

Abstract
Elastic network models are used for investigation of the p53 core domain functional dynamics. Global modes of motion indicate high positive correlations for residue fluctuations across the A-B interface, which are not observed at the B-C interface. Major hinge formation is observed at the A-B interface upon dimerization indicating stability of the A-B dimer. These findings imply A-B as the native dimerization interface, whereas B-C is the crystal interface. The A-B dimer exhibits an opening-closing motion about DNA, supporting the previously suggested clamp-like model of nonspecific DNA binding followed by diffusion. Monomer A has limited positive correlations with DNA, while monomer B exhibits high positive correlations with DNA in the functionally significant slow modes. Thus, monomer B might seem to maintain the stability of the dimer-DNA complex by forming the relatively fixed arm of the dimer clamp, whereas the other arm of the clamp, monomer A, might allow sliding via continuous association/dissociation mechanisms.
INTRODUCTION
The p53 tumor suppressor is a key transcriptional factor that activates the transcription of genes responsible in cell cycle arrest, apoptosis, and DNA repair. In normal cells, p53 level is kept low. In case of cellular stresses such as DNA damage, oncogene activation, etc., p53 is activated and facilitates the repair and survival of damaged cells before further rounds of replication or permanently removes the severely damaged cells through apoptosis. Either response would prevent replication of cells undergoing oncogenic changes and thus would inhibit tumor development (1–3). Studies show that mutation or deletion of the p53 gene favors tumor development and that p53 is mutated in half of all human cancers (4–7).
To transactivate its target genes, p53 binds to specific DNA sequences in tetramer form, i.e., dimer of dimers (8,9). p53 with 393 residues is divided into three functional domain regions, namely the N-terminal domain (residues 1–93) including the transactivation (residues 1–42) and proline-rich regions (residues 64–92); the DNA-binding core domain (residues 102–292) that binds directly to the DNA sequence and where most of the mutations in p53 are located (10,11); and the C-terminal domain including the tetramerization (residues 323–356) and negative regulatory regions (residues 363–393) (8–14). Although the structure of full-length p53 is not yet known, the crystal structures have been resolved for the DNA binding domain (10), tetramerization domain (9), a short sequence of 11 amino acids from the N-terminal domain in complex with p53 inhibitor protein MDM2 (15), and C-terminal negative regulatory domain (16). However, there are other functionally important domains and in between flexible linkers that have not yet been explored and thus the existing information is still not sufficient to deduce the overall arrangement of the domains relative to each other (17).
The crystal structure of p53 DNA binding core domain includes three asymmetric monomers A, B, and C in complex with DNA. P53 binds to a double-stranded DNA consensus binding site, which contains two copies of the 10-basepair motif 5′-Pu.Pu.Pu.C.(A/T).(T/A).G.Py.Py.Py-3′ (Pu = A/G; Py = T/C) (10). However, the solution characteristics of p53 and high-resolution NMR and crystal structures of the tetramerization domain (8,9) have elucidated the fact that p53 functions in tetramer form, i.e., a dimer of dimers. As a result of this finding, the mechanism of tetramer formation, interdimer/intradimer interactions and the identification of the native dimer pair has been the subject of much recent research (18–20). In the crystal structure of p53 core domain in complex with DNA, most of the interactions with DNA are made by monomer B and C. Monomer A is in contact with B, having little interaction with DNA. Thus, the native dimer pair was believed to be B-C and monomer A was thought to exist due to crystal packing (10). However, a recent MD simulation focused on the A-B dimer more likely being the stable biological interface than the B-C dimer (20).
We present here the vibrational mode characteristics of isolated monomers and dimer pairs of p53 core domain present in p53 trimer-DNA crystal structure (Protein Data Bank (PDB) code, 1tsr (10)). We use coarse-grained elastic network models, namely Gaussian network model (21) (GNM) and anisotropic network model (22) (ANM) to obtain the normal modes of monomers and dimers. Our objective is to see whether any significant differences between A-B and B-C dimer pairs exist in their cooperative fluctuations, which could provide insight about the native dimer pair. Moreover, for the native pair identified, we aim to suggest a mechanism of action for p53 core domain dimerization and DNA binding.
METHODS
Gaussian network model
GNM is a simple but powerful analytical approach developed for modeling the vibrational dynamics of folded proteins, providing information on the mechanism of global motions related to biological function (21,23,24). The nodes of the network are generally taken as the Cα atoms in the protein, which are considered to undergo Gaussianly distributed fluctuations. The interactions between close-neighboring nodes/residues that fall within a specified cutoff distance are described by identical harmonic springs.
In GNM, the correlations between residue fluctuations, 〈ΔRi · ΔRj〉 are evaluated from the partial inverse of the Kirchhoff matrix (Γ) of contacts between neighboring residue pairs, which describes the characteristic topology of the investigated structure, using:
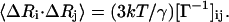 |
(1) |
Here, Ri is the position vector of the ith α carbon, k is the Boltzmann constant, T is the absolute temperature, and γ is the force constant. Cross correlations 〈ΔRi · ΔRj〉 and the mean-square fluctuations 〈(ΔRi)2〉 are thus simply given by the off-diagonal [Γ−1]ij and diagonal elements [Γ−1]ii, respectively. Residue correlations are actually the dot product of the fluctuation vectors of residues i and j. The cutoff distance including all residue pairs within a first interaction shell has been adopted as 7.0 Å in previous applications of the GNM to proteins (21,23,25). Hence, same value is used in this work. The total residue fluctuations can be decomposed into high- and low-frequency fluctuations, namely fast and slow modes. The slow modes are reported to be related to global motions, i.e., collective dynamics of the overall structure (25–28). The minima of the slowest modes correspond to the hinge regions that modulate the collective motions of the protein. On the other hand, the residues active in the fast modes (corresponding to peaks), also referred to as hot spot residues, have resistance to conformational changes, hence play an important role in maintaining the stability of the structure. These are tightly packed residues trapped in constraint minima on the conformational energy landscape (25). Previous studies (25–28) have indicated that these kinetically hot residues play a key role in maintaining the structure and stability of the global molecule with applications to proteins such as HIV-1 protease, chymotrypsin inhibitor 2, cyctocrome c, and transfer RNAs. These studies suggest that the hot spot residues and the hinge residues are associated and evolutionary conserved.
Anisotropic network model
GNM has been successful in providing information on the magnitudes of fluctuations. An extension of GNM is the anisotropic network model, which considers the anisotropy of the residue fluctuations, i.e., incorporating the directional preferences of collective motions. Thus, ANM is useful in investigating collective mode shapes of proteins, although GNM is more robust in prediction of the magnitude of the fluctuations and the correlations between them. 3N-dimensional Hessian matrix is adopted in ANM, instead of the N-dimensional Kirchhoff matrix in GNM (22), where N represents the total number of modes/residues. The cutoff distance has been adopted as 13 Å for consistency with the previous application of the ANM to proteins (22).
Previous studies have demonstrated that essential fluctuation characteristics and important collective mode shapes could be successfully reproduced by coarse-grained GNM and ANM with high efficiency, i.e., the required computational time being several orders of magnitude less than that for atom-based simulation techniques like molecular dynamics simulations (21,26,29,30).
RESULTS
Fluctuation dynamics by GNM
Isolated monomers
GNM calculations yield almost identical results for isolated monomers A, B, and C extracted from the p53 core domain-DNA structure (PDB code, 1tsr). Fig. 1, a and b, show residue mean-square (ms) fluctuations, 〈(ΔRi)2〉, for monomer A in fast and slow modes, respectively. Cumulative ms fluctuations of the fastest 10 modes and the slowest two modes are plotted. First two modes correspond to 16.7% of the total motion/fluctuations. It is observed that the peaks of the fast modes and in certain cases the hinges of the slow modes, i.e., the minima of ms fluctuations, correspond to the four conserved regions (CRs) determined to be present in core domain (10). In addition to these four CRs, another sharp peak is also identified by GNM, denoted with an asterisk (*) in Fig. 1 a, approximately between residues T155 and T170, which will also appear to have a functional importance in later sections.
FIGURE 1.

(a) High (fastest 10 modes average) and (b) low frequency (slowest first and second modes average) fluctuations of isolated monomer A. Similar graphs are obtained for the isolated monomers B and C. The peaks of the high-frequency fluctuations and most of the hinges obtained from the minima of low-frequency fluctuations correspond to the four conserved, functionally important regions. Another peak outside the four conserved regions, which is indicated with an asterisk (*) in Fig. 1 a, will be shown to have a functional importance in subsequent analysis.
Fig. 2 a points out the functionally important sections in the p53 core domain structure, which correspond to the CRs as described in literature. In this figure, L1 loop (residues 112–124) and S2 and S2′ sheets (residues 124–141), which together correspond to CR II, are colored in orange; part of L2 loop and H1 helix (residues 171–181) together corresponding to CR III are magenta; L3 loop (residues 236–251) corresponding to CR IV is blue; and end of S10 sheet (residues 271–274) and H2 helix (residues 278–286), together corresponding to CR V are red. These conserved regions have functional importance in maintaining the global structure of the protein and participating in DNA binding. It should be noted that CR I is not positioned in the p53 core domain (located in the N-terminus domain).
FIGURE 2.

(a) Functionally important parts in the p53 core domain structure resolved by Cho et al. (10) with PDB code, 1tsr. Monomer B is displayed in complex with DNA (DNA axis perpendicular to the page). Four conserved regions (CR) are indicated on the figure. CR II is colored in orange corresponding to L1 loop (112–124) and S2 and S2′ sheets (124–141), CR III is colored in magenta corresponding to part of L2 loop and H1 helix (171–181), CR IV is colored in blue corresponding to L3 loop (236–251), and CR V is colored in red corresponding to end of S10 sheet (271–274) and H2 helix (278–286). These conserved regions have functional importance in maintaining the global structure of the protein and participating in DNA binding. (b) Cross-correlation map for isolated monomer A (including first three modes, which corresponds to 23% of the global motion). Regions colored in brown, red, and orange indicate positive correlations in order of highest to lowest positive correlation values. Blue regions indicate negative correlations, similarly, dark blue and light blue reflecting higher and lower negative correlation values, respectively. Two regions that are known to have functional importance (10) are emphasized with black circles on the figure. One of these two regions demonstrates the positive correlation between the residues of the loop-sheet-helix (L1 loop-S2 and S2′ sheets-H2 helix) motif of p53 that is responsible for the direct contact with DNA major groove. The other positive correlation reflects a relation between L2 and L3 loops; i.e., L3 loop makes direct contact with DNA minor groove and L2 has a stabilizing effect on L3 via coordination of a Zn atom. Thus, residues involved in similar functions are fluctuating in a cooperative manner.
Cross correlations between residues of monomer A are also extracted by GNM. The correlations between the fluctuations of residues, 〈ΔRi · ΔRj〉, within monomer A are calculated via Eq. 1. by taking into account the slowest three modes that correspond to 23% of the total motion. The correlation map is demonstrated in Fig. 2 b. Correlation values close to one and minus one indicate residues that move in the same or opposite direction, respectively. In the figure, the residues exhibiting positive correlations are labeled as brown, red, and orange in order of decreasing positive correlation and negative correlations are shown with dark and light blue in order of decreasing negative correlation. The top left black circle on the map indicates high positive correlations between residues F112 and C135 (L1 loop + S2 and S2′ sheets, CR II) residues F270–L289 (S10 sheet + H2 helix, CR V). This region encompasses the loop-sheet-helix (L1 loop-S2 and S2′ sheets-H2 helix) motif of p53 that is responsible for the direct contact with DNA major groove (10). Similarly, residues between T163 and P195 (L2 loop + H1 helix, CR III) and between T236 and P251 (L3 loop, CR IV) possess high positive correlation (indicated with the other black circle on the figure). This correlation supports the evidence that a functional relation exists between the loops L2 and L3. In fact, L3 loop makes direct contact with DNA minor groove and L2 has a stabilizing effect on L3 via coordination of a Zn atom. Residues that are involved in a specific function are expected to be fluctuating in cooperative manner, i.e., involved in the coupled network of fluctuations, as also implied here. Similar cross-correlation maps are obtained for the isolated monomers B and C (not shown here).
Dimers
GNM calculations are also carried out with dimer pairs A-B and B-C both in the presence and absence of DNA to identify the native dimer pair by investigating the differences in their vibrational behaviors. The residue ms fluctuations are compared in isolated and dimer forms for all three monomers. Because the slowest modes (low-frequency fluctuations) reproduce the most significant characteristics of the global motion, the weighted average of the two slowest modes (amounting to 16.7 and 25.4% of the total fluctuations for isolated monomer A and dimer A-B, respectively), is considered here. Fig. 3, a and d, show the differences between the normalized fluctuations of residues in isolated monomers and in dimer form, in the absence of DNA. In Fig. 3 a, two distinct regions in monomer A (I162–R175 and Y205–S215) indicated by arrows exhibit high mobility in isolated monomer form but diminished motion when present in the dimer form with B. Residues I162–R175 also correspond to a high-frequency fluctuation region that was denoted with and asterisk (*) in Fig. 1 a. Other than these two regions, the distribution of the fluctuations is similar. Similar observation is made for monomer B. As depicted in Fig. 3 b, there is a significant decrease in the mobility of region R175–G187 of isolated monomer B when it is present in dimer form A-B (indicated by arrow). This region becomes a hinge region in dimer A-B implying the gain of stability. However, there is no distinguishable new hinge formation in the B-C dimer for either monomer B or C (Fig. 3, c and d). In the case of fast modes, no distinct difference is observed upon dimerization for both A-B and B-C dimer pairs (not shown here). Existence of a hinge axis and associated hinge residues at the dimerization, oligomerization domains of proteins during complex formation, or at the interfacial regions between proteins was previously reported in literature (27,28,31,32). These hinge residues located at the interfaces display restricted fluctuations in slow modes of the protein hence being involved in stabilizing and modulating the global motion (27,32). As an example, in the case of bacterial ribonuclease barnase experimental and computational studies have revealed the essential role of hinge regions for the stability and activity of the enzyme (33–35).
FIGURE 3.

Comparison of the average of the first two slowest modes across monomers and dimers. (a) Mean-square fluctuation of monomer A in isolated form (dotted line) and in A-B dimer complex (solid line). Two distinct regions indicated by arrows emphasize the decrease of mobility of these residues of monomer A upon dimerization; i.e., these regions gain stability when present in dimer form. (b) Mean-square fluctuation of monomer B in isolated form (dotted line) and in A-B dimer complex (dark solid line). One region exists, indicated by an arrow, where the mobility is decreased upon dimerization. (c) Mean-square fluctuation of monomer B in isolated form (dotted line) and in B-C dimer complex (dark solid line). No significant new hinge formation is observed upon dimerization. (d) Mean-square fluctuation of monomer C in isolated form (dotted line) and in B-C dimer complex (dark solid line). No significant new hinge formation is observed upon dimerization.
Cross-correlation maps including the slowest three modes in the absence and presence of DNA are displayed for dimers A-B and B-C in Fig. 4. Panels a and b are for dimers A-B and B-C, respectively, in the absence of DNA, and panels c and d are in the presence of DNA. Residue numbers colored in green belongs to monomer A, violet to monomer B, and blue to monomer C. The color code in the correlation map is the same as Fig. 2 b. Because the correlation between A-B and B-A is the same, i.e., symmetric, negative correlations between A and B are not plotted (but they are plotted in B-A) to emphasize the existing positive correlations. But it should be noted that the correlation map is actually symmetric for A-B or B-A. The difference between the A-B and B-C pairs can be clearly observed here. From Fig. 4 a, it is observed that monomers A and B are negatively correlated except a distinct positively correlated line, corresponding to the interface residues. These positively correlated interface residues also have high conservation. In general, this result together with our unpublished data on several proteins suggests that interface residues having correlated fluctuations correspond to the binding hotspots. Other than the positive correlation observed in the binding interface, monomers A and B exhibit negative correlation, meaning the global monomers move in opposite directions as in an opening-closing type of motion but the interface residues exhibit correlated fluctuations, i.e., move in accordance. Especially, as indicated with red circles, monomer A has two distinct regions showing high positive correlation (value close to 1.0) with a certain region in monomer B. In fact, these two regions exactly correspond to the previously identified hinge regions in Fig. 3, where the mobility has dramatically decreased upon dimerization, indicating the gain of stability upon dimer formation. This possibly implies functionally important fluctuations across the interface, i.e., the presence of a native dimerization interface. This synchronized motion at the dimer interface would bring about stability to the dimeric complex. On the other hand, no significant positive correlation exists between monomers B and C (Fig. 4 b). This observation suggests that the A-B dimer pair is more probable to be the native dimer than B-C, with possible dimerization interface at I162–R175 and Y205–S215 in monomer A and R175–G187 in monomer B, while monomer C may exist due to crystal packing. R175–G187 in monomer B corresponds to the H1 helix and part of L2 loop. This H1 helix seems to coordinate both of the two regions in monomer A that are the I162–R175 region corresponding to the L2 loop and part of H1 helix and Y205–S215 corresponding to S6–S7 sheets. It should also be noted that this H1 helix region of monomer B has negative correlation with the rest of the residues in monomer B as indicated with the black ellipse on the figure.
FIGURE 4.

Cross-correlation maps for dimers presenting positive/negative correlations in the absence (panels a and b) and presence (panels c and d) of DNA. (including first three modes). In the axis, residue numbers colored in green belongs to monomer A, violet to monomer B, and blue to monomer C. Positive correlations are plotted in brown, red, and orange in decreasing correlation values and negative correlations are plotted in dark and light blue, similarly in decreasing correlation values. Map is symmetric for A-B and B-A, hence, negative correlations between A and B are not shown (but they are shown in B-A) to emphasize the existing positive correlations. (a) Correlation map for dimer A-B in the absence of DNA. Dimer A-B exhibits a positively correlated interface. Highest positive correlation exists between residues I162 and R175 and Y205 and S215 of monomer A with residues R175–G187 of monomer B (shown with red circles). Except the interface residues, monomers A and B display negative correlation in the rest of the structure implying motion in opposite directions; i.e., an opening-closing type of motion, while the dimer interface move in the same direction. The positive correlation at the interface would bring about stability to the dimeric structure. Although R175–G187 of monomer B exhibit positive correlation with monomer A at the interface, this region is negatively correlated with the rest of the monomer B residues (shown with a black oval), implying this interface region of monomer B moving in opposite direction with respect to the global monomer. (b) Correlation map for dimer B-C in the absence of DNA. No positive correlation is present at the B-C interface. (c) Correlation map for dimer A-B + DNA complex. DNA binding enhances the existing positive correlation at the A-B interface (the interface line is darker red than in panel a). Monomer A has dominantly negative correlation with DNA (minor positive correlation with DNA is observed in higher modes). Monomer B has mostly positive correlation with DNA except the A-B dimer interface (indicated with black circle). This indicates that binding affinities of the two monomers to DNA are not the same, i.e., monomer A probably binds less tightly to DNA whereas monomer B maintains stronger interaction with DNA. (d) Correlation map for dimer B-C + DNA complex: The lack of any positive correlation between B-C is still valid upon DNA binding. (e) Crystal structure of p53 (1tsr) dimers A and B in complex with DNA (monomer C not shown). The suggested dimerization interface, which possesses high positive correlation, is colored: I162-R175 (orange) and Y205-S215 (violet) of monomer A interact with R175-G187 (blue) of monomer B at the interface.
Previous studies performed, both molecular dynamics (20) and NMR spectroscopy (36,37), pointed out the importance of H1 helix in dimerization. Ma et al. (20) obtained that, helix 1 residues of monomer B (R181, C182) forms hydrogen bonding with several residues in monomer A (V172, R174, R175, G244) and S185 of monomer B also interacts with T211 of monomer A. These findings map into our suggested dimerization interface region. In this work, we identified that L2 loop also takes part in dimerization interactions in addition to H1 helix, which has not been mentioned in literature so far. Additionally, we found another region in monomer A (Y205–S215) that also interacted with monomer B in the dimerization interface. On the other hand, no significant positive correlation (higher than 0.5) exists between the interface residues of monomers B and C.
GNM calculations performed on dimers A-B and B-C in the presence of DNA illustrate the effect of DNA binding. In incorporating DNA into the calculations, each nucleotide is represented with three atoms namely the P of the phosphate, C2 of the base, and C4′ of the sugar (38). Same cutoff values as for Cα atoms are adopted for each of these nodes representing DNA bases. The correlation maps for A-B (Fig. 4 c) and B-C (Fig. 4 d) dimers in complex with DNA demonstrate the difference between the residue fluctuations at the dimerization regions. As before, residues colored in green belong to monomer A, violet to monomer B, blue to monomer C, and black to DNA sections, and also making use of the symmetry, we did not display the negative correlations for A-B to emphasize the positive ones (they are shown in B-A). The results show that the positive correlation between A-B dimers is enhanced, implying A-B dimer to be more stabilized in the presence of DNA (Fig. 4 c). The absence of any positive correlation between monomers B and C is still valid in the presence of DNA (Fig. 4 d). Monomer A is highly negatively correlated with DNA, while monomer B has positive correlation with DNA except the suggested dimerization interface as indicated with a black circle in the figure (Fig. 4 c). This may be explained by the fact that the A and B subunits in the dimer may exhibit movement in opposite directions to each other, like an opening-closing clamp-like motion around DNA. Monomer B would keep closer contact with DNA as compared to monomer A. Hence, in such a clamp-like motion around DNA, the opening-closing arm of the clamp may be the negatively correlated monomer A. The correlation graphs include the first three modes, but it should be noted that monomer A exhibits minor positive correlation with DNA in higher modes (after fifth mode). Nevertheless, the positive interaction of monomer B with DNA is much more dominating. In Fig. 4 e, monomers A-B in complex with DNA are shown (monomer C not shown) with indication of the suggested dimerization interface as a result of GNM fluctuation and correlation analysis. The two regions in monomer A (orange, I162–R175; and purple, Y205–S215) that are found to be coordinated with a single region in monomer B (blue, R175–G187) are emphasized.
Mode shape analysis by ANM
The validity of the residue fluctuation results is further tested by ANM, for both isolated monomers and dimers. The cross correlations obtained by GNM are in conformity with those from ANM, i.e., monomers A-B have positively correlated interface residues, whereas B-C have not. Because ANM can provide information about the direction of fluctuations in addition to magnitudes in GNM, the motion of the suggested dimerization interface will be investigated next.
Fig. 5 demonstrates the alternative conformations of dimer A-B in the absence and presence of DNA. Panels a–d belong to the mode shapes of A-B dimer in the absence of DNA, and panels e–h belong to the ones in complex with DNA. Fig. 5, a and b, represent the conformations that describe the fluctuations in the first slowest mode; Fig. 5, c and d, in the second mode. The two alternative conformations displayed for each mode can be viewed as the positive and negative deviations from the native structure, i.e., the average structure. It should be stated that the deformations are amplified here for clarity. The interface residues are indicated by orange, violet, and blue colors as before and to clarify the direction of motion some regions are shown by rods (red and green residues). The first mode (Fig. 5, a and b) is characterized to be a “twisting” motion around interface. When the positions of red and green rods are compared between the two figures, the two monomers seem to exhibit twisting in opposite directions, while the interface residues do not lose their close proximity hence maintain a synchronized motion. The arrows indicate the directions of twisting of the two monomers with respect to their native structure. The second mode (Fig. 5, c and d) can be described with a distinct “bending” motion around interface. The two monomers bend in opposite directions, i.e., monomer A bends in counterclockwise direction, whereas monomer B bends clockwise. Similarly, the global motion of the dimer can be understood by investigating the positions of the residues shown by red and green rods. This motion may also be regarded as an opening-closing type clamp movement around interface. As before, the interface residues act together without undergoing much conformational change even at such exaggerated rescaling and their close proximity (within 7 Å neighborhood) is preserved by the existence of positive correlations and interactions. The arrows indicated on the figure better explain the possible directions of global motions of the monomers and the interface residues. It should be noted that, helix 1 (blue residues) of monomer B tend to move in opposite direction with respect to the rest of the monomer as this was an outcome of the correlation analysis.
FIGURE 5.

Alternative conformations of dimer A-B as a result of ANM calculations in absence (a–d) and presence (e–h) of DNA. The dimerization interface is indicated with orange, violet, and blue regions as in Fig. 4 e and red and green residues represented by rods are to distinguish between the two conformations and visualize the directions of global motion. The deformations are amplified for clarity. (a and b) Positive-negative deviations from the native structure obtained in first mode. The two monomers exhibit “twisting” around the interface in opposite directions whereas the interface residues preserved their close proximity. The directions of twisting are indicated on the figure for each monomer with respect to the native structure. (c and d) Positive-negative deviations from the native structure obtained in second mode. The monomers exhibit “bending” around interface in opposite directions. The global dimer can be viewed as an opening (d)-closing (c) clamp. The directions of motions are shown with arrows. The dimer interface residues move in the same direction. (e and f) Positive-negative deviations from the native structure obtained in the first mode for simulations including DNA. Interaction of monomer B, which keeps close contact with DNA in both conformations, is the dominant motion in the first mode. DNA limits the conformational flexibility of the system. (g and h) Positive-negative deviations from the native structure obtained in the second mode for simulations including DNA. In addition to monomer B, interaction of monomer A with DNA comes into the picture. Monomer A moves toward (g) and away (h) from DNA. Similar clamp-like motion is observed around DNA, opening/closing arm of the clamp being monomer A, which is known to have little positive correlation and hence minor contact with DNA. This characteristic motion of the dimer may enable a sliding mechanism along DNA.
Similarly, in panels e–h the first two mode shapes of dimer A-B in the presence of DNA are displayed. The dimer A-B exhibits motions associated with DNA interactions in addition to the characteristic synchronized motion at the dimerization interface. Presence or absence of DNA does not seem to alter the interactions valid between monomers A and B as demonstrated by correlation analysis (Fig. 4, a and c). The interface residues are observed to be the closest in all four slowest modes (only first two are shown here) both in the presence and absence of DNA and they fluctuate in the same direction. When the dimer motions in the presence and absence of DNA are compared, it is concluded that the mode shapes are shifted by the introduction of DNA. An overlap matrix demonstrating the overlap, i.e., the match of modes across given structures (in this case the modes without DNA and modes with DNA) is formed. The dot products of the normalized eigenvectors of each structure after superimposition indicate the degree of overlap of their motion deformation. Hence, best (100%) overlap produces an overlap value of 1.0. Based on this matrix, the first mode associated with twisting around the interface observed for the calculations without DNA (Fig. 5, a and b), best matches with the second mode of the calculations including DNA with an overlap value of 0.71, and the second mode without DNA corresponds to third mode with DNA, having an overlap value of 0.72.
When the alternative conformations for the most dominating first mode are investigated (Fig. 5, e–f), the presence of DNA seems to limit the flexibility of the dimeric system. The system is quite stable that monomers do not exhibit major conformational changes such as twisting, bending, etc. Though not so pronounced, the motion of the system is mostly observed to be associated with the interaction of monomer B (right) and DNA, while no significant change occurs in monomer A (left). In both figures, monomer B maintains close interaction with DNA, (approach is more in Fig. 5 e and little separation occurs in Fig. 5 f as indicated with arrows) and the dimer seems to bind DNA initially with monomer B. The motion in the second mode is more noteworthy. It reveals the interaction of monomer A with DNA and the clamp-like motion of the global dimer. As indicated by arrows, monomer A approaches to DNA (Fig. 5 g) and separates from DNA (Fig. 5 h) through its helix region; while monomer B continues to keep close contact with DNA. Due to existing interactions in the A-B dimer interface and close contact of monomer B and DNA, the global sense of the A-B dimer resembles a clamp-like molecule around DNA, mainly opening from one arm. One arm of the clamp, monomer B, mostly keeps close contact with DNA, the other arm of the clamp, monomer A, exhibits opening-closing movement around DNA. In fact, based on the previous correlation analysis it was shown that monomer A was negatively correlated with DNA, indicating monomer A moving in opposite direction with DNA most of the time. Thus, this may be the outcome of motion of the monomer A as an opening-closing clamp arm. Previous literature studies confirm such clamp-like motions of proteins binding to nucleic acids. DNA-dependent protein kinases exhibit a closure around DNA from one arm of the clamp, very similar to the one described here (39). Similar opening-closing clamp motion has been observed by ANM for both bacterial and yeast RNA polymerases (40), in conformity with experimental results.
In case of dimer B-C (figures not given here), the number of neighboring residues across the interface within 7 Å is just four in the native structure. As a result of ANM simulations, in first four slowest modes, even if some new contacts seem to occur, the residues that fall into 7 Å distance do not lie in the dimerization interface of the crystal structure, instead they either belong to the DNA binding site, A-B interface or an area with no importance so far known. These residues give topological approach probably due to the absence of DNA and monomer A, i.e., due to the absence of interactions of monomer B with DNA and/or monomer A. Moreover, characteristic opening/closing (approaching and separating) or bending motion observed in dimer A-B is not seen in dimer B-C. This result is also in accordance with the correlation profiles by GNM in which no positive correlation was obtained between B-C dimers in the absence or presence of DNA.
DISCUSSION
Coarse-grained elastic network models have been utilized to analyze the vibrational dynamics of p53-DNA complex, both in monomer and dimer forms. As p53 crystal structure consists of three identical monomers (A, B, and C) and it is known to function as a dimer of dimers, our aim is to possibly reveal the native dimerization interface (either A-B or B-C) based on structure-function relationships extracted from computationally efficient elastic networks. In general, our findings are in conformity with the results of previous studies performed by using molecular dynamics (20) and NMR techniques (36,37).
Both in the presence and absence of DNA, A-B dimer comprises interfacial regions showing high positive correlations, which do not exist in the B-C dimer. Based on these correlations, residues I162–R175 (L2 loop and part of H1 helix) and Y205–S215 (S6–S7 sheets) of monomer A and residues R175–G187 (H1 helix and part of L2 loop) of monomer B seem to form the native dimerization interface. Moreover, a significant hinge formation is observed at these regions upon dimerization indicating a stable dimer formation. These positive correlated interface residues also possess conservation. A-B dimer interactions are valid even in the absence of DNA and the conformation of dimer A-B is not changed much upon DNA binding. The characteristics of this interface are similar in the absence and presence of DNA. This implies that the structures evolve in such a way that their elastic modes facilitate their function. Consequently, our results suggest a plausible, efficient approach by using elastic network models to shed light on the native dimers in a protein's x-ray structure. The existence of correlated fluctuations in the slowest modes of motion between monomers can be a clue for functional association.
The fact that A-B dimer interactions are valid even in the absence of DNA and that the conformation of dimer A-B is not changed much with DNA supports the fact that first the dimers are formed and then DNA binding occurs. ANM calculations revealed that the dimer exhibits similar clamp motion both in the absence and presence of DNA, which is in agreement with the study of Klein et al. (37). In the study of Klein et al. (37), the authors used NMR spectroscopy to show that p53 core domain itself does not undergo major conformational changes upon addition of DNA. In a recent study (41), the authors emphasized that the proteins possess intrinsic, structure-encoded abilities necessary to achieve their functions. This preexisting equilibrium is also validated in the case of p53-DNA interactions. The dimeric p53 possesses the relevant modes of motion related to DNA binding even before the binding occurs. Hence the dominant modes of the dimer-DNA complex seem to evolve from the modes of the dimer with some additional modes of motion associated with dimer-DNA interactions. As a result, DNA binding does not change much the existing characteristics of the dimer.
It should be noted, however, that although the computational work presented here intended to unravel p53-DNA interactions, computations were performed for only the core domain structure. However the entire structure of p53 is not completely resolved yet, the results presented here can be further validated with new structural information. Nevertheless, as presented in a recent review (42), the structural characteristics, conformational changes, and mode shapes of substructures and/or monomers facilitate complex formation and determine binding properties. Hence, the results presented here are plausible from the aspect of modeling the core domain substructure both in the absence and presence of DNA.
Our findings are also in agreement with recent studies in literature reporting evidences of nonspecific DNA binding of p53 followed by diffusion, i.e., sliding toward the specific binding site where tetramerization is more likely to occur (18,20,36,37,43–45). In the experimental study of McLure and Lee (18) who first proposed the clamp-like motion of the p53 dimer, a model was proposed in which one of the monomers of the dimer first binds to DNA, followed by the other. In their experimental work, Jiao et al. (43) reported observations of dynamic interactions of p53 with DNA including continuous association/dissociation and sliding. Ma et al. (20) proposed models for p53 dimer binding to DNA including one that is a sliding mechanism. However, no distinction was made in any of these models between the monomers A, B, or C with respect to their interactions with the DNA. However, in our study for the identified native dimer (A-B) the motions of the monomers A and B are shown to be rather different from each other. We observe a clamp-like opening-closing motion in dimer A-B similar to the model postulated by McLure and Lee (18). It was observed that the positively correlated dimer interface residues move in the same direction. Moreover, monomer B binds more tightly to DNA having more close contacts, which suggests that monomer B may be the first monomer binding to DNA and then followed by monomer A. Monomer A binding less tightly to DNA (mainly negatively correlated with DNA), exhibits an opening-closing motion about the DNA so as to enable and control the sliding mechanism. Thus, we think that dimers A-B bind nonspecifically to DNA and by diffusion, i.e., sliding along DNA they are transferred toward the specific binding site where tetramerization is more likely to take place.
Sliding proteins, including transcriptional factor proteins like p53, and their diffusion-driven mechanisms in search for specific binding sites are deeply investigated in literature (46–52). Several mechanisms are proposed explaining the diffusion of the protein to the target site such as, full (macroscopic) and microscopic dissociation, sliding, and intersegmental transfers (46,47). During the sliding process, it is reported that the protein, A-B dimer in this case, should neither completely dissociate from DNA (46,47) nor bind too tightly (48). The protein should have stable interactions with DNA but the connection must temporally break down for it to slide over the DNA (49). Many proteins bind concurrently to two sites in DNA forming complex structures such as dimers or tetramers (52). These proteins first bind to one target DNA binding surface and then the other subunit binds at a second site. The protein is fixed at one site via one binding surface, and then it might seem possible for the second surface to guide the sliding until the specific site is located (52). This finding is in good agreement with the model proposed here. In such a clamp-like motion we observed in this work, the sliding process can be achieved by one arm of the clamp (monomer B) binding more tightly to DNA, while the other (monomer A) having weaker interaction with DNA may fluctuate around DNA like an opening-closing arm of the clamp.
In summary, we suggest that the initial association mechanism of the dimer complex to DNA, may start with monomer B having high correlation with DNA, and then the interactions, i.e., positive fluctuations shown to exist in the suggested dimer interface lead to the formation of stable A-B dimer + DNA complex. Through sliding, the dimer may reach the specific binding site and hence can transactivate its target proteins. In the case of a complete dissociation of the dimer from DNA, we suggest that the dissociation mechanism may start with monomer A, shown to have negative correlation with DNA.
Last of all, we believe that here we displayed mainly two novel points in regard to use of elastic network models. The dynamic modes and correlated fluctuations could be used to distinguish the native dimers from crystal dimers, which has been also confirmed by the results of the anlaysis on several other cases (our unpublished results). This approach can be used as a powerful tool for analyzing multimonomer protein structures and differentiating native/crystal interfaces. Also, by combined analysis of two elastic network models (GNM and ANM), we could suggest a binding mechanism of p53 to DNA, describing a sliding motion together with clamp-like behavior, which could also be a plausible approach for identifying similar mechanisms in other protein-DNA systems.
Acknowledgments
This work has been supported by the Bogazici University B.A.P. (projects 06A508 and 04A502), DPT Project (03K120250), the Turkish Academy of Sciences in the framework of the Young Scientist Award Program (PD-TUBA-GEBIP/2002-1-9 and TH-TUBA-GEBIP/2001-1-1), and EU-FP6-ACC-2004-SSA-2 contract No. 517991.
References
- 1.el-Diery, W. S. 1998. Regulation of p53 downstream genes. Semin. Cancer Biol. 8:345–357. [DOI] [PubMed] [Google Scholar]
- 2.Ryan, K. M., A. C. Phillips, and K. H. Vousden. 2001. Regulation and function of the p53 tumor suppressor protein. Curr. Opin. Cell Biol. 13:332–337. [DOI] [PubMed] [Google Scholar]
- 3.Hofseth, L. J., S. P. Hussain, and C. C. Harris. 2004. p53: 25 years after its discovery. Trends Pharmacol. Sci. 25:117–181. [DOI] [PubMed] [Google Scholar]
- 4.Donehower, L. A., M. Harvey, B. L. Slagle, M. J. McArthur, C. A. Montgomery, J. S. Butel, and A. Bradley. 1992. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumors. Nature. 356:215–221. [DOI] [PubMed] [Google Scholar]
- 5.Hollstein, M., K. Rice, M. S. Greenblatt, T. Soussi, R. Fuchs, T. Sorlie, E. Hovig, B. Smith-Sorensen, R. Montesano, and C. C. Harris. 1994. Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res. 22:3551–3555. [PMC free article] [PubMed] [Google Scholar]
- 6.Hollstein, M., B. Shomer, M. S. Greenblatt, T. Soussi, E. Hovig, R. Montesano, and C. C. Harris. 1996. Somatic point mutations in the p53 gene human tumors and cell lines: updated compilation. Nucleic Acids Res. 24:141–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soussi, T., K. Dehohuche, and C. Beroud. 2000. p53 website and analysis of p53 gene mutations in human cancer: forging a link between epidemiology and carcinogenesis. Hum. Mutat. 15:105–113. [DOI] [PubMed] [Google Scholar]
- 8.Clore, G. M., J. G. Omichinski, K. Sakaguchi, N. Zambrano, H. Sakamoto, E. Appella, and A. M. Gronenborn. 1994. High-resolution structure of the oligomerization domain of p53 by multidimensional NMR. Science. 265:386–391. [DOI] [PubMed] [Google Scholar]
- 9.Jeffrey, P. D., S. Gorina, and N. P. Pavletich. 1995. Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 Angstroms. Science. 267:1498–1502. [DOI] [PubMed] [Google Scholar]
- 10.Cho, Y., S. Gorina, P. D. Jeffrey, and N. P. Pavletich. 1994. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 265:346–355. [DOI] [PubMed] [Google Scholar]
- 11.Friend, S. 1994. p53: A glimpse at the puppet behind the shadow play. Science. 265:334–335. [DOI] [PubMed] [Google Scholar]
- 12.Bargonetti, J., J. J. Manfredi, X. Chen, D. R. Marshak, and C. Prives. 1993. A proteolytic fragment from the central region of p53 has marked sequence-specific DNA-binding activity when generated from wild-type but not from oncogenic mutant p53 protein. Genes Dev. 7:2565–2574. [DOI] [PubMed] [Google Scholar]
- 13.Pavletich, N. P., K. A. Chambers, and C. O. Pabo. 1993. The DNA binding domain of p53 contains the four conserved regions and the major mutation hot spots. Genes Dev. 7:2556–2564. [DOI] [PubMed] [Google Scholar]
- 14.Wang, Y., M. Reed, P. Wang, J. E. Stenger, G. Mayr, and M. E. Anderson. 1993. p53 domains: identification and characterization of two autonomous DNA-binding regions. Genes Dev. 7:2575–2586. [DOI] [PubMed] [Google Scholar]
- 15.Kussie, P. H., S. Gorina, V. Marechal, B. Elenbaas, J. Moreau, A. J. Levine, and N. P. Pavletich. 1996. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 274:948–953. [DOI] [PubMed] [Google Scholar]
- 16.Rustandi, R. R., D. M. Baldisseri, and D. J. Weber. 2000. Structure of the negative regulatory domain of p53 bound to S100B(ββ). Nat. Struct. Biol. 7:570–574. [DOI] [PubMed] [Google Scholar]
- 17.Lebrun, A., R. Lavery, and H. Weinstein. 2001. Modeling multi-component protein-DNA complexes: the role of the bending and dimerization in the complex of p53 dimers with DNA. Protein Eng. 14:233–243. [DOI] [PubMed] [Google Scholar]
- 18.McLure, K., and P. W. K. Lee. 1998. How p53 binds DNA as a tetramer. EMBO J. 17:3342–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chong, L. T., C. D. Snow, Y. M. Rhee, and V. S. Pande. 2005. Dimerization of the p53 oligomarization domain: identification of a folding nucleus by molecular dynamics simulations. J. Mol. Biol. 345:869–878. [DOI] [PubMed] [Google Scholar]
- 20.Ma, B., Y. Pan, K. Gunasekaran, R. B. Venkatarahavan, A. J. Levine, and R. Nussinov. 2005. Comparison of the protein-protein interfaces in the p53-DNA crsytal structures: towards elucidation of the biological interface. Proc. Natl. Acad. Sci. USA. 102:3988–3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bahar, I., A. R. Atilgan, and B. Erman. 1997. Direct evaluation of thermal fluctuations in proteins using a single parameter harmonic potential. Fold. Des. 2:173–181. [DOI] [PubMed] [Google Scholar]
- 22.Atilgan, A. R., S. R. Durell, R. L. Jernigan, M. C. Demirel, O. Keskin, and I. Bahar. 2001. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 80:505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haliloglu, T., I. Bahar, and B. Erman. 1997. Gaussian dynamics of folded proteins. Phys. Rev. Lett. 79:3090–3093. [Google Scholar]
- 24.Bahar, I. 1999. Dynamics of proteins and biomolecular complexes. Rev. Chem. Eng. 15:319–347. [Google Scholar]
- 25.Demirel, M. C., A. R. Atilgan, R. L. Jernigan, B. Erman, and I. Bahar. 1998. Identification of kinetically hot residues in proteins. Protein Sci. 7:2522–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bahar, I., A. R. Atilgan, M. C. Demirel, and B. Erman. 1998. Vibrational dynamics of folded proteins: significance of slow and fast motions in relation to function and stability. Phys. Rev. Lett. 80:2733–2736. [Google Scholar]
- 27.Bahar, I., and R. L. Jernigan. 1998. Vibrational dynamics of transfer RNAs: comparison of the free and synthease bound forms. J. Mol. Biol. 281:871–885. [DOI] [PubMed] [Google Scholar]
- 28.Jernigan, R. L., M. C. Demirel, and I. Bahar. 1999. Relating structure to function through the dominant modes of motion of DNA topoisomerase II. Int. J. Quantum Chem. 75:301–312. [Google Scholar]
- 29.Doruker, P., A. R. Atilgan, and I. Bahar. 2000. Dynamics of proteins predicted by molecular dynamics simulations and analytical approaches: application to α-amylase inhibitor. Proteins. 40:512–524. [PubMed] [Google Scholar]
- 30.Doruker, P., R. L. Jernigan, and I. Bahar. 2002. Dynamics of large proteins through hierarchical levels of coarse-grained structures. J. Comput. Chem. 23:119–127. [DOI] [PubMed] [Google Scholar]
- 31.Haliloglu, T., and I. Bahar. 1999. Structure-based analysis of protein dynamics: comparison of theoretical results for hen lysozyme with X-ray diffraction and NMR relaxation data. Proteins. 37:654–667. [DOI] [PubMed] [Google Scholar]
- 32.Dehocuk, Y., C. Biot, D. Gilis, J. M. Kwasigroch, and M. Rooman. 2003. Sequence-structure signals of 3D domain swapping in proteins. J. Mol. Biol. 330:1215–1225. [DOI] [PubMed] [Google Scholar]
- 33.Axe, D. D., N. W. Foster, and A. R. Fersht. 1998. A search for single substitutions that eliminate enzymatic function in a bacterial ribonuclease. Biochemistry. 37:7157–7166. [DOI] [PubMed] [Google Scholar]
- 34.Axe, D. D., N. W. Foster, and A. R. Fersht. 1999. An irregular beta-bulge common to a group of bacterial RNases is an important determinant of stability and function in barnase. J. Mol. Biol. 286:1471–1485. [DOI] [PubMed] [Google Scholar]
- 35.Nolde, S. B., A. S. Arseniev, V. Y. Orekhov, and M. Billeter. 2002. Essential domain motions in barnase revealed by MD simulations. Proteins. 46:250–258. [DOI] [PubMed] [Google Scholar]
- 36.Rippin, T. M., S. M. V. Freund, and D. B. Veprintsev. 2002. Recognition of DNA by p53 core domain and location of intermolecular contacts of cooperative binding. J. Mol. Biol. 319:351–358. [DOI] [PubMed] [Google Scholar]
- 37.Klein, C., E. Planker, T. Diercks, H. Kessler, K. P. Künkele, K. Lang, S. Hansen, and M. Schwaiger. 2001. NMR spectroscopy reveals the solution dimerization interface of p53 core domains bound to their consensus DNA. J. Biol. Chem. 276:49020–49027. [DOI] [PubMed] [Google Scholar]
- 38.Delarue, M., and Y. H. Sanejouand. 2002. Simplified normal mode analysis of conformational transitions in DNA-dependent polymerases: the elastic network model. J. Mol. Biol. 320:1011–1024. [DOI] [PubMed] [Google Scholar]
- 39.Boskovic, J., A. Rivera-Calzada, J. D. Maman, P. Chacon, K. R. Willison, L. H. Pearl, and O. Llorca. 2003. Visualization of DNA-induced conformational changes in the DNA repair kinase DNA-PKcs. EMBO J. 22:5875–5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yildirim, Y., and P. Doruker. 2004. Collective motions of RNA polymerases. Analysis of core enzyme, elongation complex and holoenzyme. J. Biomol. Struct. Dyn. 22:267–280. [DOI] [PubMed] [Google Scholar]
- 41.Tobi, D., and I. Bahar. 2005. Structural changes involved in protein binding correlate with intrinsic motions of proteins in the unbound state. Proc. Natl. Acad. Sci. USA. 102:18908–18913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Changeux, J. P., and S. J. Edelstein. 2005. Allosteric mechanisms of signal transduction. Science. 308:1424–1428. [DOI] [PubMed] [Google Scholar]
- 43.Jiao, Y., D. I. Cherny, G. Heim, T. M. Jovin, and T. E. Schaffer. 2001. Dynamic interactions of p53 with DNA in solution by time-lapse atomic force microscopy. J. Mol. Biol. 314:233–243. [DOI] [PubMed] [Google Scholar]
- 44.Weinberg, R. L., D. B. Veprintsev, and A. R. Fersht. 2004. Cooperative binding of tetrameric p53 to DNA. J. Mol. Biol. 341:1145–1159. [DOI] [PubMed] [Google Scholar]
- 45.McKinney, K., M. Mattia, V. Gottfredi, and C. Prives. 2004. p53 linear diffusion along DNA requires its C terminus. Mol. Cell. 16:413–424. [DOI] [PubMed] [Google Scholar]
- 46.Berg, O. G., R. B. Winter, and P. H. von Hippel. 1981. Diffusion-driven mechanisms of protein translocation on nucleic acids. I. Models and theory. Biochemistry. 20:6929–6948. [DOI] [PubMed] [Google Scholar]
- 47.Winter, R. B., O. G. Berg, and P. H. von Hippel. 1981. Diffusion-driven mechanisms of protein translocation on nucleic acids. III. The Escherichia coli lac repressor-operator interaction: Kinetic measurements and conclusions. Biochemistry. 20:6961–6977. [DOI] [PubMed] [Google Scholar]
- 48.Sokolov, I. M., R. Metzler, K. Pant, and M. C. Williams. 2005. Target search of N-sliding proteins on a DNA. Biophys. J. 89:895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsurimoto, T. 1998. PCNA, a multifunctional ring on DNA. Biochim. Biophys. Acta. 1443:23–29. [DOI] [PubMed] [Google Scholar]
- 50.Jen-Jacobson, L. 1997. Protein-DNA recognition complexes: conservation of structure and binding energy in the transition state. Biopolymers. 44:153–180. [DOI] [PubMed] [Google Scholar]
- 51.Shimamato, N. 1999. One-dimensional diffusion of proteins along DNA. J. Biol. Chem. 274:15293–15296. [DOI] [PubMed] [Google Scholar]
- 52.Halford, S. E., and J. F. Marko. 2004. How do site-specific DNA-binding proteins find their targets? Nucleic Acids Res. 32:3040–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]