
|---|---|---|---|---|---|
| Whole | |||||
| All | 19,540 | 79.2 (10.9) | 67.6 (17.1) | 69.2 (17.6) | 77.6 (12.6) |
| α | 15,350 | 80.0 (10.4) | 67.5 (17.6) | 69.7 (18.3) | 78.5 (12.4) |
| β | 4190 | 74.1 (13.0) | 68.1 (13.4) | 65.6 (12.0) | 71.8 (12.9) |
| TMDET | |||||
| All | 5441 | 87.3 (16.7) | 65.2 (30.2) | 71.1 (27.9) | 82.3 (20.3) |
| α | 4386 | 89.6 (13.9) | 65.9 (31.7) | 73.6 (28.8) | 84.7 (19.1) |
| β | 1055 | 72.2 (24.5) | 61.1 (18.8) | 54.4 (11.2) | 66.5 (21.3) |
Average (and standard deviation) of the three-state accuracy, Q3, for several secondary-structure prediction methods. Q3 is measured as the percentage of residues that are correctly predicted as helix, strand, or coil relative to the DSSP assignment. Results were averaged either over the whole structure or over the hydrophobic regions as defined by TMDET and separated into helix bundles (α) and β-barrels (β).
Number of residues in each subset.
Composite prediction used by HMAP.
Sequence-based profile alignments
We compare the accuracy of membrane-protein sequence alignments and the models based thereon using the methodologies described in the Methods section. Comparing the two ClustalW methods using the AL0 scores of the respective models (Fig. 3 and Table 2), the sequence-to-profile alignments are more accurate than sequence-to-sequence alignments at low sequence identities. This is in line with results for nonmembrane proteins (47,48). However, in the range of 40–50% sequence identity, the sequence-to-profile alignments are less accurate than the sequence-to-sequence alignments. This has previously been observed for globular protein alignments with ClustalW (28,49).
FIGURE 3.

Accuracy of membrane protein sequence alignments/homology models obtained from different sequence alignment methods as a function of sequence identity. Results are given for (A) the whole protein and (B) the transmembrane regions. The average AL0 score is given over all alignments/models within a window of 10% sequence identity, and error bars indicate the standard deviation over that window. Numbers correspond to the number of alignments in each window and apply to both plots. Abbreviations: seq-seq, sequence-to-sequence alignment; seq-profile, sequence-to-profile alignment; and MSA, multiple sequence alignment. The two HMAP labels indicate profile-to-profile alignments.
TABLE 2.
The number of HOMEP alignments out of 90 for which a method gives a higher score for the whole/transmembrane regions
| AL0 | CW seq-seq | CW seq-prof | CW MSA | Probcons | Muscle | T-coffee | HMAP [1,2] | HMAP [1,2,3] |
|---|---|---|---|---|---|---|---|---|
| CW seq-seq | – | 56/53 | 58/56 | 77/64 | 78/71 | 79/67 | 77/64 | 80/69 |
| CW seq-prof | 33/24 | – | 59/54 | 73/57 | 72/67 | 75/64 | 77/63 | 80/67 |
| CW MSA | 19/13 | 28/20 | – | 57/46 | 61/58 | 65/53 | 69/55 | 73/61 |
| Probcons | 10/9 | 17/18 | 29/26 | – | 32/29 | 56/38 | 52/34 | 55/39 |
| Muscle | 10/5 | 15/12 | 25/18 | 55/31 | – | 60/43 | 52/36 | 58/44 |
| T-Coffee | 10/7 | 14/12 | 23/20 | 32/15 | 23/17 | – | 41/31 | 44/36 |
| HMAP[1,2] | 12/11 | 13/14 | 20/17 | 36/23 | 35/27 | 40/26 | – | 34/32 |
| HMAP[1,2,3] | 7/6 | 9/10 | 16/11 | 32/18 | 29/21 | 35/21 | 15/12 | – |
| Q | CW seq-seq | CW seq-prof | CW MSA | Probcons | Muscle | T-coffee | HMAP [1,2] | HMAP [1,2,3] |
| CW seq-seq | – | 49/53 | 50/54 | 67/60 | 65/64 | 63/64 | 68/57 | 71/62 |
| CW seq-prof | 36/18 | – | 57/50 | 72/58 | 67/59 | 74/59 | 76/58 | 78/65 |
| CW MSA | 28/14 | 29/19 | – | 56/44 | 57/48 | 59/48 | 64/52 | 70/55 |
| Probcons | 19/10 | 18/14 | 30/22 | – | 32/34 | 47/34 | 54/36 | 58/41 |
| Muscle | 22/9 | 21/11 | 28/22 | 51/31 | – | 55/43 | 62/43 | 69/51 |
| T-Coffee | 17/2 | 16/10 | 23/18 | 35/28 | 26/22 | – | 44/32 | 55/41 |
| HMAP[1,2] | 13/10 | 10/13 | 20/15 | 29/22 | 25/20 | 39/26 | – | 39/35 |
| HMAP[1,2,3] | 11/7 | 8/9 | 14/13 | 27/20 | 17/17 | 28/19 | 11/6 | – |
Number of times that the alignments from the method in a given column have higher scores than the method in the corresponding row for whole protein/transmembrane regions, using the AL0 score and the Q-score. The total number of query-template pairs used was 90, i.e., excluding alignments with 1l0v. Abbreviations: CW, ClustalW; seq-seq, sequence-to-sequence; seq-prof, sequence-to-profile; MSA, multiple-sequence alignment. For example, the upper right cell in a) reads as follows: The HMAP [1,2,3] alignments give better scores than ClustalW sequence-to-sequence alignments 80 times using the AL0 score for the whole protein, and 69 times for the transmembrane regions only. When only the transmembrane regions are considered, two methods are more likely to give exactly the same result than when the whole sequence is considered since these regions are less variable, and thus the differences tend to be smaller in the former case.
We also compare the ClustalW alignment results with those of other recently developed multiple-sequence alignment algorithms, namely, Probcons, T-Coffee, and Muscle, which have been reported to be more accurate than ClustalW for globular protein sequence alignments (31–33). Not all of these methods were able to align single sequences to a sequence profile; thus, for each method, we generated multiple-sequence alignments using the PSI-BLAST hits for both query and template (see Methods). The ClustalW multiple-sequence alignments were more accurate than the sequence-to-profile alignments, based on the AL0 scores of the corresponding models (Table 2). Comparing ClustalW multiple-sequence alignments with those of other methods using the signed rank test, the newer methods appear to offer significant improvement over ClustalW. Closer inspection reveals that this difference is due to alignments at sequence identities around 40%.
Structure-based profile-profile alignments
The use of the HMAP [1,2] structure-based profile-to-profile alignment method improves the AL0 scores of the models compared with the ClustalW sequence-to-profile alignments and multiple-sequence alignments (Fig. 3 and Table 2). However, the improvement is less obvious when comparing against the newer multiple-sequence alignment methods and in particular with T-Coffee. The most significant improvement in AL0 obtained from HMAP is seen for the most difficult alignments, with sequence identities of <10%. HMAP [1,2,3] alignments are better than the HMAP [1,2] alignments, especially for pairs of sequences with identities of 0–30%. Three-dimensional information is incorporated here using structural alignment of the available templates to guide the combination of their sequence information, as well as the assignment of weights to the core regions (see Methods). Clearly the higher precision achieved by combining template information in this way leads to greater accuracy in the alignments.
In summary, the HMAP [1,2] and HMAP [1,2,3] structure-based profile-to-profile alignments result in the most accurate models of all the methods compared here. However, the alignments obtained from HMAP are not optimal as defined by the structure-based alignments, which obviously limits the accuracy of the models built on these alignments.
Bipartite alignments
All the alignments presented so far, whether sequence- or profile-based, were calculated using the BLOSUM62 amino acid substitution matrix, which was developed for globular proteins (50). It has been suggested that bipartite alignments, which use different substitution matrices for the transmembrane and water-soluble regions, might be more appropriate for membrane proteins (10,16). We tested the effect of using a bipartite approach in a sequence-to-sequence alignment scheme (10,16) on the HOMEP data set using a simple dynamic programming algorithm where the PHAT matrix (16) was applied to the known transmembrane regions in the template and the BLOSUM62 substitution matrix was used for the remaining residues. Note that in contrast to the STAM method (17), we do not align the transmembrane segments separately and then add the loop regions, but rather align the whole sequence and choose the substitution matrix depending on the assignment of each position (10,16). The bipartite alignments result in models with lower AL0 scores than when BLOSUM62 is used throughout (Fig. 4 and Table 3); similar results are observed using Q scores. Using the TM definition of the transmembrane region (see Methods), the bipartite alignments were worse still, which reflects the unsuitability of the PHAT matrix for residues in the bilayer interfacial region.
FIGURE 4.

Accuracy of bipartite sequence-to-sequence alignments of membrane proteins obtained with different substitution matrices. See legend to Fig. 3 for more details.
TABLE 3.
Signed rank test using AL0 values for BLOSUM62-only against bipartite alignments for the whole/transmembrane regions
| Transmembrane definition* | Better† | Worse‡ | Total§ |
|---|---|---|---|
| TMDET | 57/53 | 23/21 | 94 |
| TM | 63/54 | 19/26 | 94 |
| TMDET: helix bundles | 19/17 | 17/19 | 46 |
| TMDET: β-barrels | 38/36 | 6/2 | 48 |
| TM: helix bundles | 27/21 | 13/18 | 46 |
| TM: β-barrels | 36/33 | 6/8 | 48 |
Definition of residues treated as transmembrane in the bipartite scheme (see Methods).
Number of times that BLOSUM62-only alignments are better.
Number of times that BLOSUM62-only alignments are worse.
Number of alignments tested.
Since PHAT was developed using transmembrane helices and not β-strands, we also separated the results by fold type (Table 3). As expected, the bipartite scheme worsens the alignments of the β-barrels, whereas the alignments of the helical bundles are very similar to when BLOSUM62 alone is used. Overall, in the most basic bipartite implementation, the PHAT substitution matrix does not appear to improve sequence-to-sequence alignments of membrane proteins.
Errors in individual alignments
For a few models we observe that the alignments generated using either HMAP [1,2] or HMAP [1,2,3] profiles were less accurate than the ClustalW sequence-to-profile alignments. The largest differences are found for the TonB-coupled receptor family, most strikingly in the models where BtuB (PDB code: 1nqe) is the query or where FepA (PDB code: 1fep) is the query. These errors are likely caused by the low secondary-structure prediction accuracy for the long β-strands in the TonB-coupled receptor family, which is 65.1% with PSIPRED. Other poor quality alignments are found for the seven transmembrane helix models (see Opsins in Supplementary Material Table 1), when rhodopsin (PDB code: 1u19) is either the query or the template, although the HMAP alignments are usually better than the ClustalW sequence-to-profile alignments. The structure of bovine rhodopsin is significantly different from that of the three bacterial opsins: the transmembrane helices of rhodopsin are more distorted and it contains an additional (interfacial) helix, a small β-sheet, and much longer loops and termini. These differences, along with extremely low sequence identities, combine to yield relatively poor quality alignments and models for this family.
DISCUSSION
Membrane protein homology modeling benchmark
In this study, we have presented a detailed analysis of the applicability of sequence alignment and homology modeling methods to integral membrane proteins. The HOMEP data set is key to the analysis, since it covers a range of fold types and sequence identities and thus provides a comprehensive benchmark of realistic modeling situations. Using this benchmark we show that similar trends exist with respect to the sequence-structure relationship (43,44) and to alignment accuracy (28) as are observed for water-soluble proteins. In addition, with this benchmark, it is possible to predict the likely accuracy of a homology model, assuming that an accurate alignment can be achieved and that no refinement is attempted. We find that the relationship between sequence identity and structure similarity is similar to that observed for water-soluble proteins, so that experience based on model accuracy for soluble proteins should be applicable to membrane proteins as well. For the transmembrane regions the expected model accuracy is higher than for the whole protein. For example, at 50% sequence identity to the template, a model is expected to have a Cα-RMSD of ∼1 Å from the native structure (∼95% GDT_TS) in the transmembrane regions. Indeed, an acceptable model of, say, 2 Å Cα-RMSD in the transmembrane regions (∼85% GDT_TS) is possible for most proteins above 30% sequence identity. In contrast, below ∼25% sequence identity, which is the similarity of many G-protein-coupled receptors to bovine rhodopsin—the only available template—a model may have a transmembrane Cα-RMSD from the native above 3.0 Å (∼75% GDT_TS). The accuracy of the complete model, including all extramembranous regions, will be expected to be lower than that of the transmembrane region alone.
This analysis indicates the accuracy of a model assuming that the conformation of the template structure reflects the desired conformation of the query protein. However, many membrane proteins are believed to undergo conformational changes during functional processes. Homology models cannot be expected to accurately predict such conformational changes per se: only the conformation closest to that of the chosen template will be adequately represented. Thus, the accurate prediction of many different functional conformations of a membrane protein will require template structures in equivalent conformations to be solved.
Membrane protein sequence alignments
Our analysis of sequence alignment algorithms indicates that those methods that have proved effective for water-soluble proteins work for membrane proteins as well. There is a clear progression in alignment accuracy when recently developed multiple sequence alignment (MSA) algorithms are used and additional improvements are obtained with HMAP's profile-to-profile alignment algorithm. Moreover, the increased use of structural information in the HMAP [1,2,3] alignments yields improvements relative to the HMAP [1,2] alignments. We note that ClustalW (29) is widely used to create sequence alignments for membrane proteins (51–56). Our results suggest that future work would benefit from the use of profile-to-profile methods and/or more advanced MSA techniques.
Our results on a simple bipartite sequence-to-sequence alignment method using the membrane-protein-specific substitution matrix PHAT show no significant improvement in the alignment quality over a traditional alignment using BLOSUM62. Originally, PHAT was shown to improve sensitivity in sequence database searches of membrane proteins (16). However, since database searching aims to best discriminate between similar and dissimilar proteins, rather than to achieve the correct global alignment of two sequences, the optimal parameters for the two applications may differ. There have also been some reported improvements in alignment accuracy using PHAT within the program STAM (17), which might be attributable to the separation and independent alignment of the transmembrane and nontransmembrane regions and to differences in gap penalties, rather than to the choice of substitution matrix. Clearly, the usefulness of membrane-protein-specific substitution matrices is dependent on the context, suggesting that the contribution of the choice of matrix should be carefully assessed in future applications.
Many other strategies have been presented for the alignment of membrane protein sequences (17,57–59) and for database searches (60,61). For example, probable transmembrane regions and loop regions have been aligned separately as independent segments (17,58) and then reassembled. Alignment of hydropathy profiles, rather than of primary sequences, has also been proposed (57). These methods have not been assessed here, either because they are not automated or because they were only suitable for helical proteins. However, it would be interesting to see how these methods compare with the profile-to-profile methods in terms of membrane protein alignment accuracy. Indeed, comparison of models from fully automated methods with those generated by experts in the field (with manual adjustment of alignments, for example) suggests that the manual approaches can lead to higher model accuracies (62). This has relevance to the alignments used in, e.g., G-protein-coupled receptor modeling (63), which have often required manual intervention. Nevertheless, a poor initial alignment may introduce errors that are missed during manual adjustment, particularly at low sequence identities, emphasizing the importance of accurate alignment algorithms.
Secondary-structure prediction
The success of the profile-to-profile methods is dependent on the accurate prediction of secondary structures in the query protein. We have shown that current secondary-structure prediction algorithms, and in particular PSIPRED, are only slightly less accurate for membrane proteins than they are for water-soluble proteins. This is rather surprising, since amino acids in membranes are reported to have different secondary-structure propensities (13–15) and because early prediction methods (64) gave results in poor agreement with experimental data for membrane proteins (65). Our results, which instead assess more recent, neural-network-based approaches using a larger set of high-resolution data, are supported by a previous study of membrane protein β-barrel prediction (66) in which similar results were obtained using PSIPRED (73%). (To our knowledge, no similar study has previously been attempted for helical membrane proteins.)
Neural networks derived from soluble proteins might have been expected to perform poorly on membrane proteins for two reasons: the membrane region imposes different secondary-structure propensities on amino acids, and the algorithms were not trained on membrane protein structures. Their success for membrane proteins may be due to the detection of the periodicity that is present in both sets of proteins. Even though the periodicity is effectively inverted, i.e., the surface of transmembrane regions is more hydrophobic than the interior whereas the surface of water-soluble proteins is more hydrophilic than the interior, the existence of a regular periodic pattern alone may be sufficient to obtain good prediction accuracy. In membrane protein β-barrels, the strands often extend far beyond the hydrophobic bilayer core where their properties are likely to strongly resemble the alternating patterns of water-soluble protein β-strands. However, the five to seven residues that comprise the membrane-spanning part of the strands may have a more complex pattern: the outer face of the barrel will be predominantly hydrophobic, whereas the interior face properties will depend on whether the barrel is filled with protein or water. This might explain the lower accuracy seen for the predictions on the hydrophobic TMDET regions of the β-barrels compared with the whole structures, although definitive interpretations are difficult due to the small number of structures (Table 1).
Secondary structure versus transmembrane prediction
Since they do not predict the same property, it is somewhat specious to directly compare the accuracies of secondary-structure predictions with those of transmembrane predictions. For reference, however, we note that the best-performing transmembrane-helix predictors have two-state per-residue accuracies (i.e., whether a residue is in the membrane or not) of ∼80% (67,68). Their accuracy at the segment level (i.e., whether a membrane-spanning helix is detected or not) is generally higher, between 85 and 99%. In the case of the β-barrel predictors, per-residue accuracies of ∼82% have been achieved (69). Thus, both the transmembrane helix and transmembrane strand methods are only slightly more accurate than the secondary-structure prediction algorithms. It is noteworthy, though, that as a consequence of the low number of structures available, accuracies for transmembrane predictions may be inflated by overtraining or by tests using proteins that were also included within the training set (68). In contrast, the secondary-structure prediction algorithms were solely trained on water-soluble proteins.
CONCLUSIONS
Using the HOMEP data set, we show that the construction of membrane protein homology models follows similar general rules to the construction of water-soluble models. That is, the expected accuracy of a membrane protein model will be similar to that of a water-soluble protein, assuming that similar alignment accuracy can be achieved. However, as a result of the low numbers of experimental structures of membrane proteins currently available, many candidate proteins for modeling are likely to have low sequence identities to their templates, so that accurate alignment of their sequences will be especially challenging. Our results suggest that more accurate alignments for such proteins can be achieved using structure-based profile alignment methods that have been developed for water-soluble proteins. In the future, however, it may be possible to incorporate information specific to membrane proteins—such as the location of hydrophobic transmembrane regions—within these methods to make alignments and homology models of membrane proteins even more accurate.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
We thank Shoshana Posy, Donald Petrey, Henry Bigelow, and Andrew Kernytsky for helpful discussions, and José Faraldo-Gómez for useful comments on the manuscript.
This work was supported by the National Science Foundation under grant No. MCB-0416708.
References
- 1.Jones, D. T. 1998. Do transmembrane protein superfolds exist? FEBS Lett. 423:281–285. [DOI] [PubMed] [Google Scholar]
- 2.Wallin, E., and G. von Heijne. 1998. Genome-wide analysis of integral membrane proteins from eubacterial, archean, and eukaryotic organisms. Protein Sci. 7:1029–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krogh, A., B. Larsson, G. von Heijne, and E. L. L. Sonnhammer. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305:567–580. [DOI] [PubMed] [Google Scholar]
- 4.Drews, J. 2000. Drug discovery: a historical perspective. Science. 287:1960–1964. [DOI] [PubMed] [Google Scholar]
- 5.Berman, H. M., J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov, and P. E. Bourne. 2000. The protein data bank. Nucleic Acids Res. 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petrey, D., and B. Honig. 2005. Protein structure prediction: inroads to biology. Mol. Cell. 20:811–819. [DOI] [PubMed] [Google Scholar]
- 7.Wallin, E., T. Tsukihara, S. Yoshikawa, G. von Heijne, and A. Elofsson. 1997. Architecture of helix bundle membrane proteins: an analysis of cytochrome c oxidase from bovine mitochondria. Protein Sci. 6:808–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu, Y., D. M. Engelman, and M. Gerstein. 2002. Genomic analysis of membrane protein families: abundance and conserved motifs. Genome Biol. 3:research0054.0051–0054.0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donnelly, D., J. P. Overington, S. V. Ruffle, J. H. A. Nugent, and T. L. Blundell. 1993. Modelling α-helical transmembrane domains: the calculation and use of substitution tables for lipid-facing residues. Protein Sci. 2:55–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones, D. T., W. R. Taylor, and J. M. Thornton. 1994. A mutation data matrix for transmembrane proteins. FEBS Lett. 339:269–275. [DOI] [PubMed] [Google Scholar]
- 11.Rees, D. C., L. DeAntonio, and D. Eisenberg. 1989. Hydrophobic organization of membrane proteins. Science. 245:510–513. [DOI] [PubMed] [Google Scholar]
- 12.Eyre, T. A., L. Partridge, and J. M. Thornton. 2004. Computational analysis of alpha-helical membrane protein structure: implications for the prediction of 3D structural models. Protein Eng. Des. Sel. 17:613–624. [DOI] [PubMed] [Google Scholar]
- 13.Li, S.-C., and C. M. Deber. 1994. A measure of helical propensity for amino acids in membrane environments. Nat. Struct. Biol. 1:368–373. [DOI] [PubMed] [Google Scholar]
- 14.Blondelle, S. E., B. Forood, R. A. Houghten, and E. Pérez-Payá. 1997. Secondary structure induction in aqueous vs membrane-like environments. Biopolymers. 42:489–498. [DOI] [PubMed] [Google Scholar]
- 15.Monné, M., I. Nilsson, A. Elofsson, and G. von Heijne. 1999. Turns in transmembrane helices: determination of the minimal length of a “helical hairpin” and derivation of a fine-grained turn propensity scale. J. Mol. Biol. 293:807–814. [DOI] [PubMed] [Google Scholar]
- 16.Ng, P. C., J. G. Henikoff, and S. Henikoff. 2000. PHAT: a transmembrane-specific substitution matrix. Bioinformatics. 16:760–766. [DOI] [PubMed] [Google Scholar]
- 17.Shafrir, Y., and H. R. Guy. 2004. STAM: simple transmembrane alignment method. Bioinformatics. 20:758–769. [DOI] [PubMed] [Google Scholar]
- 18.Granseth, E., G. von Heijne, and A. Elofsson. 2005. A study of the membrane-water interface region of membrane proteins. J. Mol. Biol. 346:377–385. [DOI] [PubMed] [Google Scholar]
- 19.Riek, R. P., I. Rigoutsos, J. Novotny, and R. M. Graham. 2001. Non-α-helical elements modulate polytopic membrane protein architecture. J. Mol. Biol. 306:349–362. [DOI] [PubMed] [Google Scholar]
- 20.Bowie, J. U. 2005. Solving the membrane protein folding problem. Nature. 438:581–589. [DOI] [PubMed] [Google Scholar]
- 21.Thompson, J. D., P. Koehl, R. Ripp, and O. Poch. 2005. BAliBASE 3.0: latest developments of the multiple sequence alignment benchmark. Proteins. 61:127–136. [DOI] [PubMed] [Google Scholar]
- 22.Kabsch, W., and C. Sander. 1983. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 22:2577–2637. [DOI] [PubMed] [Google Scholar]
- 23.Tusnady, G. E., Z. Dosztanyi, and I. Simon. 2005. TMDET: web server for detecting transmembrane regions of proteins by using their 3D coordinates. Bioinformatics. 21:1276–1277. [DOI] [PubMed] [Google Scholar]
- 24.Tusnady, G. E., Z. Dosztanyi, and I. Simon. 2005. PDB_TM: selection and membrane localization of transmembrane proteins in the protein data bank. Nucleic Acids Res. 33:D275–D278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones, D. T. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292:195–202. [DOI] [PubMed] [Google Scholar]
- 26.Cuff, J. A., and G. J. Barton. 1999. Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins. 40:502–511. [DOI] [PubMed] [Google Scholar]
- 27.Rost, B. 1996. PHD: predicting 1D protein structure by profile-based neural networks. Methods Enzymol. 266:525–539. [DOI] [PubMed] [Google Scholar]
- 28.Tang, C. L., L. Xie, I. Y. Y. Koh, S. Posy, E. Alexov, and B. Honig. 2003. On the role of structural information in remote homology detection and sequence alignment methods using hybrid sequence profiles. J. Mol. Biol. 334:1043–1062. [DOI] [PubMed] [Google Scholar]
- 29.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL_W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410. [DOI] [PubMed] [Google Scholar]
- 31.Notredame, C., D. G. Higgins, and J. Heringa. 2000. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302:205–217. [DOI] [PubMed] [Google Scholar]
- 32.Edgar, R. C. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Do, C. B., M. S. P. Mahabhashyam, M. Brudno, and S. Batzoglou. 2005. ProbCons: probabilistic consistency-based multiple sequence alignment. Genome Res. 15:330–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ohlson, T., B. Wallner, and A. Elofsson. 2004. Profile-profile methods provide improved fold recognition: a study of different profile-profile alignment methods. Proteins. 57:188–197. [DOI] [PubMed] [Google Scholar]
- 35.Yang, A. S., and B. Honig. 2000. An integrated approach to the analysis and modeling of protein sequences and structures. I. Protein structural alignment and a quantitative measure for protein structural distance. J. Mol. Biol. 301:665–678. [DOI] [PubMed] [Google Scholar]
- 36.Petrey, D., A. Nicholls, and B. Honig. 2003. GRASP2: visualization, surface properties and electrostatics of macromolecular structures and sequences. Methods Enzymol. 374:492–509. [DOI] [PubMed] [Google Scholar]
- 37.Sali, A., and T. L. Blundell. 1993. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234:779–815. [DOI] [PubMed] [Google Scholar]
- 38.Moult, J., K. Fidelis, A. Tramontano, B. Rost, and T. Hubbard. 2005. Critical assessment of methods of protein structure prediction (CASP)—round 6. Proteins. 61:3–7. [DOI] [PubMed] [Google Scholar]
- 39.Zemla, A. 2003. LGA: a method for finding 3D similarities in protein structures. Nucleic Acids Res. 31:3370–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moult, J., K. Fidelis, A. Zemla, and R. E. Hubbard. 2001. Critical assessment of methods of protein structure prediction (CASP): round IV. Proteins. 45:2–7. [PubMed] [Google Scholar]
- 41.Venclovas, C., A. Zemla, K. Fidelis, and J. Moult. 2003. Assessment of progress over the CASP experiments. Proteins. 53:585–595. [DOI] [PubMed] [Google Scholar]
- 42.Wilson, C. A., J. Kreychman, and M. Gerstein. 2000. Assessing annotation transfer for genomics: quantifying the relations between protein sequence, structure and function through traditional and probabilistic scores. J. Mol. Biol. 297:233–249. [DOI] [PubMed] [Google Scholar]
- 43.Flores, T. P., C. A. Orengo, D. S. Moss, and J. M. Thornton. 1993. Comparison of conformational characteristics in structurally similar protein pairs. Protein Sci. 2:1811–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chothia, C., and A. M. Lesk. 1986. The relation between the divergence of sequence and structure in proteins. EMBO J. 5:823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rost, B., and V. A. Eyrich. 2001. EVA: large-scale analysis of secondary structure prediction. Proteins. 45:192–199. [DOI] [PubMed] [Google Scholar]
- 46.Rost, B. 2001. Review: protein secondary structure prediction continues to rise. J. Struct. Biol. 134:204–218. [DOI] [PubMed] [Google Scholar]
- 47.Thompson, J. D., F. Plewniak, and O. Poch. 1999. A comprehensive comparison of multiple sequence alignment programs. Nucleic Acids Res. 27:2682–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wallace, I. M., G. Blackshields, and D. G. Higgins. 2005. Multiple sequence alignments. Curr. Opin. Struct. Biol. 15:261–266. [DOI] [PubMed] [Google Scholar]
- 49.Elofsson, A. 2002. A study on protein sequence alignment quality. Proteins. 46:330–339. [DOI] [PubMed] [Google Scholar]
- 50.Henikoff, S., and J. G. Henikoff. 1992. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA. 89:10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ogawa, H., and C. Toyoshima. 2002. Homology modeling of the cation binding sites of Na+K+-ATPase. Proc. Natl. Acad. Sci. USA. 99:15977–15982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Casadio, R., I. Jacoboni, A. Messina, and V. De Pinto. 2002. A 3D model of the voltage-dependent anion channel (VDAC). FEBS Lett. 520:1–7. [DOI] [PubMed] [Google Scholar]
- 53.Yang, Q., X. Wang, L. Ye, M. Mentrikoski, E. Mohammadi, Y.-M. Kim, and P. C. Maloney. 2005. Experimental tests of a homology model for OxlT, the oxalate transporter of Oxalobacter formigenes. Proc. Natl. Acad. Sci. USA. 102:8513–8518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuhlbrandt, W., J. Zeelen, and J. Dietrich. 2002. Structure, mechanism, and regulation of the Neurospora plasma membrane H+-ATPase. Science. 297:1692–1696. [DOI] [PubMed] [Google Scholar]
- 55.Bostina, M., B. Mohsin, W. Kühlbrandt, and I. Collinson. 2005. Atomic model of the E. coli membrane-bound protein translocation complex SecYEG. J. Mol. Biol. 352:1035–1043. [DOI] [PubMed] [Google Scholar]
- 56.Oyedotun, K. S., and B. D. Lemire. 2004. The quaternary structure of the Saccharomyces cerevisiae succinate dehydrogenase: homology modeling, cofactor docking and molecular dynamics simulation studies. J. Biol. Chem. 279:9424–9431. [DOI] [PubMed] [Google Scholar]
- 57.Lolkema, J. S., and D. J. Slotboom. 1998. Estimation of structural similarity of membrane proteins by hydropathy profile alignment. Mol. Membr. Biol. 15:33–42. [DOI] [PubMed] [Google Scholar]
- 58.Bissantz, C., A. Logean, and D. Rognan. 2004. High-throughput modeling of human G-protein coupled receptors: amino acid sequence alignment, three-dimensional model building, and receptor library screening. J. Chem. Inf. Comput. Sci. 44:1162–1176. [DOI] [PubMed] [Google Scholar]
- 59.Cserzo, M., J.-M. Bernassau, I. Simon, and B. Maigret. 1994. New alignment strategy for transmembrane proteins. J. Mol. Biol. 243:388–396. [DOI] [PubMed] [Google Scholar]
- 60.Clements, J. D., and R. E. Martin. 2002. Identification of novel membrane proteins by searching for patterns in hydropathy profiles. Eur. J. Biochem. 269:2101–2107. [DOI] [PubMed] [Google Scholar]
- 61.Hedman, M., H. Deloof, G. Von Heijne, and A. Elofsson. 2002. Improved detection of homologous membrane proteins by inclusion of information from topology predictions. Protein Sci. 11:652–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tress, M. L., I. Ezkurdia, O. Graña, G. López, and A. Valencia. 2005. Assessment of predictions submitted for the CASP6 comparative modeling category. Proteins. 61:27–45. [DOI] [PubMed] [Google Scholar]
- 63.Fanelli, F., and P. G. De Benedetti. 2005. Computational modeling approaches to structure-function analysis of G protein-coupled receptors. Chem. Rev. 105:3297–3351. [DOI] [PubMed] [Google Scholar]
- 64.Chou, P. Y., and G. D. Fasman. 1974. Conformational parameters for amino acids in helical, β-sheet and random coil regions calculated from proteins. Biochemistry. 13:211–222. [DOI] [PubMed] [Google Scholar]
- 65.Wallace, B. A., M. Cascio, and D. L. Mielke. 1986. Evaluation of methods for the prediction of membrane protein secondary structures. Proc. Natl. Acad. Sci. USA. 83:9423–9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bagos, P. G., T. D. Liakopoulos, I. C. Spyropoulos, and S. J. Hamodrakas. 2004. PRED-TMBB: a web server for predicting the topology of β-barrel outer membrane proteins. Nucleic Acids Res. 32:W400–W404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen, C. P., and B. Rost. 2002. State-of-the-art in membrane protein prediction. Appl. Bioinformatics. 1:21–35. [PubMed] [Google Scholar]
- 68.Chen, C. P., A. Kernytsky, and B. Rost. 2002. Transmembrane helix predictions revisited. Protein Sci. 11:2774–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bagos, P. G., T. Liakopoulos, and S. Hamodrakas. 2005. Evaluation of methods for predicting the topology of beta-barrel outer membrane proteins and a consensus prediction method. BMC Bioinformatics. 6:7. [DOI] [PMC free article] [PubMed] [Google Scholar]