

Abstract
Quantitative in vivo measurements are essential for developing a predictive understanding of cellular behavior. Here we present a technique that converts observed fluorescence intensities into numbers of molecules. By transiently expressing a fluorescently tagged protein and then following its dilution during growth and division, we observe asymmetric partitioning of fluorescence between daughter cells at each division. Such partition asymmetries are set by the actual numbers of proteins present, and thus provide a means to quantify fluorescence levels. We present a Bayesian algorithm that infers from such data both the fluorescence conversion factor and an estimate of the measurement error. Our algorithm works for arbitrarily sized data sets and handles consistently any missing measurements. We verify the algorithm with extensive simulation and demonstrate its application to experimental data from Escherichia coli. Our technique should provide a quantitative internal calibration to systems biology studies of both synthetic and endogenous cellular networks.
INTRODUCTION
A goal of systems biology is to build a predictive, computational cellular model (1). A major challenge, however, is the lack of quantitative in vivo data for the many parameters required, such as protein concentrations and reaction rates (2). Well-established techniques that address this issue directly do exist for the confocal microscope, such as fluorescence correlation spectroscopy (3,4), image correlation spectroscopy (5), photon-counting histogram analysis (6), and fluorescence intensity distribution analysis (7), but unambiguous results usually require considerable expertise. Another approach is to construct a model and extract parameters by fitting in vivo data (8–10). Nevertheless, experimental data, such as fluorescence levels of tagged proteins or immunoblots, is usually limited to unitless ratios of expression levels that are only proportional to the actual protein concentrations. Not having direct measures can significantly hinder or complicate finding parameter values (9). In many studies (see (11,12) for reviews), the linear relation between the concentrations of fluorescent proteins and their measured fluorescence intensities has been used to measure protein levels in living cells, though only in relative terms and not in absolute numbers. Here we present a fluctuation method for measuring and calculating the conversion factor between the amount of a fluorescent protein and the measured fluorescence level. We will denote this conversion factor by ν; it is measured in fluorescence units per fluorescent protein (or, more generally, fluorescence units per fluorescent particle).
For a cell or a cellular compartment with a fluorescence intensity of y, the number of protein molecules, n, is given by y/ν. The measured fluorescence is, however, perturbed by measurement error. Assuming that this error is additive, each fluorescence measurement f actually satisfies
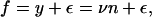 |
(1) |
with the magnitude of ε reflecting the size of the measurement error.
Analyzing the partitioning of proteins or other molecules in daughter cells upon cell division provides, in principle, a means to quantify fluorescence data (13,14). We constructed a synthetic network in Escherichia coli that enables control of the expression levels of a reporter protein (13). The λ-phage protein, CI, was fused to yellow fluorescent protein (YFP) to make a fluorescent reporter CI-YFP. The reporter was placed on a plasmid under the control of the tetracycline promoter, pTet: a promoter tightly repressed by the tetracycline repressor, TetR. The tetR gene, itself, was chromosomally inserted into the bacterial genome where it is constitutively expressed. Consequently, only the presence of the inducer, anhydrotetracycline (aTc), which inhibits the DNA binding properties of TetR, allows fluorescent protein production. By washing out aTc, fluorescent gene expression is cut off. Fig. 1 shows time-lapse images of an E. coli microcolony. The colony originates from one cell taken from a population that was briefly induced by aTc and therefore contains a fixed amount of CI-YFP. No synthesis or significant degradation or photobleaching of CI-YFP takes place (see Fig. 1 C), and its concentration only dilutes through microcolony growth (the average number of molecules per cell halving at each division). Analysis of such movies gives not only quantitative fluorescence levels, but also the lineage tree shown in Fig. 1 B. We shall denote by f2i and f2i+1 the fluorescence levels in the two daughter cells that originate from a mother cell with fluorescence fi; see Fig. 1 D.
FIGURE 1.

Partitioning of a fluorescent protein during microcolony growth. (A) Snapshots from a typical dilution experiment. Images are taken using yellow fluorescence filters, and the time between the frames shown is ∼36 min. (B) The lineage tree extracted from the same movie. Time increases downwards as more and more divisions occur. Measurements are marked by a dot and were taken approximately every 9 min (giving a total of just over 700). (C) Cellular fluorescence only decreases significantly at cell division. For this dataset, fluorescence measurements were taken every second frame, i.e., every 18 min. The first cell divides after ∼20 min, and its daughters in turn both divide at 60 min. Notice the different fluorescence values in each daughter cell become more apparent at later divisions. For clarity, only the initial part of the movie is shown. (D) Schematic of a lineage tree generated from an initial cell with fluorescence value f1. Second-generation cells have fluorescence f2 or f3, while third-generation cells have f4, f5, f6, or f7.
We present two methods to infer ν from the fluorescence data of such lineage trees. Assuming that fluorescent proteins are distributed with equal probability to either daughter cell at division, daughter cells will only have, on average, equal fluorescence levels. Our technique gathers information on ν by examining the deviation of actual daughter fluorescence levels from this average behavior. Such fluctuation analyses, although perhaps uncommon in molecular biology, are well established in neuroscience (15). For example, fluctuations in membrane current through a patch have long been used to infer the numbers of conducting ion channels in the patch (16). Although the mathematics of our analysis is different, we follow the same philosophy.
METHOD I: AN APPROXIMATE SOLUTION
Method I ignores the structure of the lineage tree and assumes no measurement error, i.e., ε = 0 in Eq. 1. The data is collected into triads, each triad containing fluorescence from a mother cell and its two daughters. A triad is represented as (yi, y2i, y2i+1), where i denotes a mother cell and runs from 1 to L, say, and y2i and y2i+1 are the fluorescence levels in the daughters.
Given a triad, we wish to infer the most probable value of ν. Using Bayes's rule, the probability of ν given the data, y, is
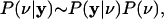 |
(2) |
where P(y|ν) is the likelihood of the data given a value of ν and P(ν) is the prior distribution for ν. We assume the prior distribution to be constant over a range of ν and zero elsewhere, so that, a priori, ν is equally likely to be found anywhere between a minimum (1, say) and a maximum (100, for example). As the fluorescent protein is neither synthesized nor degraded, the number of proteins in the parent cell, ni, is equal to the sum of the numbers in the daughters, ni = n2i + n2i+1. With no measurement error, conservation of proteins implies that the fluorescence of the mother cell must also equal the sum of the fluorescent values of the two daughters, yi = y2i + y2i+1. In reality, measurement error causes this relationship to hold only approximately; Method I ignores these errors.
Calculation of P(ν|y) involves evaluating the likelihood, P(y|ν). Considering one triad, yi, y2i, and y2i+1, denoted by the vector t, the likelihood obeys
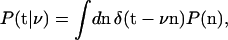 |
(3) |
where the ni are approximated to be continuous, and the vector notation implies three δ-functions, one for each member of the triad. The probability P(n) = P(ni, n2i, n2i+1) for the protein numbers can be factorized
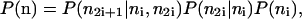 |
(4) |
where P(n2i+1|ni, n2i) is set by the constraint n2i+1 = ni – n2i, and P(n2i|ni) is an even binomial distribution: each fluorescent molecule has the same chance of going to either daughter upon cell division.
An even binomial distribution can be approximated by a normal distribution with mean ni/2 and standard deviation 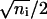 (17). Using a δ-function to enforce the conservation of protein numbers, Eq. 4 then becomes
(17). Using a δ-function to enforce the conservation of protein numbers, Eq. 4 then becomes
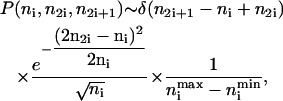 |
(5) |
where the prior distribution, P(ni), is a uniform bounded distribution such that ni lies anywhere between 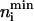 and
and  a priori. Inserting Eq. 5 into Eq. 3, and using the relation δ(y – νn) = δ(n – y/ν)/ν to carry out the integrations, gives
a priori. Inserting Eq. 5 into Eq. 3, and using the relation δ(y – νn) = δ(n – y/ν)/ν to carry out the integrations, gives
 |
(6) |
Assuming independent measurements of each triad, P(y|ν) for the full set of L triads is a product of terms like Eq. 6,
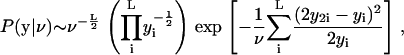 |
(7) |
assuming that the conservation constraints are satisfied. With a constant prior distribution, P(ν), the posterior (see Eq. 2) has the same form as Eq. 7.
The most probable value of ν maximizes the posterior probability, and is found by differentiation. The maximum occurs at ν = ν*, with
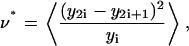 |
(8) |
where angled brackets denote an average over all L triads, and is equal to the average of the square of the difference in fluorescence of the two daughters divided by the value of the fluorescence in the mother cell. By evaluating the second derivative of Eq. 7 at ν = ν*, the error in the inferred value, Eq. 8, is estimated as 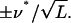
Method I, exemplified by Eq. 8, is equivalent to the more ad hoc approach used previously (13,14). Although it does ignore measurement error, it involves only a few simple computations.
METHOD II
We assume that each fluorescence measurement satisfies Eq. 1, where the measurement error term, εi, or equivalently fi–yi, has a normal distribution with zero mean and standard deviation σ. The size of σ sets the magnitude of the measurement error. The posterior for both ν and σ satisfies P(ν, σ|f) ∼ P(f|ν, σ)P(ν, σ), or
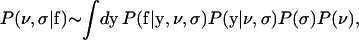 |
(9) |
using the product rule of probability theory and noting that the prior distribution for σ is independent of ν.
Including measurement error
The probability of the data f given y depends only on the measurement error, P(f|y, ν, σ) = P(f|y, σ). Using the normal distribution model for εi, we have
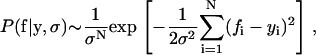 |
(10) |
assuming that the errors in each measurement are independent and that there are N measurements.
Including the tree
The second probability in Eq. 9, P(y|ν, σ), is independent of the measurement error σ. From conservation of proteins, the yi obey P(y2i+1|yi, y2i) = δ(y2i+1 – yi + y2i) for a mother cell and its daughters. Considering, for example, Fig. 1 D, factorizing P(y|ν) implies
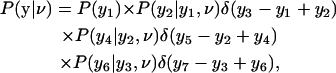 |
(11) |
where P(y1) is the prior distribution for the fluorescence level in the first cell. For one mother-daughter pair, P(y2i|yi, ν) is given by the exponential term of Eq. 6, and so
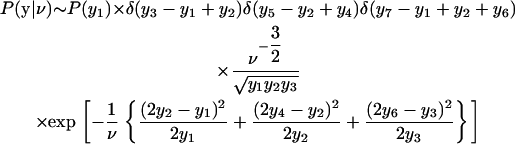 |
(12) |
after rearranging the δ-functions.
The posterior distribution
Assuming constant (but bounded) prior distributions for ν, y1, and σ, the posterior distribution satisfies
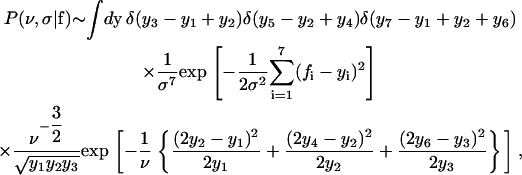 |
(13) |
for the tree of Fig. 1 D (a more general expression is given in the Appendix). Partly integrating Eq. 13, y3 is replaced by y1 – y2, y5 by y2 – y4, and y7 by y1 – y2 – y6.
It is instructive to consider only three cells, i.e., just one division event, and a given σ: All the integrals in the equivalent of Eq. 13 can be evaluated analytically, and the value of ν that maximizes this posterior is
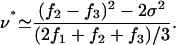 |
(14) |
In the limit of σ → 0, f1 exactly equals f2 + f3, and Eq. 14 recovers Eq. 8. Notice that the best estimate for y1, the denominator of Eq. 14, is now a weighted sum of f1 and f2 + f3, the latter being a second estimate of y1 in a data set with measurement errors.
To evaluate Eq. 13 in general, we use the variable elimination method (18) to numerically find the integral for any given ν and σ. Let ys denote the set of independent y variables—those not defined by the conservation of number constraints. As the integrand factorizes into a product of terms, one for each triad in the tree, the M-dimensional integral over ys transforms into a series of tractable two-dimensional computations. Typically, we perform this calculation over a grid defined by a priori ranges of ν and σ, thus giving a two-dimensional posterior distribution (see Fig. 2 A for an example). For the case of a normal distribution model of measurement error, however, we can derive an accurate estimate of σ (see Appendix),
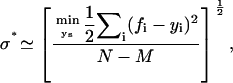 |
(15) |
where the δ-function constraints in Eq. 13 hold. There are N data points and M (<N) independent y variables. We use this estimate with 20 steps of Golden section search to efficiently explore the a priori interval for ν. The results of Fig. 3 were generated with this method. For such simulated data, we compared the posteriors found with those generated using the true value of σ instead of σ*. Their difference is negligible.
FIGURE 2.

Results of Method II applied to the data of Fig. 1. (A) A contour plot of the posterior probability of σ and ν. The distribution has a single peak with a most probable value of ν ≃ 15 ± 4 fluorescence units per fluorescent particle and of σ ≃ 156 ± 5 fluorescence units. (B) The posterior marginalized over σ. The most probable value of ν is given at the maximum and the error in this estimate by the peak width at half-maximum. The inset shows the marginalized posteriors for the data of Fig. 1 (left curve) and for three other data sets taken on the same microscope on the same day. Inferred values of ν are consistent.
FIGURE 3.

Evaluation of the inference methods for simulated data using the inference score iε = | log 2(ν/ν0)| (a perfect fit has an inference score of zero), with simulated ν0 = 25. (A) Inference improves with more data, but is sensitive to signal/noise ratios; Method II is robust. Twenty different data sets, each of seven generations, were created from an initial cell having either 500 (first two bars) or 5000 fluorescent proteins (last two bars). For generation numbers below seven, the appropriate lower part of the data tree was discarded. Measurement error was added with σ = 150, and three measurements were taken per cell. (B) The performance of Method II as the size of the measurement error grows; inference improves by increasing the number of measurements per cell. Notice the new y-axis scale. Twenty different data sets with 500 proteins in the initial cell were fit, and the results averaged to generate each bar. For both figures, we used the analytical estimate for σ (Eq. 15), and took the maximum of the posterior as the best estimate for ν. Error bars are standard errors.
Extra and missing data
It is often possible to measure fluorescence levels in a cell several times before it divides (Fig. 1 B). The data is then stored in a matrix, rather than a vector, where fij is the jth measurement of fluorescence in cell i. Each measurement for cell i improves the estimate of yi. The number of yi variables does not change, and Eq. 10 just gains more terms. For example, with C measurements per cell,
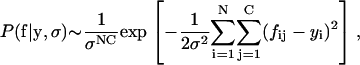 |
(16) |
and the general form of Eq. 13 (see Appendix) remains essentially unchanged.
Cells do not all divide synchronously (Fig. 1 B), and so for some we have more measurements than for others. Consequently, the C-values in Eq. 16 vary from cell to cell. Fig. 1 B also shows that data is missing from parts of the lineage tree, particularly at the extremities. Usually these cells are obscured by surrounding cells, as large microcolonies no longer grow in a plane and terrace. Excluding the missing data corresponds to deleting the unnecessary terms (including those generated by the daughters of missing cells) in the general form of Eq. 13. These cases are automatically handled, our code generating as many terms as is appropriate for each cell and only when the corresponding data exist.
Experimental methods
Cultures of λ-cascade strains (13) were grown overnight in LB + 15 μg/mL kanamycin at 37°C from single colonies and diluted 1:100 in MSC media (M9 minimal medium + 0.6% succinate + 0.01% casamino acids + 0.15 μg/ml biotin + 1.5 μM thiamine). Cultures were grown to OD600 ∼ 0.1 at 32°C and then induced by adding aTc to a concentration of 100 ng/mL for 3 min at ambient temperature, followed by two washes with MSC to remove aTc. Cells were allowed to grow and then diluted to give ≃1 cell per visual field when placed between a coverslip and 1.5% low-melt MSC agarose. Growth of microcolonies was observed at 32°C using a Leica DMIRB/E automated fluorescence microscope at 100× magnification with a mercury light source (Leica, Bannockburn, IL) and YFP filter cube Chroma #41028 (Chroma Technologies, Brattleboro, VT).
Custom software was used to control the microscope and related equipment (Ludl motorized stage, Ludl, Hawthorne, NY; and Hamamatsu Orca ER CCD camera, Hamamatsu, Shizuoka, Japan), via ImagePro Plus and ScopePro packages (Media Cybernetics, Silver Springs, MD). Fluorescence background values were estimated from regions of the fluorescent images containing no cells. One background value was chosen for each movie (the minimum of the measured background levels for the first 10–20 frames). Cellular autofluorescence was measured in cells containing no YFP reporters and was low, having values similar to the change in background fluorescence from image to image. An autofluorescence value was therefore selected within this range that led to the most constant YFP signal for the entire microcolony and to constant YFP levels in cell divisions (so that the sum of YFP fluorescence in the daughters would equal that in the mother cell). Flat-field corrections were found to be negligible using an analytic correction method developed previously (19).
With this normalization, the total colony YFP remains constant (to within 5%) during the first 4 h of growth. For later times, there is more variation (arising from crosstalk with the cyan fluorescent protein designed to be induced at low CI-YFP levels in the λ-cascade strains (13)). The measurement error model incorporated in Method II automatically attaches less weight to those cells whose fluorescence values are of order σ or lower, and the inferred ν is not significantly changed when data from the last generations of the lineage tree in Fig. 1 B is ignored. Cellular YFP levels are given in Fig. 1 C.
Software written in MatLab (The MathWorks, Natick, MA) identified and tracked cells from the phase contrast images and quantified their fluorescence levels using the segmented images to identify the appropriate pixels for each cell (13). Cellular fluorescence was measured by summing the fluorescence intensities of all pixels within a particular cell. Typical intervals between exposures were 9 min (for accurate tracking), but YFP fluorescence images were taken on alternate frames to reduce photobleaching. Images were usually acquired for ∼8 h, with colonies reaching eight or nine generations.
RESULTS
The performance of the algorithm is determined by two competing effects: first, as the number of molecules increases binomial deviations become less significant relative to mean values, and so the numbers of molecules in daughter cells become effectively indistinguishable; second, low numbers of molecules have small signal/noise ratios and measurement error can potentially swamp binomial deviations.
Fig. 2 A shows the two-dimensional posterior inferred by Method II from the data of Fig. 1. The most probable values of ν and σ are given by the posterior maximum. The distribution is orientated approximately parallel to the coordinate axes, implying that the two parameters can be inferred independently, and that the errors in such inference will be given by the width of the distribution at half-maximum along the appropriate axis. Summing over all values of σ gives the marginalized distribution for ν shown in Fig. 2 B. For this data set, ν* ≃15 ± 4 fluorescence units per fluorescent particle, implying that the predicted number of fluorescent proteins ranges from ≃840 in the initial cell to ≃10 for the generation-eight cells. Method I predicts ν* ≃15 ± 1. The inferred value of ν is consistent (within a factor of two) over four separate experiments, where a microcolony was grown from a different initial cell (Fig. 2 B). This level of accuracy is certainly high enough to provide parameters for cellular models, where only ballpark estimates are usually required (9), and could be improved by increasing the number of measurements per cell or the levels of intracellular fluorescent proteins (see below).
To illustrate the importance of the signal/noise ratio (fi/σ for a cell with fluorescence fi), we simulated data for an eight-generation tree. Starting from an initial number of proteins in the first cell, a microcolony was created by equally binomially partitioning protein into daughter cells. Each data point was multiplied by ν0, arbitrarily set to 25. Normally distributed samples with zero mean and standard deviation σ were added to each data point to include measurement errors. If, for example, three measurements per cell were desired, three different normal samples were added to the original data point to give three final data points.
Fig. 3 A shows that Method II performs robustly: its accuracy increases steadily as the number of data points grows. Method I, which ignores measurement error, performs as well as Method II only for those lineage trees whose cells all have a high signal/noise ratio. We simulated two types of data: one generated from an initial cell having 500 molecules (low signal/noise ratios) and the other generated from an initial cell containing 5000 molecules (high signal/noise). Although lineage trees with seven generations were initially created, the inference algorithms were run on data sets with just the first three generations and then new data added generation by generation to explore the inclusion of additional layers of the lineage tree. In all cases, the true value of ν, ν0, was set to 25. We use a relative measure, iε = |log2(v/v0)|, to score an inferred value of ν. An inference score iε = 0 is thus an exact inference, while iε = 1 implies that the inferred value is either twice or half the true value.
Although large numbers of proteins increase the signal/noise ratio, too many, as mentioned earlier, can degrade the inference. Large numbers lead to very tight binomial distributions and so to potentially immeasurable differences between the number of proteins in each daughter cell. For 5000 molecules (∼3 μM; see Fig. 3 B) and 50,000 molecules (not shown), accurate inference is not significantly affected, at least for simulated data: the inference score for the 50,000 case increases by ≃20% for colonies of six or seven generations. Such high numbers of molecules may be more frequent in eukaryotic cells.
Higher σ degrades inference, but this degradation is reduced by increasing the number of measurements per cell (Fig. 3 B). Six measurements gives accurate inference for σ as high as 200 (i.e., 8ν). For the same data, Method II performed as well as or better than Method I 78% of the time, increasing to 93% of the time when σ = 200.
DISCUSSION
The difference in fluorescence levels between two daughter cells after cell division is determined by the number of fluorescent proteins in the mother cell: the difference is on average larger if the number of proteins in the mother cell increases. We exploit this phenomenon to deduce in vivo numbers of molecules by following a cell that has transiently expressed fluorescent protein and recording the daughter cell fluorescence levels as the fluorescent protein is diluted out during growth. Our method assumes equal binomial partitioning of proteins at cell division, so that each protein has the same chance of going to either daughter cell.
We have introduced two algorithms to infer ν. Method I, exemplified by Eq. 8, is fast and easy to compute. It is reliable when the signal/noise is significantly greater than one. Method II, shown in Eq. 13, although more computationally demanding, is valid for both high and low signal/noise ratios. It returns both the posterior probability for ν and the measurement error σ. MatLab (The MathWorks) code for both methods is available on request.
Although we use a normal distribution model for measurement error, other distributions can be adopted providing they allow Eq. 13 to be factored into triad terms. For example, multiplicative measurement error with a corresponding log normal distribution only changes the middle term of Eq. 13 to
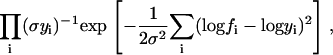 |
(17) |
with the variable elimination method working as before.
The fluorescent protein of interest must only be transiently expressed, and then its expression fully repressed once fluorescence measurements are begun. The copy number of the protein's gene is therefore not important, providing repression remains tight after the transient expression. Consequently, the fluorescent protein can be conveniently added on a plasmid. Given the value of ν of YFP, other fluorescent proteins can be quantified by comparing their expression to that of YFP in a bacterial strain that expresses both proteins from identical promoters (13).
Our analysis assumes even partitioning of fluorescent proteins into daughter cells, i.e., a protein is as equally likely to go to one daughter as to the other. This assumption is reasonable for a repressor protein: nonspecific DNA binding presumably causes most to be carried from mother to daughters by (evenly partitioning) chromosomes. For cytosolic proteins in cells that divide asymmetrically, for example by budding rather than by fission, the situation may be more complicated. Our algorithm could be adapted to include such asymmetries, where, for example, the probability of a protein going to a particular daughter cell could be proportional to the volume of the daughter cell. Alternatively, it may be possible to preprocess the data, restricting the analysis to daughter cells of equal size where division events are, presumably, even. In our movies, >85% of division events generated daughters with a 5% difference in cell volume or less.
A protein that exists in several different multimer forms can cause additional difficulties. If the distribution of the protein between multimers changes at each measurement and particularly at each cell division, not only must our algorithm be extended but many more measurements will be needed to gain reliable statistics. The fluorescent protein considered here, CI-YFP, dimerizes. Nevertheless, given that its dissociation constant is ∼10 nM (20,21), we assume that it only exists in dimer form, and so only need to halve the inferred value of ν to find the proportionality constant per YFP molecule.
By calibrating fluorescence measurements, our method allows parameters fit to network output to be expressed in absolute rather than relative units, and so enables information from different experiments to be easily combined into a larger, predictive framework. The technique can be applied in parallel to measurements of network function (13) and properties (14), and potentially to eukaryotic cells.
Acknowledgments
We thank Derek Bowie, Robert Sidney Cox III, Jon Young, and the anonymous referees for useful comments. M.B.E. acknowledges a CASI award from the Burroughs Wellcome Fund and the Searle Scholars Program. U.A. and M.B.E. are supported by the Human Frontiers Science Program. P.S.S. is supported by the National Sciences and Engineering Research Council (Canada) and by a Tier II Canada Research Chair.
APPENDIX
General expression for the posterior probability
For a complete lineage tree with N cells, Eq. 13 becomes
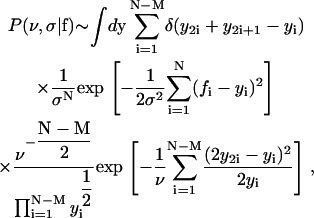 |
(18) |
with N – M = (N – 1)/2.
Derivation of an estimate for the measurement error
The posterior for σ satisfies
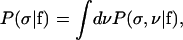 |
(19) |
where P(σ, ν|f) is given by Eq. 18. After integrating out the δ-functions in Eq. 18, the number of y variables in the integral drops to M because of the conservation of numbers constraints. We denote this set of M variables by ys = {y1, y2, ···, yM}.
To estimate the remaining integral in Eq. 18 (and so evaluate Eq. 19), the exponent of the middle term of Eq. 18 can be rearranged into a quadratic form in the ys, i.e., as 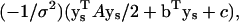 where A is a symmetric M × M matrix and b is a M × 1 vector. This quadratic form can be diagonalized through the transformation
where A is a symmetric M × M matrix and b is a M × 1 vector. This quadratic form can be diagonalized through the transformation
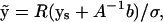 |
(20) |
with R the matrix of eigenvectors of A. The exponent then becomes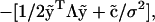 where Λ is the diagonal matrix of eigenvalues λi of A and
where Λ is the diagonal matrix of eigenvalues λi of A and 
Once diagonalized, the middle term of Eq. 18 can be written as a product of normal distributions, one for each eigenvalue λi, which we can integrate. Defining 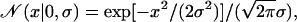 the middle term satisfies
the middle term satisfies
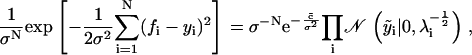 |
(21) |
remembering Eq. 20.
Integrating out the δ-functions in Eq. 18 leads to its last term becoming a function of ys rather than y,
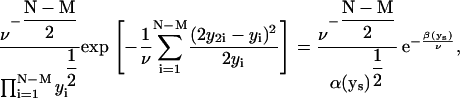 |
(22) |
where α(ys) and β(ys) are algebraic functions of ys.
The posterior distribution for σ, from Eq. 19, is thus
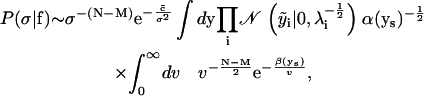 |
(23) |
from Eqs. 21 and 22. Evaluating the integral over ν gives
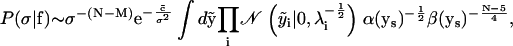 |
(24) |
where
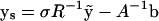 |
(25) |
from Eq. 20.
Most of the contribution to the integral in Eq. 24 will come from the maxima of the normal distributions at 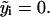 At these values, Eq. 25 shows that ys will have little σ-dependence and consequently that the entire integral can be approximated as being independent of σ. The term before the integral dominates, and maximizing this term with respect to σ gives the estimate in Eq. 15. For 5000 simulated data sets with randomly assigned measurement errors of σ0, the mean of log2(σ*/σ0) was ≃ −0.009, a negligible difference for accurately inferring ν.
At these values, Eq. 25 shows that ys will have little σ-dependence and consequently that the entire integral can be approximated as being independent of σ. The term before the integral dominates, and maximizing this term with respect to σ gives the estimate in Eq. 15. For 5000 simulated data sets with randomly assigned measurement errors of σ0, the mean of log2(σ*/σ0) was ≃ −0.009, a negligible difference for accurately inferring ν.
References
- 1.Ideker, T., T. Galitski, and L. Hood. 2001. A new approach to decoding life: systems biology. Annu. Rev. Genomics Hum. Genet. 2:343–372. [DOI] [PubMed] [Google Scholar]
- 2.Gilman, A., and A. P. Arkin. 2002. Genetic “code”: representations and dynamical models of genetic components and networks. Annu. Rev. Genomics Hum. Genet. 3:341–369. [DOI] [PubMed] [Google Scholar]
- 3.Elson, E. L., and D. Magde. 1974. Fluorescence correlation spectroscopy. I. Conceptual basis and theory. Biopolymers. 13:1–27. [DOI] [PubMed] [Google Scholar]
- 4.Magde, D., E. L. Elson, and W. W. Webb. 1974. Fluorescence correlation spectroscopy. II. An experimental realization. Biopolymers. 13:29–61. [DOI] [PubMed] [Google Scholar]
- 5.Wiseman, P. W., J. A. Squier, M. H. Ellisman, and K. R. Wilson. 2000. Two-photon image correlation spectroscopy and image cross-correlation spectroscopy. J. Microsc. 200:14–25. [DOI] [PubMed] [Google Scholar]
- 6.Chen, Y., J. D. Muller, P. T. So, and E. Gratton. 1999. The photon counting histogram in fluorescence fluctuation spectroscopy. Biophys. J. 77:553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kask, P., K. Palo, D. Ullmann, and K. Gall. 1999. Fluorescence-intensity distribution analysis and its application in biomolecular detection technology. Proc. Natl. Acad. Sci. USA. 96:13756–13761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ronen, M., R. Rosenberg, B. I. Shraiman, and U. Alon. 2002. Assigning numbers to the arrows: parameterizing a gene regulation network by using accurate expression kinetics. Proc. Natl. Acad. Sci. USA. 99:10555–10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown, K. S., and J. P. Sethna. 2003. Statistical mechanical approaches to models with many poorly known parameters. Phys. Rev. E. 68:021904. [DOI] [PubMed] [Google Scholar]
- 10.Jaeger, J., S. Surkova, M. Blagov, H. Janssens, D. Kosman, K. N. Kozlov, Manu, E. Myasnikova, C. E. Vanario-Alonso, M. Samsonova, D. H. Sharp, and J. Reinitz. 2004. Dynamic control of positional information in the early Drosophila embryo. Nature. 430: 368–371. [DOI] [PubMed] [Google Scholar]
- 11.Kaern, M., T. C. Elston, W. J. Blake, and J. J. Collins. 2005. Stochasticity in gene expression: from theories to phenotypes. Nat. Rev. Genet. 6:451–464. [DOI] [PubMed] [Google Scholar]
- 12.Sprinzak, D., and M. B. Elowitz. 2005. Reconstruction of genetic circuits. Nature. 438:443–448. [DOI] [PubMed] [Google Scholar]
- 13.Rosenfeld, N., J. W. Young, U. Alon, P. S. Swain, and M. B. Elowitz. 2005. Gene regulation at the single-cell level. Science. 307:1962–1965. [DOI] [PubMed] [Google Scholar]
- 14.Golding, I., J. Paulsson, S. M. Zawilski, and E. C. Cox. 2005. Real-time kinetics of gene activity in individual bacteria. Cell. 123:1025–1036. [DOI] [PubMed] [Google Scholar]
- 15.Traynelis, S. F., and F. Jaramillo. 1998. Getting the most out of noise in the central nervous system. Trends Neurosci. 21:137–145. [DOI] [PubMed] [Google Scholar]
- 16.Neher, E., and C. F. Stevens. 1977. Conductance fluctuations and ionic pores in membranes. Annu. Rev. Biophys. Bioeng. 6:345–381. [DOI] [PubMed] [Google Scholar]
- 17.Mathews, J., and R. L. Walker. 1970. Mathematical Methods of Physics. Addison-Wesley, New York, New York.
- 18.Zhang, N. L., and D. Poole. 1996. Exploiting causal independence in Bayesian network inference. J. Artific. Intell. 5:301–328. [Google Scholar]
- 19.Elowitz, M. B., A. J. Levine, E. D. Siggia, and P. S. Swain. 2002. Stochastic gene expression in a single cell. Science. 297:1183–1186. [DOI] [PubMed] [Google Scholar]
- 20.Johnson, A. D., C. O. Pabo, and R. T. Sauer. 1980. Bacteriophage λ-repressor and cro protein: interactions with operator DNA. Methods Enzymol. 65:839–856. [DOI] [PubMed] [Google Scholar]
- 21.Koblan, K. S., and G. K. Ackers. 1991. Energetics of subunit dimerization in bacteriophage λ-cI repressor: linkage to protons, temperature, and KCl. Biochemistry. 30:7817–7821. [DOI] [PubMed] [Google Scholar]