

Abstract
Nonperturbative monitoring of intracellular organelle transport in unstained living cells was achieved with coherent anti-Stokes Raman scattering (CARS) microscopy. To avoid possible interference with the organelle transport introduced by laser radiation, we first examined different illumination conditions. Using a new photodamage criterion based on morphological changes of the cells, we determined the threshold values of both pulse energy and average power at relevant wavelengths. Under excitation conditions much milder than the threshold levels, we were able to monitor the motions of lipid droplet (LD) organelles in steroidogenic mouse adrenal cortical (Y-1) cells with CARS microscopy in real time without perturbations to the cells. Particle tracking analyses revealed subdiffusion as well as active transport of LDs along microtubules. Interestingly, LD active transport is only present in Y-1 cells that rounded up in culture, a morphological change associated with steroidogenesis, suggesting possible involvements of LD active transport in the latter. Simultaneous imaging of LDs and mitochondria with CARS and two-photon fluorescence microscopy clearly showed that interactions between the two organelles could be facilitated by high LD motility. These observations demonstrate CARS microscopy as a powerful noninvasive imaging tool for studying dynamic processes in living cells.
INTRODUCTION
Intracellular trafficking of organelles driven by molecular motors along cytoskeletal fibers is a strongly regulated process, which is essential for cellular functioning (1). Different organelles may exhibit different trafficking properties, and small perturbations to the cells can cause profound changes in organelle transport. To monitor organelle transport events in the cell's native state, therefore, the imaging method has to meet at least two criteria: chemical selectivity and noninvasiveness. Fluorescence microscopy exhibits superior selectivity, but the toxicity and photobleaching of fluorophores are often of concern. Besides, some organelles cannot be easily labeled with fluorescence tags suitable for live cell imaging. Differential interference contrast (DIC) microscopy avoids using fluorophores at the cost of chemical specificity, which severely restricts the selectivity in probing the intracellular compounds.
For certain organelles, the limitations of available imaging techniques have precluded a reliable, nonperturbative measurement of their transport properties. Among others, the lipid droplet (LD) organelle, a cellular structure of aggregated neutral lipids, is a typical example. LDs have attracted a lot of research activities since recent studies recognized their important roles in lipid metabolism and membrane trafficking (2–4), and have been named as adiposome (2) or retinosome (5). Unlike other cellular organelles, however, the intracellular trafficking of LDs has been rarely studied, mostly due to the lack of a reliable probe in living cells (6). The few existing studies on this subject with fluorescence microscopy (7,8) yielded very different results from each other, leaving questions on the nature of LDs trafficking largely unanswered.
In contrast, coherent anti-Stokes Raman scattering (CARS) microscopy (9–11) is an ideal tool to image LDs in unstained living cells (12). A key advantage of CARS microscopy is that cellular constituents such as lipids (12–18), water (14,19,20), and protein (21) can be selectively visualized without using fluorescent labels. The chemical selectivity in CARS stems from the vibrational signatures of chemical bonds in the molecules of interest. Access to specific chemical species is easily realized by tuning the frequency difference of the two pulsed excitation beams (pump and Stokes) to match the frequency of the desired vibrational mode. CARS microscopy is a sensitive technique, and is especially suitable for imaging lipids because of the large number of C-H bonds in these molecules. In fact, even a single lipid bilayer can be visualized with CARS microscopy (22). LDs, which are densely packed lipids, generate very strong CARS signals when the Raman shift is tuned to the CH2 stretching vibration at 2845 cm−1 (12). With CARS microscopy, it is therefore possible to study the trafficking of LDs in unstained live cells, which may help in understanding its physiological relevance and regulation.
Besides chemical selectivity, noninvasiveness is another requirement in the study of organelle transport. CARS microscopy uses intense, pulsed laser sources for sample illumination. For nonperturbative imaging, laser induced effects including linear and nonlinear photodamages, have to be minimized. The integrated CARS signal intensity per pixel scales as:
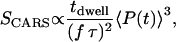 |
(1) |
where 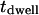 is the pixel dwell time,
is the pixel dwell time, 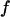 is the repetition rate of the pulse trains,
is the repetition rate of the pulse trains, 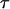 is the temporal width of the pump and Stokes pulses, and
is the temporal width of the pump and Stokes pulses, and 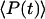 is the total time-integrated average power of the incident radiation (pump and Stokes beams of similar intensity are assumed). From Eq. 1 it follows that the same CARS signal can be obtained at higher pulse energy at a reduced f and with a lower average power 〈P(t)〉, avoiding linear damage to the sample. This, however, could introduce nonlinear photodamage once the pulse energy is above a critical value (23). To maintain a strong signal while minimizing both linear and nonlinear photodamages, proper ranges of average power and pulse energy have to be determined.
is the total time-integrated average power of the incident radiation (pump and Stokes beams of similar intensity are assumed). From Eq. 1 it follows that the same CARS signal can be obtained at higher pulse energy at a reduced f and with a lower average power 〈P(t)〉, avoiding linear damage to the sample. This, however, could introduce nonlinear photodamage once the pulse energy is above a critical value (23). To maintain a strong signal while minimizing both linear and nonlinear photodamages, proper ranges of average power and pulse energy have to be determined.
Here we first investigate the optimum excitation conditions for noninvasive imaging of live cells with CARS microscopy. A new criterion for evaluating photodamage based on laser-induced morphological changes of the cell is used to determine the thresholds of pulse energy and average power of the excitation lasers. We will show that by operating well below such thresholds, cells can be imaged continuously over a long period of time without being perturbed. Under these conditions, we studied the trafficking of LDs in mouse adrenal cortical (Y-1) cells in detail.
MATERIALS AND METHODS
Laser scanning CARS microscopy
The laser scanning CARS microscope was modified from a previously reported setup (11). To deliver a wide range of pulse energies and average powers for the pump and the Stokes beams, we implemented three different excitation schemes, all of which are based on two synchronized picosecond Ti:sapphire lasers (Tsumani, Spectra-Physics, Mountain View, CA).
In Scheme A (Fig. 1 A), a single Pockels cell (Model No. 350-160, Conoptics, Danbury, CT) was used to control the repetition rates of the temporally overlapped and collinearly propagating pump and Stokes pulse trains. This scheme retains the full pulse energy provided by the two lasers while keeping low average power levels by reducing the repetition rate. A typical extinction ratio of ∼100:1 for the two beams combined could be achieved with the Pockels cell. A final repetition rate of 4–8 MHz was typically used.
FIGURE 1.

Excitation schemes of the laser scanning CARS microscope used in this study. (A) Pulse trains from two Ti:sapphire lasers, synchronized, combined, and pulse-picked with a Pockels cell before being introduced into the microscope. (B) Passive amplifier with an external cavity used to increase the pulse energy of the pump laser. The amplified pump pulses were synchronized and combined with the pulse-picked Stokes beam before entering the microscope.
Scheme B (Fig. 1 B) was used for achieving even higher pulse energies in the pump beam. We obtained high energy pulses by employing a passive amplifier with an external cavity (24). The passive amplifier relies on coherent addition of the mode-locked pump pulses after trapping them in a cavity with a free spectral range (FSR) similar to the Ti:Saphire pump laser. Amplified pulses (up to 150 nJ) were dumped out of the cavity by a Bragg cell at tunable repetition rates. The repetition rate of the Stokes beam was adjusted by a Pockel's cell. Pulses from the Bragg cell and the Pockel's cell were synchronized by using the same photodiode signal from the pump laser cavity to trigger the corresponding drivers.
Scheme C (not shown) was for high average power excitation experiments, which is the same as previously reported (11), namely, synchronized 80-MHz pulse trains from the two lasers were collinearly combined and sent directly into the microscope.
Due to beam attenuation in the setup, ∼15% of the pump (711 nm) and 10% of the Stokes (892 nm) powers reached the objective (Olympus, Tokyo, Japan) UPlanApo/IR, 60×, water immersion, NA 1.2). Typically, the pump pulse energy at the sample is up to 0.7 nJ in scheme A and C, and 0.5–10 nJ in scheme B. The pulse energy of the Stokes beam is up to 0.4 nJ in all schemes.
All images were taken on an Olympus FV300 laser scanning microscope controlled by FluoView (Ver 3.0, Olympus). CARS signals from lipids were recorded in the forward direction through a stack of band pass filters (600 ± 20 nm).
Simultaneous CARS and two-photon fluorescence microscopy
Simultaneous CARS and two-photon fluorescence (TPF) imaging was carried out with the scheme A described above (Fig. 1 A), with the repetition rates of both lasers typically set at 8 MHz. To label mitochondria for TPF imaging, cells were incubated with 100 nM MitoTracker Red (CMXRos, Molecular Probes, Eugene, OR) for 30 min and washed with fresh medium. We found that the CMXRos dye is better excited by the Stokes (892 nm) than by the pump (711 nm) laser for TPF. To reduce photobleaching and increase the TPF signal, the average powers of 0.5 and 2 mW for the pump beam and Stokes beams, respectively, were used. We collected the TPF signals in the epi-fluorescence channel.
Cell culture and treatment
Mouse adrenal cortical tumor (Y-1) cells were obtained from ATCC and cultured in F-12K medium (ATCC, Manassas, VA), supplemented with 12.5% horse serum and 2.5% FBS. Mouse fibroblast cells (3T3-L1, ATCC) were cultured in DMEM with 10% BCS. All cell cultures were maintained at 37°C with humidified 5% CO2.
Nocodazole, cytochalasin-D, sodium vanadate, 5′-adenylyl imidodiphosphate (AMP-PNP), and bovine adrenalcoritcaltropical hormone (ACTH) were purchased from Sigma (St. Louis, MO) and administered to cell cultures at final concentrations as indicated in the results section.
All the cells used for imaging were seeded on chambered slides (Labtek, Rochester, NY) with glass coverslip bottoms. Where indicated, the chambers were incubated with 0.1% poly-lysine (Sigma) for ∼15 min to obtain a glass substrate covered with poly-lysine. During imaging, the chambers were maintained at 37°C with a stage temperature controller (Harvard Apparatus, Holliston, MA).
Particle tracking
Particle tracking was performed with home-written software in MATLAB (The MathWorks, Natick, MA). Images were first segmented by watershed transformations to recognize individual particles. Centroid positions of each particle were then calculated either by intensity-weighed averaging of pixel coordinates or by fitting to a two-dimensional Gaussian function. Tracking of the particles was undertaken by iterative comparison of their physical proximity as well as similarities in diameter and signal intensity in successive frames.
Photodamage determination
Photodamage of cells by ultrafast lasers was previously characterized with cell lyses (25), by the change of calcium indicator signal (23), or by the stop of cell division after exposure (26). These criteria are based on permanent cellular damages induced by the laser. At lower illumination doses, nonvital laser perturbations to the cells could result in cellular responses such as cell contraction or other morphological changes. In our experiments, we use these changes as a milder indicator of cell damage. Briefly, a stack of CARS images (3 s/frame) were taken at 2845 cm−1 over a 50 × 50 μm2 area containing one or two cells. The stack of images, all featuring LDs as bright particles (12), were then overlaid. Laser-induced perturbations to the cells were considered significant if in the overlaid image: a), cell boundaries became blurred, indicating morphological changes during the scan; or b), trajectories of LDs, which were clearly revealed in the overlaid image, were highly parallel to each other. The parallel movements of LDs were not likely due to collinear active transport of individual LDs, but rather due to the cell contraction during which LDs were collectively dragged along.
RESULTS
Optimum laser imaging conditions
We started to evaluate the laser effects with either high pulse energies or high average powers applied to the sample using the criterion described in the materials and methods section, which is based on morphological changes of the cells.
To determine the nonlinear photodamage threshold for the pump beam (711 nm), we used high pulse energies (>1 nJ per pulse) in this beam delivered by scheme B (Fig. 1 B), in which a passive pulse amplifier was utilized. As shown in Fig. 2 A, the cells were unaffected during a sustained scanning of 300 s (100 frames) when using 2 nJ pump pulses. The LDs and cell boundaries retained their original positions throughout the scanning. In contrast, illumination with a 3 nJ pump beam induced rapid retraction of the cell boundaries during which the LDs were dragged along in parallel paths (Fig. 2 B). The repetition rates in both cases were 1 MHz. In another experiment, 1.5 or 2.3 nJ pulses in the pump beam were used, both at a 2 MHz repetition rate. The cells appeared to be unaffected by 1.5 nJ pulses (Fig. 2 C), but the 2.3 nJ pulses induced significant responses from the cells (Fig. 2 D). The average powers of the pump beam in the above experiments were kept below 5 mW, which is much lower than the linear damage threshold determined later. The Stokes pulse energies remained constant at 0.5 nJ. These observations indicate that for the Y-1 cells a nonlinear damage threshold exists for pulse energy of the 711 nm beam, which sets in around 2 nJ.
FIGURE 2.

Laser effects on cultured Y-1 cells. (A–D) Nonlinear effects examined with pump pulse energy and repetition rate, and those of Stokes beam set at (A) 2 nJ, 1 MHz and 0.5 nJ, 1 MHz; (B) 3 nJ, 1 MHz and 0.5 nJ, 1 MHz; (C) 1.5 nJ, 2 MHz and 0.25 nJ, 2 MHz; and (D) 2.3 nJ, 2 MHz and 0.25 nJ, 2 MHz, respectively. (E–F) Linear effects observed with pump and Stokes average powers set at (E) 9 mW and 3 mW, and (F) 12 mW and 3 mW, with repetition rates all at 80 MHz. A sequence of 100 CARS images (2845 cm−1) of one or two Y-1 cells were taken at 3 s/frame (total recording time 5 min), with the above specified pulse energy and repetition rate. The images were then overlaid to visualize cellular responses. Scale bar, 10 μm.
Similarly, by keeping the pulse energies low but using a high average power, we measured the linear damage threshold of the pump beam. Scheme C was used in these experiments. Average powers below 9 mW were well tolerated by the cells during a sustained scan of 5 min (Fig. 2 E), whereas a 12-mW pump beam induced drastic morphological changes in the cells (Fig. 2 F). The Stokes average power was kept constant at 3 mW (∼0.04 nJ pulse energy). This result suggests that linear photodamage effects due to the pump laser radiation start to become important in the 9–12 mW range.
In the following experiments, both pulse energy and average power of the pump beam were kept well below the above determined thresholds. Typically, for imaging LDs at 2845 cm−1, we only needed to use 1.5 mW (8 MHz, 0.2 nJ) for the pump and 0.5 mW (8 MHz, 0.06 nJ) for the Stokes beams (scheme A, Fig. 1 A). Under these conditions, we have not observed responses from the cells during scanning.
Active transport of LDs
We next applied CARS microscopy to monitor LDs trafficking in live, unstained Y-1 cells. Originated from the mouse adrenal gland, Y-1 cells are specialized in synthesizing steroid hormones using the cholesterol stored in cytoplasmic LDs (27). Consistent with their physiological roles, Y-1 cells possess a large number of cytoplasmic LDs, as clearly visible in the CARS images taken at 2845 cm−1 (Fig. 2 A). Tracking of the LDs revealed both diffusive and rapid active transport along defined paths, and LDs often jumped from one type of motion to the other. For example, shown in Fig. 3, A and D, are trajectories of a single LD (in inset) in different periods during a 120-s scan. In the first 70 s the LD diffused in a confined radius of ∼1 μm (Fig. 3 A and shaded area in Fig. 3 B). In the next 40 s, it moved ∼8 μm along an extended track, indicative of association with molecular motors (Fig. 3 D and shaded region in Fig. 3 E). Analyses of the trajectories in Fig. 3, A and D, resulted in mean-square displacement versus time relations given by 〈Δx2〉 = 2Dt0.6 (Fig. 3 C) and 〈Δx2〉 = 2Dt1.9 (Fig. 3 F), characteristic of subdiffusion and active transport, respectively.
FIGURE 3.

Diffusive motions and active transport of LDs in living Y-1 cells. The same LD (in inset) was seen to undergo different motions at different periods within the 120 s scanning time. A–C and D–F are the x, y position trajectories (overlaid with CARS images showing the original positions for each trajectory), speed-time traces, and mean-square displacement (〈Δx2〉) time plots during the first 70 and the last 40 s, respectively. In the 〈Δx2〉 ∼ t plot, “+” represents the actual data points and the solid line is the fitted curve. Scale bar, 2 μm.
We found that the active transport of LDs is microtubule dependent. Disruption of microfilaments with 30 μM cytochalasin-D had only a slight effect on the active transport whereas disruption of microtubules with 50 μM nocodazole completely diminished it (Fig. 4 A). This suggests that kinesin and dynein motors, which move toward the plus (cell periphery) and minus (near cell nucleus) ends of the microtubules, respectively, mediate the active transport of LDs. Accordingly, incubation of cells with AMP-PNP (28) or vanadate (29), preferential inhibitors for kinesin and dynein, respectively, altered the relative frequency between plus and minus-end movements significantly (Fig. 4 B).
FIGURE 4.

Dependences of LD active transport on microtubules and the microtubule motor proteins, kinesin and dynein, in Y-1 cells. (A) Cytochalasin-D, an F-actin disruption drug, had little effect on LD active transport whereas nocodazole, a microtubule depolymerization reagent, almost completely abolished it. (B) AMP-PNP and sodium vanadate, which preferentially inhibits kinesin and dynein, respectively, increased the relative frequencies of microtubule minus- and plus-end directed movements, respectively.
LD active transport and cell morphology
Interestingly, we observed a strong dependence of LD active transport frequency on cell rounding, a characteristic phenomenon during steroid synthesis (steroidogenesis) in Y-1 cells (30,31). When cultured on a glass substrate, such as the chambered slides used in our experiments (see Materials and Methods), Y-1 cells often rounded up spontaneously. LDs in these rounded cells underwent frequent active transport. Besides the rounded ones, there was a small population of cells that appeared flat with extended edges (Fig. 5 A). In those cells LDs exhibited little motility. For example, in Fig. 3 B, none of the LDs moved more than 0.5 μm in a period as long as 180 s. On average the LDs moved <3 nm/s, and were almost completely stationary. Further more, when Y-1 cells grown on a glass substrate were treated with 10 nM ACTH, a reagent known to stimulate steroidogenesis (31), all cells rounded up within 2 h, accompanied with a twofold increase in LD active transport events (Fig. 5 C). On the contrary, if Y-1 cells were seeded on a glass substrate treated with poly-lysine, a treatment known to inhibit steroidogenesis and desensitize Y-1 cells to ACTH (30), Y-1 cells appeared mostly flat and LD active transport was virtually absent (Fig. 5 C). These observations strongly suggest that steroidogenesis, cell morphology, and LD active transport are correlated events in Y-1 cells.
FIGURE 5.

Absence of LD active transport in flat Y-1 cells. (A) Flat Y-1 cells with extending edges in culture, with a similar density of LDs in the cytoplasm as seen in previous figures. (B) The x, y position trajectories of LDs in the inset of A, during a total scanning time of 180 s. (C) Total number of observed LD active transport events in Y-1 cells under different conditions, including grown on glass and before ACTH treatment (middle bar), grown on glass and treated with 10 nM for 2 h (left bar), and grown p-lysine treated glass without ACTH treatment (right bar). Scale bars, 10 and 2 μm in A and B, respectively.
Interaction of LDs with mitochondria
To further understand the role of LD active transport in steroidogenesis, we combined CARS with two-photon fluorescence (TPF) microscopy (13,17,18) to simultaneously image LDs and mitochondria. It has been known that mitochondria inner membranes are locations where steroid synthesis takes place, with cholesterol stored in LDs as the starting material (31). To image mitochondria with TPF, we labeled the cells with MitoTracker Red, a dye efficiently excitable by the 892 nm (2-ps pulse-width) Stokes beam, which is simultaneously used for CARS imaging of LDs through two-photon excitation.
As shown in Fig. 6, A–C, TPF images revealed a blurry network of mitochondria in a rounded Y-1 cell, indicating a high density of the organelle. This distribution of mitochondria is very different from that of other nonsteroidogenic cells, such as the mouse fibroblast 3T3-L1 (Fig. 6 D). Obviously, the high densities of both LDs and mitochondria are consistent with the physiological roles of Y-1 cells. From the TPF images, we did not see significant differences of mitochondria morphology or density between flat and round Y-1 cells (data not shown). It is likely that it is the enhanced movements of LDs, rather than the intrinsic high density of both LDs and mitochondria, that determines the interactions between LDs and mitochondria in the rounded Y-1 cells. In a typical round Y-1 cell shown in Fig. 6 C, LDs undergoing active transport moved on average ∼2 μm within 24 s (the dye photobleaches afterward) of which the paths overlapped (“collided”) with mitochondria. Compared with those in flat Y-1 cells which moved at <3 nm/s (Fig. 5), LDs in rounded Y-1 cells will have more chances to interact (“collide”) with mitochondria.
FIGURE 6.

Simultaneous CARS and TPF imaging of living cells in culture. (A) TPF image of a (round) Y-1 cell labeled with MitoTracker, revealing a dense network of mitochondria. (B) Overlaid CARS and TPF images of LDs (red) and mitochondria (green). (C) Trajectories of LDs over a period of 24 s (20 frames) shown in white lines, overlaid on top of the TPF image of mitochondria, showing the “collision” between LDs and mitochondria due to the movement of LDs. (D) Overlapped CARS and TPF images of LDs and mitochondria, with the same color schemes as those of Y-1 cells shown in (A–C). Scale bars are both 2 μm. Note that although the images are of similar size, the densities and distributions of mitochondria in Y-1 and 3T3-L1 cells are very different.
DISCUSSION
Nonperturbative imaging conditions for CARS microscopy
In this study we investigated the trafficking properties of LD organelles in live, steroidogenic Y-1 cells with CARS microscopy. Although CARS avoids using exogenous fluorophores and associated issues of photobleaching and toxicity, reliable measurements of organelle transport are realized only after laser effects on the cells are also diminished. By keeping illumination doses well below the photodamaging thresholds, we were able to observe differential LDs motilities correlated with cell rounding and steroidogenesis in an unperturbed manner.
To evaluate laser-induced effects on the cells, we introduced a simple criterion based on changes of cellular morphology. Morphological changes include a cytoskeleton or extracellular matrix (ECM) breakdown seen as a collapse of the cell body or cell movements. Likely, sustained exposure to the lasers could depolymerize biopolymers in the cytoskeleton or the ECM, either by linear heating or nonlinear ionization. All these cellular responses will cause changes in LD positions, as shown in Fig. 2, and complicate analysis of LD motions. We have also observed that under high illumination doses, LDs active transport in Y-1 cells could sometimes be completely stalled by the lasers (data not shown). Obviously, nonperturbative imaging conditions are critical to study the transport of LDs.
It should be noted that when determining the photodamage thresholds, we predominately considered the effect of the 711 nm pump beam. This is because the throughput of the Stokes beam is low in our setup, and in consequence both the pulse energy (≤0.2 nJ) and the average power (≤3 mW) were much lower than those of the pump beam (∼2 nJ and >9 mW, respectively) used in the threshold measurement experiments. Also, the Stokes beam is at a near-IR wavelength which is longer than that of the pump beam and should be better tolerated by biological samples. Overall, the linear and nonlinear effects from the Stokes beam should be negligible compared with those of the pump beam. By shifting both the pump and the Stokes beams to even longer wavelengths, the linear and nonlinear laser effects can be reduced at the expense of a poorer spatial resolution.
Studies by Hopt and Neher on cellular photo damaging of mammalian cells induced by ultrafast near-infrared laser sources under typical in vitro imaging conditions, revealed that the number of scans before photo damaging occurs is empirically given by (23)
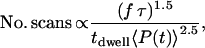 |
(2) |
Equation 2 indicates that photo damaging is a nonlinear function of the input beams, underlining that higher order light-matter interactions contribute to the damaging process. Hopt and Neher observed limited photodamage to Chinese hamster ovary cells when illuminated with a 840-nm, 190-fs pulse train with average powers up to 10 mW at a 82 MHz repetition rate and a pixel dwell time of 10 μs. Comparing these numbers with our excitation conditions (2-ps pulses, 3.3-μs dwell time) using Eq. 2, we find a tolerable average illumination power of 63 mW at 80 MHz and 4 mW at 1 MHz, corresponding to pulse energies of 0.8 nJ and 4 nJ, respectively. The fact that we observed cellular responses to laser radiation at the somewhat lower pulse energy of ∼2 nJ at 1 MHz can be explained by the shorter wavelength of the pump beam and our different criteria for determining a cellular response. Despite this difference, the photodamaging pulse energies comply very well with the range predicted by Eq. 2.
Besides adjusting pulse energy, we also found it necessary to control the average power to avoid cellular responses. In our experiments, laser-induced linear photodamage effects occurred when an average power of the pump beam higher than 9 mW was used, which was independent of the pulse energy. This threshold is close to the empirical value of 10 mW previously observed in TPF experiments (23,32,33).
Active transport of LDs
With the elimination of both laser effects and exogenous fluorophores, we achieved selective and nonperturbative imaging of LD trafficking with CARS microscopy. The advantages of CARS microscopy in this study can be better appreciated when compared with previous approaches for imaging LDs in live cells, i.e., ADRP-EGFP or Nile red. Nile red is a universal dye for lipids. Care has to be taken when assigning fluorescent features labeled by Nile red to LDs. In particular, on the same Vero (African green monkey kidney) cells, the authors using Nile red observed frequent active transport of LDs (7), whereas the others using ADRP-EGFP, an LD-targeting protein fused with EGFP, reported the opposite (8). On the other hand, although ADRP-EGFP is an LD-specific marker, increased expression of ADRP by ADRP-EGFP transfection will very likely perturb lipid metabolism of the cell.
Through a careful study with CARS microscopy, we observed a correlation between LD active transport and cell rounding, a phenomenon characteristic of steroidogenesis. Specifically, in cells that rounded up, either spontaneously or in response to hormonal (ACTH) stimulus, LDs made frequently active transport along microtubules. In contrast, in cells that exhibited extended and flat morphology, which is especially profound when cells were cultured on poly-lysine treated glass surfaces, LDs rarely moved. We speculate that Y-1 cells could regulate steroidogenesis in part by modulating the motility of LDs. Our results suggest that, like other cellular processes, the trafficking of LDs in Y-1 cells is tightly regulated. External perturbations, including hormones, culture conditions, and even laser radiations, could change the trafficking of LDs dramatically.
Further evidence for the involvement of LD active transport in steroidogenesis came from simultaneous imaging of LDs and mitochondria with combined CARS and TPF microscopy. Mitochondria inner membranes contain the enzymes needed for the initial steps of steroidogenesis that use cholesterol as the starting substrate. In Y-1 and several other adrenal gland cells, LDs are the main source of cholesterol. In accordance with their important roles, electron microscopy studies have shown that during steroidogenesis, LDs and mitochondria are often in physical contact (27). Up to date, however, there is limited understanding on how LDs and mitochondria were brought together due to the lack of an appropriate simultaneous imaging method. With combined CARS and TPF microscopy, we clearly observed that although both round and flat Y-1 cells have high densities of LDs and mitochondria, LDs that underwent active transport will have much more chances to interact (“collide”) with mitochondria.
In summary, we have demonstrated that excitation conditions with the pulse energy and average power of the pump laser around 710 nm kept under 2 nJ and 9 mW, respectively, should be used for noninvasive imaging of live cells with CARS microscopy. Under these conditions, organelle transport events can be noninvasively monitored with CARS microscopy in real time, allowing for a careful investigation of the delicately regulated LD active transport process in live steroidogenic cells. CARS microscopy offers a powerful tool for studying rapid dynamic processes in live and unstained biological systems.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
We thank Dr. Ji-Xin Cheng for the help at the early stage of this work and Paul Blainey for helpful discussions on particle tracking.
This work was supported by National Institutes of Health grant No. GM62536-01.
Eric Potma's present address is University of California, Irvine, Dept. of Chemistry, Natural Sciences II, Irvine, CA 92697.
References
- 1.Vale, R. D. 2003. The molecular motor toolbox for intracellular transport. Cell. 112:467–480. [DOI] [PubMed] [Google Scholar]
- 2.Liu, P., Y. Ying, Y. Zhao, D. I. Mundy, M. Zhu, and R. G. Anderson. 2004. Chinese hamster ovary K2 cell lipid droplets appear to be metabolic organelles involved in membrane traffic. J. Biol. Chem. 279:3787–3792. [DOI] [PubMed] [Google Scholar]
- 3.Murphy, D. J. 2001. The biogenesis and functions of lipid bodies in animals, plants and microorganisms. Prog. Lipid Res. 40:325–438. [DOI] [PubMed] [Google Scholar]
- 4.Fujimoto, Y., H. Itabe, J. Sakai, M. Makita, J. Noda, M. Mori, Y. Higashi, S. Kojima, and T. Takano. 2004. Identification of major proteins in the lipid droplet-enriched fraction isolated from the human hepatocyte cell line HuH7. Biochim. Biophys. Acta. 1644:47–59. [DOI] [PubMed] [Google Scholar]
- 5.Imanishi, Y., V. Gerke, and K. Palczewski. 2004. Retinosomes: new insights into intracellular managing of hydrophobic substances in lipid bodies. J. Cell Biol. 166:447–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fukumoto, S., and T. Fujimoto. 2002. Deformation of lipid droplets in fixed samples. Histochem. Cell Biol. 118:423–428. [DOI] [PubMed] [Google Scholar]
- 7.Pol, A., S. Martin, M. A. Fernandez, C. Ferguson, A. Carozzi, R. Luetterforst, C. Enrich, and R. G. Parton. 2004. Dynamic and regulated association of caveolin with lipid bodies: modulation of lipid body motility and function by a dominant negative mutant. Mol. Biol. Cell. 15:99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Targett-Adams, P., D. Chambers, S. Gledhill, R. G. Hope, J. F. Coy, A. Girod, and J. McLauchlan. 2003. Live cell analysis and targeting of the lipid droplet-binding adipocyte differentiation-related protein. J. Biol. Chem. 278:15998–16007. [DOI] [PubMed] [Google Scholar]
- 9.Cheng, J. X., and X. S. Xie. 2004. Coherent anti-Stokes Raman scattering nicroscopy: instrumentation, theory, and applications. J. Phys. Chem. B. 108:827–840. [Google Scholar]
- 10.Zumbusch, A., G. R. Holtom, and S. X. Xie. 1999. Three-dimensional vibrational imaging by coherent anti-Stokes Raman scattering. Phys. Rev. Lett. 82:4142–4145. [Google Scholar]
- 11.Cheng, J. X., Y. K. Jia, G. Zheng, and X. S. Xie. 2002. Laser-scanning coherent anti-Stokes Raman scattering microscopy and applications to cell biology. Biophys. J. 83:502–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nan, X., J. X. Cheng, and X. S. Xie. 2003. Vibrational imaging of lipid droplets in live fibroblast cells with coherent anti-Stokes Raman scattering microscopy. J. Lipid Res. 44:2202–2208. [DOI] [PubMed] [Google Scholar]
- 13.Evans, C. L., E. O. Potma, M. Puoris'haag, D. Cote, C. P. Lin, and X. S. Xie. 2005. Chemical imaging of tissue in vivo with video-rate coherent anti-Stokes Raman scattering microscopy. Proc. Natl. Acad. Sci. USA. 102:16807–16812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li, L., H. Wang, and J. X. Cheng. 2005. Quantitative coherent anti-Stokes Raman scattering imaging of lipid distribution in coexisting domains. Biophys. J. 89:3480–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wurpel, G. W., H. A. Rinia, and M. Muller. 2005. Imaging orientational order and lipid density in multilamellar vesicles with multiplex CARS microscopy. J. Microsc. 218:37–45. [DOI] [PubMed] [Google Scholar]
- 16.Kennedy, A. P., J. Sutcliffe, and J. X. Cheng. 2005. Molecular composition and orientation in myelin figures characterized by coherent anti-Stokes Raman scattering microscopy. Langmuir. 21:6478–6486. [DOI] [PubMed] [Google Scholar]
- 17.Wang, H., Y. Fu, P. Zickmund, R. Shi, and J. X. Cheng. 2005. Coherent anti-Stokes Raman scattering imaging of axonal myelin in live spinal tissues. Biophys. J. 89:581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Potma, E. O., and X. S. Xie. 2005. Direct visualization of lipid phase segregation in single lipid bilayers with coherent anti-Stokes Raman scattering microscopy. ChemPhysChem. 6:77–79. [DOI] [PubMed] [Google Scholar]
- 19.Potma, E., W. P. de Boeij, P. J. van Haastert, and D. A. Wiersma. 2001. Real-time visualization of intracellular hydrodynamics in single living cells. Proc. Natl. Acad. Sci. USA. 98:1577–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng, J. X., S. Pautot, D. A. Weitz, and X. S. Xie. 2003. Ordering of water molecules between phospholipid bilayers visualized by coherent anti-Stokes Raman scattering microscopy. Proc. Natl. Acad. Sci. USA. 100:9826–9830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng, J. X., L. D. Book, and S. X. Xie. 2001. Polarization coherent anti-Stokes Raman scattering microscopy. Opt. Lett. 26:1341–1343. [DOI] [PubMed] [Google Scholar]
- 22.Potma, E. O., and S. X. Xie. 2003. Detection of single lipid bilayers with coherent anti-Stokes Raman scattering (CARS) microscopy. J. Raman Spectrosc. 34:642–650. [Google Scholar]
- 23.Hopt, A., and E. Neher. 2001. Highly nonlinear photodamage in two-photon fluorescence microscopy. Biophys. J. 80:2029–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Potma, E. O., C. Evans, X. S. Xie, R. J. Jones, and J. Ye. 2003. Picosecond-pulse amplification with an external passive optical cavity. Opt. Lett. 28:1835–1837. [DOI] [PubMed] [Google Scholar]
- 25.Konig, K., U. Simon, and K. J. Halbhuber. 1996. 3D resolved two-photon fluorescence microscopy of living cells using a modified confocal laser scanning microscope. Cell. Mol. Biol. 42:1181–1194. [PubMed] [Google Scholar]
- 26.Konig, K., P. C. T. So, W. W. Mantulin, and E. Gratton. 1997. Cellular response to near-infrared femtosecond laser pulses in two-photon fluorescence microscopy. Opt. Lett. 22:135–136. [DOI] [PubMed] [Google Scholar]
- 27.Hall, P. F., and G. Almahbobi. 1997. Roles of microfilaments and intermediate filaments in adrenal steroidogenesis. Microsc. Res. Tech. 36:463–479. [DOI] [PubMed] [Google Scholar]
- 28.Brady, S. T. 1991. Molecular motors in the nervous system. Neuron. 7:521–533. [DOI] [PubMed] [Google Scholar]
- 29.Kobayashi, T., T. Martensen, J. Nath, and M. Flavin. 1978. Inhibition of dynein ATPase by vanadate, and its possible use as a probe for the role of dynein in cytoplasmic motility. Biochem. Biophys. Res. Commun. 81:1313–1318. [DOI] [PubMed] [Google Scholar]
- 30.Betz, G., and P. F. Hall. 1987. Steroidogenesis in adrenal tumor cells: influence of cell shape. Endocrinology. 120:2547–2554. [DOI] [PubMed] [Google Scholar]
- 31.Gallo-Payet, N., and M. D. Payet. 2003. Mechanism of action of ACTH: beyond cAMP. Microsc. Res. Tech. 61:275–287. [DOI] [PubMed] [Google Scholar]
- 32.Denk, W., J. H. Strickler, and W. W. Webb. 1990. Two-photon laser scanning fluorescence microscopy. Science. 248:73–76. [DOI] [PubMed] [Google Scholar]
- 33.Williams, R. M., D. W. Piston, and W. W. Webb. 1994. Two-photon molecular excitation provides intrinsic three-dimensional resolution for laser-based microscopy and microphotochemistry. FASEB J. 8:804–813. [DOI] [PubMed] [Google Scholar]