

Abstract
Several biologically important peripheral (e.g., myristoylated alanine-rich C kinase substrate) and integral (e.g., the epidermal growth factor receptor) membrane proteins contain clusters of basic residues that interact with acidic lipids in the plasma membrane. Previous measurements demonstrate that the polyvalent acidic lipid phosphatidylinositol 4,5-bisphosphate is bound electrostatically (i.e., sequestered) by membrane-adsorbed basic peptides corresponding to these clusters. We report here three experimental observations that suggest monovalent acidic lipids are not sequestered by membrane-bound basic peptides. 1), Binding of basic peptides to vesicles does not decrease when the temperature is lowered below the fluid-to-gel phase transition. 2), The binding energy of Lys-13 to lipid vesicles increases linearly with the fraction of monovalent acidic lipids. 3), Binding of basic peptides to vesicles produces no self-quenching of fluorescent monovalent acidic lipids. One potential explanation for these results is that membrane-bound basic peptides diffuse too rapidly for the monovalent lipids to be sequestered. Indeed, our fluorescence correlation spectroscopy measurements show basic peptides bound to phosphatidylcholine/phosphatidylserine membranes have a diffusion coefficient approximately twofold higher than that of lipids, and those bound to phosphatidylcholine/phosphatidylinositol 4,5-bisphosphate membranes have a diffusion coefficient comparable to that of lipids.
INTRODUCTION
The inner leaflet of the plasma membrane from a typical mammalian cell contains both monovalent and polyvalent acidic lipids. Numerous membrane proteins contain clusters of basic residues that can interact with these negatively charged lipids. Examples include peripheral proteins such as Src, K-Ras4B, myristoylated alanine-rich C kinase substrate (MARCKS), growth-associated protein43/neuromodulin (GAP43), and A kinase anchoring protein 12 (also called gravin), as well as integral proteins such as the epidermal growth factor receptor (EGFR/ErbB) family of receptor tyrosine kinases and the ion channel N-methyl-D-aspartate receptor. Earlier work showed that the basic clusters on Src (1,2), K-Ras4B (3–5), and MARCKS (6) help anchor and target these proteins to the plasma membrane; the cytoplasmic leaflet of this membrane has a more negative electrostatic surface potential than the cytoplasmic leaflets of internal membranes (7). Integral membrane proteins or doubly palmitoylated proteins (e.g., GAP43), however, do not require basic/hydrophobic regions for membrane binding, suggesting that they may serve additional functions.
Experiments using three independent techniques (fluorescence resonance energy transfer (FRET), electron paramagnetic resonance, and the phospholipase-C-hydrolysis assay) have shown that peptides corresponding to the basic domains of MARCKS, EGFR, GAP43, and N-methyl-D-aspartate reeptor laterally sequester polyvalent acidic lipids, such as phosphatidylinositol-4,5-bisphosphate (PIP2), via nonspecific electrostatics (8–10). This occurs because when basic peptides bind to the membrane, they produce a local positive electrostatic potential that attracts polyvalent acidic lipids (11,12). A detailed study of peptides with 13 basic residues—Lys-13, Arg-13, and MARCKS(151–175), a basic/hydrophobic peptide corresponding to the effector domain of MARCKS (see Table 1 for sequence)—showed that three PIP2 form an electrostatic complex with a single peptide (8), even in the presence of physiological concentrations (15–30%) of monovalent acidic lipids (9). A peptide corresponding to the MARCKS basic region is unstructured in solution and when bound to a membrane (13–17), and the other highly basic clusters may also be extended rather than helical. The targeting of structured domains to membranes is reviewed elsewhere (18).
TABLE 1.
Sequences of peptides
| Peptide | Sequence |
|---|---|
| MARCKS(151–175) | CKKKKKRFSFKKSFKLSGFSFKKNKK |
| FA-MARCKS(151–175) | CKKKKKRASAKKSAKLSGASAKKNKK |
| ErbB1(645–660) | CRRRHIVRKRTLRRLLQ |
| Lys-13 | CKKKKKKKKKKKKK |
| Arg-13 | CRRRRRRRRRRRRR |
Basic residues are shown in bold and aromatic residues are underlined. The N-terminus of each peptide is blocked with an acetyl group and the C-terminus is blocked with an amide group. Cysteine residues were added at the N-terminus to facilitate attachment of fluorescent or radioactive labels. MARCKS(151–175) corresponds to the bovine effector domain. The effector domain of human MARCKS has an identical sequence.
This sequestration of PIP2 is reversible for MARCKS(151–175); either binding of calcium/calmodulin or PKC phosphorylation of three Ser can reverse membrane binding of both the peptide and native protein, and thus PIP2 sequestration (19–22). Calcium/calmodulin or PKC phosphorylation of a Ser in the basic cluster of K-Ras4B can also reverse the binding of this protein to the plasma membrane (23–25). Proteins containing clusters enriched in basic and hydrophobic residues may control the local free concentration of PIP2 in the plasma membrane by sequestering the lipids electrostatically and releasing them in response to calcium/calmodulin (12).
Theoretical analyses also predict that the local positive potential produced by a membrane-adsorbed basic peptide will attract monovalent acidic lipids, suggesting that they, too, should accumulate adjacent to the peptide, but less strongly than polyvalent lipids (26–29). We used several techniques to test the degree to which membrane-bound basic and basic/hydrophobic peptides laterally sequester monovalent acidic lipids, first measuring the effects of simple basic peptides, then analyzing more complex basic/hydrophobic peptides.
Data from numerous binding experiments indicate that nonspecific electrostatic interactions govern the association of simple basic peptides with lipid bilayer membranes. The peptides bind outside the envelope of the lipid polar headgroups (30–32), suggesting that hydrophobic interactions are negligible. Increasing the number of basic residues increases the binding energy linearly, as does increasing the mol fraction of acidic lipid in the membrane (32), whereas increasing the salt concentration in the solution reduces binding (32). Finally, neither the chemical nature of the acidic lipids nor that of the basic amino acids affects the binding strongly (31). Binding studies with basic/hydrophobic peptides suggest that they, too, interact with membranes mainly through electrostatic interactions, provided the membrane contains a significant fraction of acidic lipids (10,15,33); the hydrophobic residues penetrate the bilayer, but generally make a smaller contribution to the binding energy (9,14,16,17).
We used three different approaches to test whether basic peptides laterally sequester monovalent acidic lipids. 1), We compared the binding of basic peptides to vesicles at temperatures below and above the gel-to-liquid-crystalline phase transition; this should vary only if the peptides perturb the initially random lipid distribution in the fluid membranes. 2), We measured the binding energy of a simple basic peptide to lipid vesicles with increasing fractions of monovalent acidic lipids; the energy should increase linearly if the peptide does not sequester the acidic lipids (34). 3), We tested whether basic or basic/hydrophobic peptides produce self-quenching of NBD-labeled phosphatidylserine (PS) in vesicles; self-quenching should occur only if the peptides sequester the lipid. Finally, we measured the diffusion constants of fluorescently labeled lipids and membrane-bound peptides using fluorescence correlation spectroscopy (FCS); the results suggest that the relatively rapid diffusion of the peptide may explain the lack of monovalent lipid sequestration.
MATERIALS AND METHODS
Materials
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine (POPC),1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylserine (POPS), 1,2-dimyristoyl-sn-glycero-3-phosphatidylcholine (DMPC), 1,2-dimyristoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (DMPG), 1-palmitoyl-2-[6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]caproyl]-sn-glycero-3-phosphatidylcholine (NBD-PC) and 1-oleoyl-2-[12-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl]-sn-glycero-3-phosphatidylserine (NDB-PS), and triammonium salt of PIP2 from porcine brain were purchased from Avanti Polar Lipids (Alabaster, AL). 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine,4-chlorobenzenesulfonate (DiD), N-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a,-diaza-s-indacene-3-propionyl)-1-hexadecanoyl-sn-glycero-3-phosphatidylcholine (BODIPY-FL-PC), and Alexa488 were purchased from Molecular Probes (Eugene, OR). Rhodamine 6G was purchased from Sigma (St. Louis, MO). Radioactively labeled [dioleoyl-1-14C]-L-α-dioleoyl-phosphatidylcholine and [ethyl-1,2-3H] N-ethylmaleimide (3H-NEM) were from PerkinElmer Life Sciences (Boston, MA). BODIPY-TMR-C16-PIP2 and NBD-PIP2 were purchased from Echelon (Salt Lake City, UT). Table 1 shows the sequences of the basic or basic/hydrophobic peptides, purchased from American Peptide Co. (Sunnyvale, CA). Peptides used for experiments were determined to be >95% pure by high-performance liquid chromatography and matrix-assisted laser desorption ionization time-of-flight mass spectroscopy.
Peptide labeling
We labeled peptides with radioactive 3H-NEM as described previously (9,15). Briefly, we placed 250 μCi of 3H-NEM in pentane on top of 20 μl of N,N′-dimethylformamide (DMF), evaporated the pentane with argon gas, then mixed the 3H-NEM in DMF with 1 ml of 1 mM peptide solution. We blocked the unlabeled cysteines on the peptide by adding an excess of nonradioactive NEM (mol ratio of 1.5:1 NEM/peptide). We modified a protocol from “Conjugation with Thiol-Reactive Probes” (Molecular Probes) to label peptides with the thiol-reactive Alexa488. Briefly, we mixed 1 ml of 1 mM peptide in 10 mM K2HPO4/KH2PO4, pH 7.0, with the probe dissolved in DMF (1:1 probe/peptide molar ratio) and incubated it for at least 1 h. Labeled peptides were purified >95% using high-performance liquid chromatography (Proteomics Center, State University of New York, Stony Brook, NY).
Vesicle preparations
We used large unilamellar vesicles (LUVs;, diameter 100 nm) for the centrifugation binding and self-quenching experiments, as described in detail elsewhere (9,15,35). Briefly, we added solutions containing the appropriate lipid mixture in chloroform to a 50-ml round-bottom flask, attached the flask to a rotary evaporator, and rotated without vacuum for ∼5 min with the flask well immersed in a 30–35°C water bath. We then applied the maximum vacuum that does not boil the chloroform until most of the solvent evaporated, followed by evaporation under full vacuum for at least 30 min to remove traces of chloroform. We added the appropriate solution (100 mM KCl, 1 mM MOPS, pH 7.0, for vesicles used in self-quenching and FCS experiments, 176 mM sucrose, 1 mM MOPS, pH 7.0, for vesicles used in centrifugation binding experiments, which require sucrose-loaded LUVs) to form multilamellar vesicles, which were then subjected to a rapid freeze-and-thaw cycle five times. LUVs were formed by extruding multilamellar vesicles through 100-nm-diameter polycarbonate filters 10 times. For vesicles used in centrifugation measurements, the outer solution of the sucrose-loaded LUVs was exchanged for 100 mM KCl, 1 mM MOPS, pH 7, via ultracentrifugation. DMPC/DMPG vesicles for centrifugation binding experiments were extruded and maintained at ∼30°C, significantly above the fluid-to-gel phase transition of ∼24°C, until the centrifugation experiment was performed.
We used both gentle hydration and rapid evaporation methods to form giant unilamellar vesicles (GUVs) for FCS measurements (36,37). The gentle hydration method involves drying the appropriate lipid mixture in chloroform (9:1 phosphatidylcholine/phosphatidylserine (PC/PS) for lipid diffusion measurements, which provides the negative charge necessary for GUV formation in high-salt solutions; 0.01 mol % DiD for confocal imaging of the GUVs, and 0.0001 mol % fluorescent lipids for FCS measurements) under vacuum for 30 min to form a thin film, then prehydrating the dried film with argon-saturated water vapor at 35–40°C for 1 h. We then added 1–2 ml of buffer solution (100 mM KCl, 1 mM MOPS, pH 7.0, warmed to the same temperature as hydrated lipids) containing 10–100 nM Alexa488-labeled peptides and incubated the sealed flask for 5–12 h at room temperature. As the GUVs form they trap the peptide inside and, because basic peptides bind to glass with high affinity, essentially all peptides bound to the GUVs are on the inner leaflet of the vesicles We harvested 100–200 μl of the upper part of the solution and transferred the GUVs to a chamber composed of glass coverslip and a microscope glass slide sandwiched with thermoplastic material. Only 10–20% of the GUVs were useful for FCS measurements (5–30 μm diameter, unaggregated, single-walled GUVs). We also used the gentle hydration method to prepare GUVs directly on a microscope coverglass held in a stainless steel chamber with Teflon insert designed by R. Galneder in the Ludwig-Maximilians-Universität, Sektion für Physik, Münich, Germany; the lipid mixture was exposed only to Teflon and glass. We deposited small drops of the appropriate lipid mixture in chloroform (∼0.01 mg/ml) on the coverglass to form thin film. Further steps were similar to previous methods. Due to the problems with incorporation of PIP2, GUVs containing this lipid were prepared in round-bottom flasks using the gentle hydration method. To measure diffusion of Alexa488-labeled peptides bound to the outer leaflet of the GUVs, we used GUVs prepared directly on the glass coverslip and added 10 nM Alexa488-labeled peptide 5 min before the FCS data recording. The rapid evaporation method (37) involves carefully adding 2 ml of Alexa488-labeled peptide solution (100 mM KCl, 1 mM MOPS, pH 7.0) to a 50-ml round-bottom flask containing 20 μl of 0.1 M lipid mixture dissolved in 320 μl of chloroform and 70 μl of methanol. Two minutes under vacuum on a rotary evaporator produces an opalescent fluid containing ∼10% single-walled GUVs, as reported previously (38). This method had the advantage of producing GUVs rapidly. Diffusion measurements with vesicles produced by both methods yielded identical results.
Centrifugation binding experiments
We measured binding of 3H-NEM-labeled peptides to sucrose-loaded DMPC/DMPG, POPC/POPG, and POPC/POPS LUVs using the centrifugation technique described in detail elsewhere (9,15,35). Briefly, we mixed trace concentrations of labeled peptide (2–10 nM) with sucrose-loaded LUVs and centrifuged at 100,000 g for 1 h. (We added 100 μM of PC in the form of sonicated vesicles to the experimental mixture to minimize loss of the peptide onto the walls of the centrifuge tubes.) For the temperature dependence experiments (Fig. 1), both mixing and centrifugation were done at the given temperature (10°, 17°, or 30°C). We used a substituted peptide, F-A MARCKS(151–175), rather than MARCKS(151–175) for these experiments because the aromatic Phe residues in the latter penetrate the polar headgroup region of a liquid crystalline lipid bilayer (17), but presumably would be expelled from a gel-phase bilayer; this would complicate interpretation of the results. The binding measurements reported in Fig. 2 were carried out at room temperature, 24°C. We calculated the percent peptide bound to the vesicles by comparing the radioactivity (liquid scintillation) or fluorescence (spectrofluorometry) present in the supernatant and pellet.
FIGURE 1.

Binding of Arg-13 and F-A-MARCKS(151–175) to DMPC/DMPG LUVS does not depend on the state of the lipids. (A) Binding of radioactively labeled Arg-13 to 5:1 DMPC/DMPG LUVs at 10°C. The percent of peptide bound was determined as a function of accessible lipid concentration, [lipid], using a centrifugation technique. The curve represents the least-squares fit of Eq. 1 to the data. The molar partition coefficient, K (reciprocal of accessible lipid concentration that binds 50% of the peptide), is 1.4 × 105 M−1. Experiments were conducted at 10°C, 17°C, and 30°C, and the average values of K are plotted in B. (B) Temperature dependence of the molar partition coefficient, K, for Arg-13 (•) and F-A-MARCKS(151–175) (∇) binding to 5:1 DMPC/DMPG LUVs. Each point represents an average (± SD) of at least three independent experiments similar to those shown in A. For all results in Figs. 1–4, the external solutions contained 100 mM KCl, 1 mM MOPS, pH 7.0.
FIGURE 2.

Binding of Lys-13 to PC/PS LUVS increases exponentially with the mol fraction of PS in the vesicles. (A) Binding of 3H-NEM-labeled Lys-13 to PC/PS LUVs containing either 15% (▪) or 20% (□) PS. The percent of peptide bound was determined as a function of accessible lipid concentration using a centrifugation technique. The curves represent the least-squares fit of Eq. 1 to the data. K = 1 × 104 M−1 and 1 × 105 M−1 for vesicles with 15% and 20% PS, respectively. The average value of K (± SD, n = 6) is plotted in B, together with the results of similar experiments with PC/PS vesicles containing different mol fractions of PS. (B) The molar partition coefficient, K, of 3H-NEM-labeled Lys-13 as a function of the mol % PS in the PC/PS LUVs (○). Note that K increases exponentially with mol % PS. (Control experiments with Alexa488-Lys-13 produced identical values of K from centrifugation (•), and from FCS measurements (▴).)
The fraction of peptide, P, bound through electrostatic/hydrophobic interactions to membranes is given by Eq. 1:
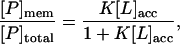 |
(1) |
where [P]mem is the concentration of membrane-bound peptide, [P]total is the total concentration of the peptide, [L]acc is the accessible lipid concentration (½ the total lipid concentration as the peptide is added to preformed vesicles), and K is the molar partition coefficient (the reciprocal of the concentration of accessible lipid that binds 50% of the peptide); see Wang et al. (15) and Peitzsch and McLaughlin (39) for details. We plot the fraction of peptide bound to the membranes versus accessible lipid concentration (e.g., Fig. 1 A) and determine the molar partition coefficient from the least-squares fit of Eq. 1 to the data points.
Fluorescence quenching experiments
We measured self-quenching using an SLM-AMINCO spectrofluorometer (Foster City, CA). Experiments using NDB labels used a 470-nm excitation wavelength and emission spectra were collected from 500 to 600 nm; experiments using BODIPY-TMR labels used a 547-nm excitation wavelength and emission spectra were collected from 560 to 660 nm. The emission spectra were collected as we added unlabeled peptide to the solution containing the LUVs. We used Eq. 2 to calculate the percentage of quenching:
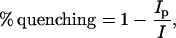 |
(2) |
where Ip is the intensity of fluorescence emitted from labeled lipid in the presence of peptide, and I is the intensity of fluorescence emitted from labeled lipid in the absence of peptide. The total lipid concentration was 0.1 mM and 0.75 mM for 5:1 PC/PS and 99:1 PC/PIP2 LUVs, respectively. These concentrations are sufficiently high (as determined using the centrifugation binding assay) to bind 99% of the MARCKS(151–175) or Lys-13 peptides in the solution.
The quenching experiments were performed with vesicles containing fluorescent lipids present at the maximal mol fractions that did not exhibit appreciable self-quenching (4% for NBD-lipids and 1% for BODIPY-TMR-PIP2 in the LUVs).
We determined the amount of self-quenching due to the proximity of neighboring NBD lipids by measuring the NBD fluorescence in LUVs containing increasing percentages of NBD lipid (from 0 to 6% NBD-PC or NBD-PS). We measured the fluorescence of NBD lipid both in LUVs (50 μM total lipid in 100 mM KCl, 1 mM MOPS, pH 7) and in a nonmembrane form in methanol, and calculated self-quenching (Fig. 3 A) using Eq. 2, substituting the fluorescence intensity of the NBD lipid in LUVs for Ip and the corresponding intensity of NBD-lipid in methanol for I.
FIGURE 3.

Binding of MARCKS(151–175) and Lys-13 does not produce self-quenching of NBD-PS. (A) Self-quenching of NDB-PS (or NBD-PC) versus mol fraction of NBD-PS (or NBD-PC) in 5:1 PC/PS 100 nm LUVs. Each point represents an average (± SD) of at least three independent vesicle preparations. (B) Neither MARCKS(151–175) (•) nor Lys-13 (○) produce significant self-quenching of NBD-PS when they bind to 83:13:4 PC/PS/NBD-PS LUVs. (C) Addition of either MARCKS(151–175) (•) or Lys-13 (○) produces strong self-quenching of BODIPY-TMR-PIP2 in 99:1 PC/BODIPY-TMR-PIP2 LUVs, because the peptides laterally sequester the PIP2 (8,9).
FCS diffusion measurements
Confocal imaging and FCS measurements were performed on a commercial Zeiss (Jena, Germany) LSM 510 Meta/confocor 2 apparatus using standard configurations. Minimal laser powers were chosen to avoid photobleaching of the fluorescent probes. We used a 40× NA 1.2 C-Apochromat water-immersion objective and adjusted pinholes at least daily. The detection volume was calibrated by measuring the diffusion of rhodamine 6G (D = 2.8 × 10−6 cm2/s (40)) in the buffer solution (100 mM KCl, 1 mM MOPS, pH 7.0). The 1/e2 radius of the detection volume for the 488-nm line was determined to be ω1 = 0.14 ± 0.01 μm. We excited Alexa488, BODIPY-FL, and BODIPY-TMR with the 488-nm line of the Argon laser and collected emission spectra through a 505-530 BP filter (Alexa488, BODIPY-FL) or a 505-600 BP filter (BODIPY-TMR). We monitored the count rate during data acquisition and rejected measurements with a visible decrease to avoid artifacts due to vesicle movements and bleaching. We used Sigma Plot and a least-squares algorithm to fit the autocorrelation curves to the model equation for free Brownian diffusion in two dimensions commonly used in FCS (41):
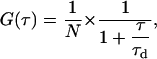 |
(3) |
where τd, is the average residence time and N is the average number of particles in the measurement volume. We calculated the diffusion coefficient, D, from the Einstein relation (Eq. 4)
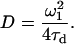 |
(4) |
For cases with i different populations of molecules (e.g., Alexa488-Lys-13 on 4:1 and 5:1 PC/PS GUVs had a small but significant fraction of unbound peptide) moving with different diffusion coefficients, Di, with fractions equal to Yi, and residence times 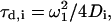 the expression becomes (assuming they have the same fluorescence quantum yields)
the expression becomes (assuming they have the same fluorescence quantum yields)
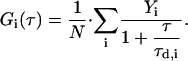 |
(5) |
GUVs were prepared with Alexa488-labeled basic peptides bound to either the inner or outer leaflet of the membrane as described above. Using the laser scanning microscope (LSM) module, we scanned the chamber and selected GUVs with a diameter of 5–30 μm attached to the coverslip. GUVs used for measurements had an average diameter ∼10 μm (see Fig. 4 A). The effective concentration of lipids in the inner leaflet of a 10-μm-diameter GUV is ∼10−3 M, assuming they are distributed uniformly in the enclosed volume). Alexa488-Lys-13 binds 3:1 PC/PS LUVs with K = 1 × 105 M−1; thus, 99% of the peptides trapped inside such GUVs are bound to the membrane. MARCKS(151–175) has higher affinity for PC/PS membranes and binds 5:1 PC/PS LUVs with a molar partition coefficient K ∼105 M−1; thus 99% of the enclosed peptides are bound to the membrane of a 10-μm-diameter 5:1 PC/PS GUV. We ensured that we were measuring the D of membrane-bound peptides by examining the relationship between D and the mol fraction of PS in the vesicles: increasing the fraction of PS both increases the peptide's affinity to and decreases its rate of dissociation from membranes. We performed an axial (z) scan through the membrane before the FCS recording and placed the laser focus on the top central membrane region of the GUV (the point of the maximum fluorescence intensity; see cartoon in Fig. 4 B). We determined the amount of fluorescence emitted by Alexa488 fluorophore in the solution for the given laser power. During data acquisition we ensured that Alexa488-labeled peptides emit the same amount of light.
FIGURE 4.

(A) A representative LSM confocal image of a GUV used for FCS measurements. The vesicle contains 0.01% red fluorescent lipid DiD for LSM imaging. Scale bar, 5 μm. (B) Cartoon showing a GUV (red circle) with Alexa488-labeled basic peptide (green circles with blue bars) bound to its inner leaflet. The confocal volume (green hourglass shape) is positioned at the top of the GUV. (C) Autocorrelation curves of Alexa488-Lys-13 diffusing on a 4:1 PC/PS GUV (black with green fit) and BODIPY-TMR-PIP2 diffusing in a 10:1 PC/PS GUV (black with red fit). The black curves represent the experimentally determined autocorrelation functions, the green curve the fit of Eq. 3, with τd = 890 μs and D = 6.5 × 10−8 cm2/s for Alexa488-Lys-13, the red curve the fit of Eq. 3, with τd = 2 ms and D = 3 × 10−8 cm2/s for BODIPY-TMR-PIP2. (D) Diffusion constants of membrane-bound Alexa488-Lys-13 and BODIPY-TMR-PIP2 in GUVS determined by FCS. Bars indicate average values ± SD. Note that Lys-13 diffuses twofold more rapidly on the 3:1 PC/PS vesicles than on the 99:1 PC/PIP2 vesicles, and that the diffusion constant of the peptide bound to the PC/PIP2 vesicles is the same as the diffusion constant of a lipid, as expected theoretically.
We selected vesicles for measurement first by discarding aggregated, visibly multiple-shelled, or very bright GUVs (in LSM mode), then selecting GUVs with sharper peaks and lower fluorescence-intensity scans (in FCS mode). We also examined several GUVs selected as putative unilamellar vesicles using phase microscopy to confirm that they were indeed unilamellar. We performed FCS measurements inside, above, and next to the GUVs to determine the background signal from unbound peptide and from a free label in the solution; we rejected samples with background >10%.
Because peptides trapped between multiple lamellae of a vesicle could exhibit anomalously slow diffusion, we measured diffusion of Alexa488-MARCKS(151–175) trapped in a visibly multiple-shelled vesicle: the signal from such peptides was high, the number of particles was large, and the diffusion was slow compared to measurements done on putative unilamellar GUVs. As an additional control, we measured diffusion of peptides bound to the outer leaflet of the GUVs prepared directly on the glass coverslips and exposed to a 10-nM Alexa488-labeled peptide solution 5 min before the measurements. We detected neither a significant concentration of free peptide in solution nor a significant change in the number of peptides bound to GUV during the measurement period. The measured value of D was identical for peptides bound to the outer or inner leaflet of the GUV. These results support our assumption that most of our measurements were made on authentic unilamellar vesicles.
FCS binding measurements
FCS binding experiments were performed as described in detail elsewhere (33). Briefly, we precoated eight-well LabTechII chambers (Nunc, Wiesbaden, Germany) with PC membranes (33) to prevent loss of the peptide. The laser focus was placed in the solution 200 μm above the top of the glass coverslip. LUVs are significantly larger than the Alexa488-labeled peptides, so single color autocorrelation measurements can distinguish between the correlation times for the free and bound peptide. We determined the correlation time of free peptide (∼60 μs) and 100-nm-diameter LUVs (∼1600 μs) separately. We used the ConfoCor2 software to fit the autocorrelation function to the collected data. In the three dimensions, Eq. 6 describes the autocorrelation function:
 |
(6) |
where N is the number of fluorescent particles in the confocal volume (typically 1), SP (structure parameter) is the ratio between the equatorial and axial radii of the confocal volume (∼6), τd is the average residence time of the particle in the confocal volume, τt is the triplet state lifetime, and F is the fraction of fluorescent particles in the triplet state. In the case of a multicomponent system, the measured correlation function G( 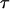 ) is a sum of the autocorrelation functions of each component.
) is a sum of the autocorrelation functions of each component.
Statistical analysis
We used SigmaStat and SigmaPlot (SPSS, Chicago, IL) for statistical analysis and curve fitting, respectively. We compared the values of diffusion coefficient of membrane-bound peptides and lipids using Kruskal-Wallis one-way analysis of variance on ranks, Dunn's method, and one-way analysis of variance, Tukey's method (42). We concluded that the values are significantly different when P < 0.05.
RESULTS
Basic peptides bind equally strongly to gel (frozen) and liquid-crystalline (fluid) lipid membranes
We measured the binding of two basic peptides, Arg-13 and F-A-MARCKS(151–175), see Table 1, to 5:1 DMPC/DMPG vesicles at 10°, 17°, and 30°C using a centrifugation technique. DMPC and DMPG have similar liquid-to-gel transition temperatures (Tc ∼ 24°C) and these lipids distribute randomly in the membrane below and above the transition (43,44). Fig. 1 A shows the results of a typical experiment using Arg-13 at 10°C. We calculated the molar partition coefficient, K, from a least-squares fit of Eq. 1 to the data. Fig. 1 B shows the values of K for binding of Arg-13 and F-A-MARCKS(151–175) to 5:1 DMPC/DMPG at the three different temperatures: K does not change significantly with temperature (i.e., the peptides bind with similar affinity to frozen and fluid membranes). We also measured Arg-13 binding to 10:1 DMPC/DMPG vesicles because reducing the mol fraction of acidic lipid should make any phase-related differences in binding more pronounced. Again, we observed no significant differences in binding: K = 1 × 104 M−1 at 10°, 17°, and 30°C (data not shown). As a control, we measured the binding of Arg-13 and F-A-MARCKS(151–175) to 5:1 POPC/POPG vesicles, which remain in a liquid crystalline state at 10°, 17°, and 30°C, and found no significant differences in K (data not shown).
The simplest explanation for the observation that changing the membrane from a gel to a liquid-crystalline state does not affect peptide binding is that the peptides do not cause lateral redistribution of monovalent acidic lipids when they bind. The rationale here is simple: the diffusion constant of the lipids decreases ∼1000-fold in the gel state (45); the diffusion constant of the peptide does not decrease markedly (not shown). Thus, a rapidly diffusing peptide will skate over the frozen surface of the membrane and not perturb the random distribution of the lipids. As the binding energy is the same when the membrane is in a liquid/crystalline state (Fig. 1 B), the simplest interpretation is that the peptide also does not perturb the random distribution of the lipids when it binds to the fluid membrane.
The molar partition coefficient, K, of basic peptides increases exponentially with increasing mol fraction of acidic lipids in the membrane
We measured the binding of the basic peptide Lys-13 (Table 1) to LUVs composed of mixtures of the zwitteronic phospholipid POPC and the monovalent acidic lipid POPS using a centrifugation technique. Fig. 2 A shows the results of two typical experiments with LUVs: K = 1 × 104 M−1 or K = 1 × 105 M−1 for vesicles containing POPC and 15% or 20% PS, respectively. Fig. 2 B is a plot of K values deduced from similar experiments for binding of Lys-13 to LUVs composed of PC and different fractions of PS. K increases exponentially with the mol % of PS in the vesicles, i.e., the binding energy increases linearly with the percent of PS in the membrane. The results are consistent both with previous observations of binding of basic/hydrophobic peptides corresponding to the basic effector domain of MARCKS or the juxtamembrane domain of the EGFR (10,33), and with theory (34), if one assumes that binding of basic and basic/hydrophobic peptides to PC/PS membrane does not produce redistribution of PS.
We validated the results of the centrifugation technique by comparing the binding of Alexa488-labeled Lys-13 using both centrifugation and an FCS technique described in detail elsewhere (33); both methods produced similar results, as shown in Fig. 2 B. (n.b., The affinity of Alexa488-labeled Lys-13 for 3:1 PC/PS vesicles is ∼10-fold weaker than the affinity of 3H NEM-labeled Lys-13, because the Alexa488 probe is negatively charged, whereas the NEM label is electrically neutral.)
Neither basic nor basic/hydrophobic peptides induce self-quenching of monovalent acidic lipids
We examined whether addition of either unlabeled Lys-13 or MARCKS(151–175) produces self-quenching of NBD-labeled PS when the fluorescent lipid is present in the vesicles at a fraction just below the level that produces self-quenching. We first determined the mol fraction of NBD-labeled PC or PS in the vesicles where the probe begins to self-quench. Fig. 3 A plots the percentage of self-quenching versus the fraction of NBD-labeled lipids in the vesicles (the results of experiments with NBD-PC and NBD-PS were averaged): >4% NBD-labeled lipid produces significant self-quenching. Previous studies showed that NBD-PS self-quenches when present at mol fractions >5% (46), because the average distance between labeled lipids is comparable to the Förster radius for NBD (Ro ∼ 35Å). Thus, for vesicles with 4% NBD-labeled PS, even a minor redistribution of acidic lipids should increase self-quenching.
Fig. 3 B plots the percentage of self-quenching of NBD-PS calculated from Eq. 2 versus peptide concentration: neither MARCKS(151–175) nor Lys-13 produce measurable self-quenching when they bind to vesicles with 4% NBD-PS. The simplest interpretation is that membrane-bound basic and basic/hydrophobic peptides do not enhance the local concentration of NBD-PS. In contrast, adding high-molecular-weight polylysine (∼2000 residues) does induce significant self-quenching of NBD-PS (e.g., 100 pM produces 20% quenching; data not shown), suggesting that it may laterally sequester monovalent lipids. An extended 2000-residue polylysine molecule could encircle the 100-nm vesicle and cross over itself; presumably such a bound molecule would diffuse only slowly on the surface of the vesicle. The difference in diffusion rates between membrane-bound Lys-13 and polylysine could account for the presumed lateral sequestration by the latter. We have not examined experimentally whether membrane-bound basic peptides that have structure (e.g., amphipathic helices) laterally sequester PS.
These self-quenching experiments suggest that small membrane-bound basic and basic/hydrophobic unstructured peptides do not sequester monovalent acidic lipids; in contrast, Fig. 3 C shows that the same peptides produce significant sequestration of the BODIPY-labeled polyvalent acidic lipid PIP2. (A control experiment shows MARCKS(151–175) produces similar quenching of NBD-PIP2 and BODIPY-PIP2; data not shown). Fig. 3 C agrees with previous self-quenching, FRET, spin-label, and kinetic measurements (8,9). We discuss elsewhere the different factors that contribute to the quenching observed in Fig. 3 C (9).
Diffusion of membrane-bound basic and basic/hydrophobic peptides
Membrane-bound Lys-13 diffuses more rapidly than lipids
We measured the lateral diffusion of Alexa-labeled Lys-13 bound to the inner leaflet of PC/PS GUVs using FCS. Fig. 4 A shows an LSM image of a 10-μm-diameter GUV labeled with DiD to aid visualization. Fig. 4 B is a cartoon showing a GUV with the confocal volume positioned on the top central region and Alexa-labeled peptides diffusing on the inner leaflet of the GUV. The FCS instrument's software produces autocorrelation curves that correspond to peptides moving in and out of the confocal volume.
Fig. 4 C (green) shows an example of an autocorrelation curve for Alexa488-Lys-13 diffusing on a 3:1 PC/PS membrane. We used the least-squares fit of Eq. 3 to the autocorrelation curve to calculate the residency time, τd ∼890 μs, in the confocal volume and the Einstein relation (Eq. 4) to calculate the diffusion coefficient, D, of the membrane-bound peptide. Fig. 4 D shows that D = 7 ± 1 × 10−8 cm2/s (± SD, n = 48) for Lys-13 bound to GUVs containing 25% PS. All diffusion measurements were made at room temperature, 21 ± 1°C.
Fig. 4 D shows the diffusion coefficient of Lys-13 bound to PC/PS GUVS is approximately twofold greater than the diffusion coefficient of a lipid (compare left and right bars), a difference that is statistically significant (P < 0.05). Specifically, we measured the diffusion coefficient of BODIPY-FL-PC in PC/PS GUVs, D = 2.9 ± 0.4 × 10−8 cm2/s (n = 11), and of BODIPY-TMR-PIP2 incorporated into PC/PS GUVs, D = 3.3 ± 0.8 × 10−8 cm2/s (n = 33), as shown in Fig. 4, C (red) and D. (As a control, we measured the diffusion of BODIPY-TMR-PIP2 incorporated into the outer leaflet of preformed GUVs (by exposing them to PIP2 micelles), D = 3.3 ± 0.7 × 10−8 cm2/s (n = 23).) These values (∼3 × 10−8 cm2/s) agree qualitatively with the diffusion coefficients of unsaturated chain lipids reported previously (47–50). Specifically, Wagner and Tamm (51) showed that NBD-labeled PE and PIP2 had similar diffusion coefficients in planar supported bilayers.
We confirmed that we were measuring true lateral diffusion of Alexa488-Lys-13 (rather than desorption of the peptide and aqueous diffusion) by repeating the diffusion measurements on PC/PS GUVs containing different mol fractions of acidic lipid. (The forward rate constant of a basic peptide with a PC/PS membrane is diffusion-limited (52), and the equilibrium association constant increases exponentially with the mol fraction of acidic lipid (Fig. 2 B). Thus, both the rate constant for the peptide moving off the membrane and the fraction of peptide free in the aqueous phase inside the vesicle will decrease markedly as the mol fraction of PS increases.) Table 2 shows that the average diffusion coefficient of the membrane-bound peptide is 6.5 × 10−8 cm2/s with 5:1, 4:1, 3:1, and 2:1 PC/PS vesicles, i.e., it is independent of the mol fraction of PS in the vesicle. Hence, the measured diffusion coefficient does not depend on the rate at which the peptide moves off the membrane.
TABLE 2.
Diffusion coefficients of basic and basic/hydrophobic peptides bound to GUVs composed of POPC and either POPS or porcine PIP2 as determined by FCS
| Diffusion coefficient of membrane-bound peptide (10−8 cm2/s)
|
|||||
|---|---|---|---|---|---|
| Peptide | 83:17 PC/PS | 80:20 PC/PS | 75:25 PC/PS | 67:33 PC/PS | 99:1 PC/PIP2 |
| Alexa488- Lys-13 | |||||
| On inner leaflet | 7 ± 1 | 6 ± 2 | 7 ± 1 | 6 ± 2 | 3 ± 1 |
| n = 17 | n = 12 | n = 48 | n = 13 | n = 15 | |
| On outer leaflet | 6.5 ± 0.9 | 6 ± 1 | |||
| n = 16 | n = 18 | ||||
| Alexa488-MARCKS(151–175) | |||||
| On inner leaflet | 4 ± 1 | 4.4 ± 0.6 | 4 ± 1 | 4.1 ± 0.8 | 2.3 ± 0.7 |
| n = 59 | n = 15 | n = 17 | n = 12 | n = 52 | |
| On outer leaflet | 4.1 ± 0.6 | 5.2 ± 0.9 | 4.3 ± 0.5 | ||
| n = 4 | n = 10 | n = 14 | |||
| Alexa488-ErbB1(645–660) | |||||
| On inner leaflet | 5 ± 1 | 7 ± 1 | 5 ± 2 | ||
| n = 14 | n = 51 | n = 18 | |||
Values are presented as mean ± SD. n, number of experiments.
Finally, we measured the diffusion of peptides bound to the outer leaflet of the GUVs to ensure that our measurements did not reflect data on peptides trapped between multiple lamellae of the vesicle: D = 6 × 10−8 cm2/s for peptides bound to either the inner or outer leaflet of the membrane (Table 2). In summary, Fig. 4 D shows Alexa488-Lys-13 bound to PC/PS vesicles diffuses twofold more rapidly than lipids.
Basic peptides bound to PC/PIP2 vesicles diffuse as slowly as lipids
We measured the lateral diffusion of Alexa488-Lys-13 bound to 99:1 PC/PIP2 GUVs. Fig. 4 D and Table 2 show D = 3 ± 1 × 10−8 cm2/s (n = 15), a value identical, within experimental error, with diffusion measurements of BODIPY-TMR-PIP2 incorporated into PC/PS GUVs, D = 3.3 ± 0.8 × 10−8 cm2/s (n = 33). Why does Alexa488-Lys-13 diffuse twofold more slowly when bound to 99:1 PC/PIP2 versus 3:1 PC/PS vesicles? Lys-13 has an identical affinity for 3:1 PC/PS (Fig. 2) and 99:1 PC/PIP2 (8) vesicles, K = 106 M−1. Previous studies showed that when Lys-13 binds to PC/PIP2 membranes, three PIP2 molecules diffuse toward the peptide to form an electrostatic binding site (8); the peptide cannot diffuse laterally at a speed faster than that of the lipid without desorbing from the binding site. Thus it is the nature of the binding, rather than the magnitude of the affinity for the PC/PIP2 vesicle, that determines the value of D. Our FCS result thus agrees with the expectation: the peptide diffuses together with the laterally sequestered PIP2.
Basic/hydrophobic peptides also diffuse more rapidly on PC/PS than PC/PIP2 membranes
We measured the lateral diffusion of two basic/hydrophobic peptides, Alexa488-MARCKS(151–175) and Alexa488-ErbB1(645–660) (Table 1), which corresponds to the juxtamembrane region of the EGFR/ErbB receptor (10) bound to PC/PS GUVs: D ∼4 × 10−8 cm2/s and ∼6 × 10−8 cm2/s, respectively (Table 2), and is independent of the mol fraction of PS in the vesicle (or binding affinity). These values are slightly but significantly (P < 0.05) higher than the D of lipids. Measurements with Alexa488-MARCKS(151–175) bound to 99:1 PC/PIP2 GUVs (Table 2) show that D = 2.3 ± 0.7 × 10−8 cm2/s (n = 52), slightly but significantly (P < 0.05) slower than the D of the same peptide bound to PC/PS vesicles.
Incorporating physiological (1%) levels of PIP2 into PC/PS GUVS slows the diffusion of basic and basic/hydrophobic peptides
Table 3 shows the diffusion coefficients of the three Alexa488-labeled peptides bound to PC/PS/PIP2 GUVs. Comparison of the value in Tables 2 and 3 shows that the peptide diffusion constants are slightly faster on PC/PS versus PC/PS/PIP2 membranes. The latter composition is a better reflection of the cytoplasmic leaflet of a plasma membrane, which contains both monovalent (e.g., PS) and polyvalent (e.g., PIP2) acidic lipids. Previous experiments (e.g., FRET) showed that basic and basic/hydrophobic peptides sequester PIP2 even when the membrane contains physiological mol fractions (15–30%) of monovalent acidic lipids and 100-fold less PIP2 (9).
TABLE 3.
Diffusion coefficients of basic and basic/hydrophobic peptides bound to the inner leaflet of PC/PS/PIP2 GUVs, as determined by FCS
| Diffusion coefficient of membrane-bound peptide (10−8 cm2/s)
|
||||
|---|---|---|---|---|
| Peptide | 82:17:1 PC/PS/PIP2 | 83:17:0.1 PC/PS/PIP2 | 73:25:1 PC/PS/PIP2 | 74:26:0.1 PC/PS/PIP2 |
| Alexa488-Lys-13 | 4.0 ± 0.7 | 4.0 ± 0.9 | ||
| n = 20 | n = 12 | |||
| Alexa488-MARCKS (151–175) | 3.5 ± 0.8 | 3.1 ± 0.6 | ||
| n = 10 | n = 22 | |||
| Alexa488-ErbB1 (645–660) | 3.3 ± 0.6 | 3.2 ± 0.7 | ||
| n = 9 | n = 16 | |||
Values are presented as mean ± SD. n, number of experiments.
DISCUSSION
Three different types of experiments strongly suggest that binding of either basic (e.g., F-A-MARCKS(151–175), Lys-13, Arg-13) or basic/hydrophobic (e.g., MARCKS(151–175)) peptides to membranes produces no significant lateral sequestration of monovalent acidic lipids. First, Arg-13 and F-A-MARCKS(151–175) bind with the same affinity to DMPC/DMPG vesicles in the gel and liquid-crystalline state (Fig. 1 B). As discussed above, the simplest interpretation is that the peptide also does not perturb the random distribution of the lipids when it binds to the fluid membrane. Earlier measurements of the effect of Lys-5 on the electrophoretic mobility (ζ potential) of DMPC/DMPG multilamellar vesicles supports our interpretation: the peptide binds to the same degree at temperatures above and below the transition temperature (31). Second, the binding affinity of Lys-13 (Fig. 2 B) for PC/PS vesicles increases exponentially with the mol fraction of monovalent acidic lipids, as was observed previously with basic/hydrophobic peptides (10,33). This is the theoretically predicted result if the peptides do not perturb the initial random distribution of acidic lipids in the membrane (34). Specifically, if binding of Lys-13 to the PC/PS membrane produced lateral sequestration of PS, a simple theoretical analysis of the entropy price paid for this redistribution suggests that Fig. 2 B would be a curve, steep at the low mol fractions, less steep at high mol fractions of PS (J. Nagle, Carnegie Mellon University, personal communication, 2005). Third, Fig. 3 B shows that neither Lys-13 nor MARCKS(151–175) produces self-quenching of NBD-PS when it binds to the membrane, in agreement with a previous report that membrane-adsorbed Lys-5 does not laterally sequester spin-labeled monovalent acidic lipids (53).
Our interpretation of these results is that membrane-adsorbed basic or basic/hydrophobic peptides concentrate or sequester monovalent acidic lipids minimally, if at all. In contrast, membrane-bound basic or basic/hydrophobic peptides laterally sequester polyvalent PIP2 strongly (9). Theoretical analyses indicate that peptide binding should concentrate monovalent acidic lipids in the electrical double layer adjacent to the binding site, albeit less strongly than polyvalent lipids (26,27,29). Why do our experimental results indicate no significant sequestration of PS next to an adsorbed basic peptide? We suspect that our diffusion measurements provide a clue: a membrane-bound basic peptide diffuses twofold more rapidly than the lipids in a PC/PS membrane (Fig. 4 D, Table 2). We admit, however, that we do not understand the phenomenon fully.
Structural studies show that the five Phe residues of MARCKS(151–175) insert into the membrane to the level of the acyl chains, suggesting that the diffusion coefficient of the bound peptide might be expected to be the same as that of the membrane lipids. Our measurements, however, indicate that this basic/hydrophobic peptide diffuses slightly faster than the lipids in a PC/PS membrane (14,16,17). One possible, albeit speculative, explanation is that the Phe residues move in and out of the membrane rapidly (e.g., approximately every microsecond). This is not unreasonable, because electrostatic interactions provide most of the binding energy: MARCKS(151–175) binds to 5:1 PC/PS membranes with K = 106 M−1, four orders of magnitude higher than K = 102 M−1 for neutral PC membranes (33). Thus the Phe residues could hop out of the bilayer but if the peptide remains close to the surface it would retain its electrostatic binding energy, and could diffuse relatively rapidly parallel to the membrane. Once the Phe residues repenetrate, the peptide should diffuse at about the same speed as a lipid. This mechanism could explain both why the peptide diffuses more rapidly than a lipid, and why it does not sequester PS. Alternatively, the Phe residues could simply slide rapidly between the lipid molecules.
Why is the diffusion constant of Lys-13 bound to the membrane much slower than the diffusion in a bulk aqueous solution? We know that basic peptides (e.g., Lys-13) adsorb outside the envelope of the polar headgroup region (they do not increase the surface pressure of a monolayer when they bind), but we don't know if they are located zero, one, or two water layers away from the surface. If there is no water between the bound peptide and the lipids, perhaps a “no-slip” boundary condition results in a relatively slow diffusion. Faxen's law (54) predicts that the drag exerted by a surface on a sphere extends for several diameters from the surface. The molecular roughness of the surface, as revealed by MD simulations and experiments (55,56), presumably also plays a role in slowing the diffusion constant of the membrane-bound peptides.
Why does incorporating PIP2 into the membrane slow the diffusion of membrane-bound peptides? Previous studies show that binding of basic and basic/hydrophobic peptides redistributes or sequesters polyvalent acidic lipids such as PIP2 in both PC/PIP2 (8) and PC/PS/PIP2 membranes (9). Atomic models show that the sequestered PIP2 produces an “electrostatic well” at the binding site (8,9). Our results show that basic and basic/hydrophobic peptides bound to PC/PIP2 or PC/PS/PIP2 membranes have diffusion coefficients comparable to those of lipids (Table 3). To diffuse laterally at a rate faster than lipids, the peptide would have to desorb from the electrostatic well, a process that would require significant energy. In contrast, a peptide adsorbed to a PC/PS membrane experiences an essentially uniform electrostatic potential and can thus diffuse laterally without significant electrostatic energy cost.
PIP2 sequestration and raft formation on the inner leaflet of a plasma membrane
The existence of cholesterol-enriched rafts in cell membranes remains highly controversial and several recent reviews concluded that the evidence supporting raft formation is “not yet compelling” (57–60). Formation of rafts in the cytoplasmic leaflet is particularly problematical: Silvius (61) showed that membranes composed of physiological levels of cholesterol and other lipids found in the cytoplasmic leaflet do not form coexisting lipid domains. Munro (57) concluded that “there is as yet no clear evidence for a mechanism for the formation of lipid domains in the inner leaflet of the plasma membrane.” Anderson and Jacobson (62), however, have suggested that proteins may nucleate raft formation by attracting lipid shells enriched in cholesterol. The work reported here, together with earlier structural studies (13–17), suggests a mechanism by which basic/hydrophobic clusters on proteins may attract shells of lipids enriched in cholesterol. MARCKS, the EGFR, and gravin all contain basic/hydrophobic clusters that may interact with the plasma membrane.
There is indirect evidence that MARCKS (63,64), the EGFR (65–67), and gravin (68) associate with noncaveolar cholesterol-enriched lipid rafts. The hydrophobic Phe residues in the MARCKS basic effector domain insert into the membrane to the level of the acyl chains, dragging the adjacent residues into the polar headgroup region (see Fig. 1 of Gambhir et al. (9) or Fig. 3 a of McLaughlin and Murray (12)). This increases the local lateral surface pressure, which could attract lipids with a small polar headgroup, such as cholesterol, and nucleate raft formation. (McIntosh and Simon (69) and Lee (70) review authoritatively how lipid properties can effect membrane lateral organization and the effects of lateral pressure profiles in membranes.) This could occur, however, only if basic/hydrophobic clusters on proteins diffuse sufficiently slowly to allow cholesterol sequestration. One way to retard lateral diffusion of the protein is for a basic/hydrophobic cluster to bind PIP2; our results suggest this could reduce lateral diffusion of the cluster to a value that would allow sequestration of other lipids. (Cross-linking or aggregration could also slow lateral diffusion: caveolin, which also contains a basic/hydrophobic cluster, forms self-aggregates in caveolae.) In agreement with this suggestion, noncaveolar cholesterol-enriched rafts in biological membranes appear to contain an enhanced mol fraction of PIP2 (71). Sid Simon and Tom McIntosh (Duke University, personal communication, 2006) suggested another mechanism by which basic/hydrophobic clusters might nucleate the formation of rafts. They point out that when a basic/hydrophobic cluster penetrates the polar headgroup region of the cytoplasmic leaflet, the thickness of this leaflet immediately below the cluster will decrease (72) as the lipid chains curl into the region to avoid forming a vacuum. This locally slimmed-down region of the cytoplasmic leaflet should attract lipids with longer-than-average chains, e.g., sphingomyelin, to the corresponding region of the extracellular leaflet; sphingomyelin could then nucleate local raft formation on the extracellular leaflet. This Simon/McIntosh mechanism could act in parallel with the lateral pressure mechanism. The hypothesis that basic/hydrophobic clusters could nucleate raft formation is highly speculative, but could be tested by experimental approaches such as magic angle spinning NMR, fluorescence, and diffraction.
Acknowledgments
Note added in proof: Our observation that small unstructured basic and basic/hydrophobic peptides diffuse more rapidly than lipids when bound to PC/PS vesicles cannot be extrapolated to all amphipathic helical peptides. Frey and Tamm (73) showed that a cytochrome c oxidase subunit IV signal peptide diffuses ∼1.5-fold more rapidly than lipids when bound to POPC membranes but ∼1.5-fold less rapidly than lipids when bound more strongly to 4:1 POPC/POPG membranes. The degree to which these amphipathic, helical, membrane-bound peptides laterally concentrate monovalent acidic lipids has apparently not been investigated to date.
We thank John Nagle for a valuable theoretical contribution to the interpretation of Fig. 2 B; Avinon Ben-Shaul, Nir Ben-Tal, Sylvio May, Tom McIntosh, Diana Murray, Kim Sharp, and Sid Simon for helpful discussions; and William Woturski for excellent technical input with respect to the FCS measurements.
This work was supported by National Institutes of Health grant R-37 GM24971 to S.M. and Deutsche Forschungsgemeinschaft SFB 563-A4 to J.R.
Urszula Golebiewska and Alok Gambhir contributed equally to this article.
References
- 1.Sigal, C. T., W. Zhou, C. A. Buser, S. McLaughlin, and M. D. Resh. 1994. Amino-terminal basic residues of Src mediate membrane binding through electrostatic interaction with acidic phospholipids. Proc. Natl. Acad. Sci. USA. 91:12253–12257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buser, C. A., C. T. Sigal, M. D. Resh, and S. McLaughlin. 1994. Membrane binding of myristylated peptides corresponding to the NH2 terminus of Src. Biochemistry. 33:13093–13101. [DOI] [PubMed] [Google Scholar]
- 3.Hancock, J. F., K. Cadwallader, H. Paterson, and C. J. Marshall. 1991. A CAAX or a CAAL motif and a second signal are sufficient for plasma membrane targeting of ras proteins. EMBO J. 10:4033–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghomashchi, F., X. Zhang, L. Liu, and M. H. Gelb. 1995. Binding of prenylated and polybasic peptides to membranes: affinities and intervesicle exchange. Biochemistry. 34:11910–11918. [DOI] [PubMed] [Google Scholar]
- 5.Leventis, R., and J. R. Silvius. 1998. Lipid-binding characteristics of the polybasic carboxy-terminal sequence of K-Ras4B. Biochemistry. 37:7640–7648. [DOI] [PubMed] [Google Scholar]
- 6.McLaughlin, S., and A. Aderem. 1995. The myristoyl-electrostatic switch: a modulator of reversible protein-membrane interactions. Trends Biochem. Sci. 20:272–276. [DOI] [PubMed] [Google Scholar]
- 7.Okeley, N. M., and M. H. Gelb. 2004. A designed probe for acidic phospholipids reveals the unique enriched anionic character of the cytosolic face of the mammalian plasma membrane. J. Biol. Chem. 279:21833–21840. [DOI] [PubMed] [Google Scholar]
- 8.Wang, J., A. Gambhir, G. Hangyas-Mihalyne, D. Murray, U. Golebiewska, and S. McLaughlin. 2002. Lateral sequestration of phosphatidylinositol 4,5-bisphosphate by the basic effector domain of myristoylated alanine-rich C kinase substrate is due to nonspecific electrostatic interactions. J. Biol. Chem. 277:34401–34412. [DOI] [PubMed] [Google Scholar]
- 9.Gambhir, A., G. Hangyas-Mihalyne, I. Zaitseva, D. S. Cafiso, J. Wang, D. Murray, S. N. Pentyala, S. O. Smith, and S. McLaughlin. 2004. Electrostatic sequestration of PIP2 on phospholipid membranes by basic/aromatic regions of proteins. Biophys. J. 86:2188–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McLaughlin, S., S. O. Smith, M. J. Hayman, and D. Murray. 2005. An electrostatic engine model for autoinhibition and activation of the epidermal growth factor receptor (EGFR/ErbB) family. J. Gen. Physiol. 126:41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang, J., A. Gambhir, S. McLaughlin, and D. Murray. 2004. A computational model for the electrostatic sequestration of PI(4,5)P2 by membrane-adsorbed basic peptides. Biophys. J. 86:1969–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McLaughlin, S., and D. Murray. 2005. Plasma membrane phosphoinositide organization by protein electrostatics. Nature. 438:605–611. [DOI] [PubMed] [Google Scholar]
- 13.Qin, Z., and D. S. Cafiso. 1996. Membranes structure of protein kinase C and calmodulin binding domain of myristoylated alanine rich C kinase substrate determined by site-directed spin labeling. Biochemistry. 35:2917–2925. [DOI] [PubMed] [Google Scholar]
- 14.Victor, K., J. Jacob, and D. S. Cafiso. 1999. Interactions controlling the membrane binding of basic protein domains: phenylalanine and the attachment of the myristoylated alanine-rich C-kinase substrate protein to interfaces. Biochemistry. 38:12527–12536. [DOI] [PubMed] [Google Scholar]
- 15.Wang, J., A. Arbuzova, G. Hangyas-Mihalyne, and S. McLaughlin. 2001. The effector domain of myristoylated alanine-rich C kinase substrate binds strongly to phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 276:5012–5019. [DOI] [PubMed] [Google Scholar]
- 16.Ellena, J. F., M. C. Burnitz, and D. S. Cafiso. 2003. Location of the myristoylated alanine-rich C-kinase substrate (MARCKS) effector domain in negatively charged phospholipid bicelles. Biophys. J. 85:2442–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang, W., E. Crocker, S. McLaughlin, and S. O. Smith. 2003. Binding of peptides with basic and aromatic residues to bilayer membranes: phenylalanine in the myristoylated alanine-rich C kinase substrate effector domain penetrates into the hydrophobic core of the bilayer. J. Biol. Chem. 278:21459–21466. [DOI] [PubMed] [Google Scholar]
- 18.Cho, W., and R. V. Stahelin. 2005. Membrane-protein interactions in cell signaling and membrane trafficking. Annu. Rev. Biophys. Biomol. Struct. 34:119–151. [DOI] [PubMed] [Google Scholar]
- 19.Taniguchi, H., and S. Manenti. 1993. Interaction of myristoylated alanine-rich protein kinase C substrate (MARCKS) with membrane phospholipids. J. Biol. Chem. 268:9960–9963. [PubMed] [Google Scholar]
- 20.Kim, J., T. Shishido, X. Jiang, A. Aderem, and S. McLaughlin. 1994. Phosphorylation, high ionic strength, and calmodulin reverse the binding of MARCKS to phospholipid vesicles. J. Biol. Chem. 269:28214–28219. [PubMed] [Google Scholar]
- 21.Swierczynski, S. L., and P. J. Blackshear. 1995. Membrane association of the myristoylated alanine-rich C kinase substrate (MARCKS) protein. J. Biol. Chem. 270:13436–13445. [DOI] [PubMed] [Google Scholar]
- 22.Ohmori, S., N. Sakai, Y. Shirai, H. Yamamoto, E. Miyamoto, N. Shimizu, and N. Saito. 2000. Importance of protein kinase C targeting for the phosphorylation of its substrate, myristoylated alanine-rich C-kinase substrate. J. Biol. Chem. 275:26449–26457. [DOI] [PubMed] [Google Scholar]
- 23.Fivaz, M., and T. Meyer. 2005. Reversible intracellular translocation of KRas but not HRas in hippocampal neurons regulated by Ca2+/calmodulin. J. Cell Biol. 170:429–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sidhu, R. S., R. R. Clough, and R. P. Bhullar. 2003. Ca2+/calmodulin binds and dissociates K-RasB from membrane. Biochem. Biophys. Res. Commun. 304:655–660. [DOI] [PubMed] [Google Scholar]
- 25.Bivona, T. G., S. E. Quatela, B. O. Bodemann, I. M. Ahearn, M. J. Soskis, A. Mor, J. Miura, H. H. Wiener, L. Wright, S. G. Saba, D. Yim, A. Fein, et al. 2006. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol. Cell. 21:481–493. [DOI] [PubMed] [Google Scholar]
- 26.May, S., D. Harries, and A. Ben-Shaul. 2000. Lipid demixing and protein-protein interactions in the adsorption of charged proteins on mixed membranes. Biophys. J. 79:1747–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haleva, E., N. Ben-Tal, and H. Diamant. 2004. Increased concentration of polyvalent phospholipids in the adsorption domain of a charged protein. Biophys. J. 86:2165–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mbamala, E. C., A. Ben-Shaul, and S. May. 2005. Domain formation induced by the adsorption of charged proteins on mixed lipid membranes. Biophys. J. 88:1702–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tzlil, S., and A. Ben-Shaul. 2005. Flexible charged macromolecules on mixed fluid lipid membranes: theory and Monte Carlo simulations. Biophys. J. 89:2972–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Montich, G., S. Scarlata, S. McLaughlin, R. Lehrmann, and J. Seelig. 1993. Thermodynamic characterization of the association of small basic peptides with membranes containing acidic lipids. Biochim. Biophys. Acta. 1146:17–24. [DOI] [PubMed] [Google Scholar]
- 31.Kim, J., M. Mosior, L. A. Chung, H. Wu, and S. McLaughlin. 1991. Binding of peptides with basic residues to membranes containing acidic phospholipids. Biophys. J. 60:135–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ben-Tal, N., B. Honig, R. M. Peitzsch, G. Denisov, and S. McLaughlin. 1996. Binding of small basic peptides to membranes containing acidic lipids: theoretical models and experimental results. Biophys. J. 71:561–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rusu, L., A. Gambhir, S. McLaughlin, and J. Radler. 2004. Fluorescence correlation spectroscopy studies of peptide and protein binding to phospholipid vesicles. Biophys. J. 87:1044–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arbuzova, A., L. Wang, J. Wang, G. Hangyas-Mihalyne, D. Murray, B. Honig, and S. McLaughlin. 2000. Membrane binding of peptides containing both basic and aromatic residues. Experimental studies with peptides corresponding to the scaffolding region of caveolin and the effector region of MARCKS. Biochemistry. 39:10330–10339. [DOI] [PubMed] [Google Scholar]
- 35.Buser, C. A., and S. McLaughlin. 1998. Ultracentrifugation technique for measuring the binding of peptides and proteins to sucrose-loaded phospholipid vesicles. Methods Mol. Biol. 84:267–281. [DOI] [PubMed] [Google Scholar]
- 36.Akashi, K., H. Miyata, H. Itoh, and K. Kinosita, Jr. 1996. Preparation of giant liposomes in physiological conditions and their characterization under an optical microscope. Biophys. J. 71:3242–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moscho, A., O. Orwar, D. T. Chiu, B. P. Modi, and R. N. Zare. 1996. Rapid preparation of giant unilamellar vesicles. Proc. Natl. Acad. Sci. USA. 93:11443–11447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bagatolli, L. A., T. Parasassi, and E. Gratton. 2000. Giant phospholipid vesicles: comparison among the whole lipid sample characteristics using different preparation methods: a two photon fluorescence microscopy study. Chem. Phys. Lipids. 105:135–147. [DOI] [PubMed] [Google Scholar]
- 39.Peitzsch, R. M., and S. McLaughlin. 1993. Binding of acylated peptides and fatty acids to phospholipid vesicles: pertinence to myristoylated proteins. Biochemistry. 32:10436–10443. [DOI] [PubMed] [Google Scholar]
- 40.Hess, S. T., and W. W. Webb. 2002. Focal volume optics and experimental artifacts in confocal fluorescence correlation spectroscopy. Biophys. J. 83:2300–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schwille, P., U. Haupts, S. Maiti, and W. W. Webb. 1999. Molecular dynamics in living cells observed by fluorescence correlation spectroscopy with one- and two-photon excitation. Biophys. J. 77:2251–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zar, J. H. 1998. Biostatistical Analysis. Prentice Hall, Englewood Cliffs, NJ.
- 43.Findlay, E. J., and P. G. Barton. 1978. Phase behavior of synthetic phosphatidylglycerols and binary mixtures with phosphatidylcholines in the presence and absence of calcium ions. Biochemistry. 17:2400–2405. [DOI] [PubMed] [Google Scholar]
- 44.Lewis, R. N., Y. P. Zhang, and R. N. McElhaney. 2005. Calorimetric and spectroscopic studies of the phase behavior and organization of lipid bilayer model membranes composed of binary mixtures of dimyristoylphosphatidylcholine and dimyristoylphosphatidylglycerol. Biochim. Biophys. Acta. 1668:203–214. [DOI] [PubMed] [Google Scholar]
- 45.Derzko, Z., and K. Jacobson. 1980. Comparative lateral diffusion of fluorescent lipid analogues in phospholipid multibilayers. Biochemistry. 19:6050–6057. [DOI] [PubMed] [Google Scholar]
- 46.Hoekstra, D. 1982. Fluorescence method for measuring the kinetics of Ca2+-induced phase separations in phosphatidylserine-containing lipid vesicles. Biochemistry. 21:1055–1061. [DOI] [PubMed] [Google Scholar]
- 47.Schwille, P., J. Korlach, and W. W. Webb. 1999. Fluorescence correlation spectroscopy with single-molecule sensitivity on cell and model membranes. Cytometry. 36:176–182. [DOI] [PubMed] [Google Scholar]
- 48.Korlach, J., P. Schwille, W. W. Webb, and G. W. Feigenson. 1999. Characterization of lipid bilayer phases by confocal microscopy and fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. USA. 96:8461–8466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kahya, N., D. Scherfeld, K. Bacia, B. Poolman, and P. Schwille. 2003. Probing lipid mobility of raft-exhibiting model membranes by fluorescence correlation spectroscopy. J. Biol. Chem. 278:28109–28115. [DOI] [PubMed] [Google Scholar]
- 50.Zhang, L., and S. Granick. 2005. Slaved diffusion in phospholipid bilayers. Proc. Natl. Acad. Sci. USA. 102:9118–9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wagner, M. L., and L. K. Tamm. 2001. Reconstituted syntaxin1A/SNAP25 interacts with negatively charged lipids as measured by lateral diffusion in planar supported bilayers. Biophys. J. 81:266–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arbuzova, A., J. Wang, D. Murray, J. Jacob, D. S. Cafiso, and S. McLaughlin. 1997. Kinetics of interaction of the myristoylated alanine-rich C kinase substrate, membranes, and calmodulin. J. Biol. Chem. 272:27167–27177. [DOI] [PubMed] [Google Scholar]
- 53.Kleinschmidt, J. H., and D. Marsh. 1997. Spin-label electron spin resonance studies on the interactions of lysine peptides with phospholipid membranes. Biophys. J. 73:2546–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Svoboda, K., and S. M. Block. 1994. Biological applications of optical forces. Annu. Rev. Biophys. Biomol. Struct. 23:247–285. [DOI] [PubMed] [Google Scholar]
- 55.Tieleman, D. P., S. J. Marrink, and H. J. C. Berendsen. 1997. A computer perspective of membranes: molecular dynamics studies of lipid bilayer systems. Biochim. Biophys. Acta. 1331:235–270. [DOI] [PubMed] [Google Scholar]
- 56.White, S. H., and W. C. Wimley. 1998. Hydrophobic interactions of peptides with membrane interfaces. Biochim. Biophys. Acta. 1376:339–352. [DOI] [PubMed] [Google Scholar]
- 57.Munro, S. 2003. Lipid rafts: elusive or illusive? Cell. 115:377–388. [DOI] [PubMed] [Google Scholar]
- 58.Edidin, M. 2003. The state of lipid rafts: from model membranes to cells. Annu. Rev. Biophys. Biomol. Struct. 32:257–283. [DOI] [PubMed] [Google Scholar]
- 59.McMullen, T. P. W., R. N. A. H. Lewis, and R. N. McElhaney. 2004. Cholesterol-phospholipid interactions, the liquid-ordered phase and lipid rafts in model and biological membranes. Curr. Opin. Colloid Interface Sci. 8:459–468. [Google Scholar]
- 60.Nichols, B. 2005. Without a raft. Nature. 436:638–639. [DOI] [PubMed] [Google Scholar]
- 61.Silvius, J. R. 2003. Role of cholesterol in lipid raft formation: lessons from lipid model systems. Biochim. Biophys. Acta. 1610:174–183. [DOI] [PubMed] [Google Scholar]
- 62.Anderson, R. G. W., and K. Jacobson. 2002. A role for lipid shells in targeting proteins to caveolae, rafts, and other lipid domains. Science. 296:1821–1825. [DOI] [PubMed] [Google Scholar]
- 63.Laux, T., K. Fukami, M. Thelen, T. Golub, D. Frey, and P. Caroni. 2000. GAP43, MARCKS, and CAP23 modulate PI(4,5)P2 at plasmalemmal rafts, and regulate cell cortex actin dynamics through a common mechanism. J. Cell Biol. 149:1455–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Caroni, P. 2001. Actin cytoskeleton regulation through modulation of PI(4,5)P2 rafts. EMBO J. 20:4332–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carpenter, G. 2000. The EGF receptor: a nexus for trafficking and signaling. Bioessays. 22:697–707. [DOI] [PubMed] [Google Scholar]
- 66.Pike, L. J. 2005. Growth factor receptors, lipid rafts and caveolae: An evolving story. Biochim. Biophys. Acta. 1746:260–273. [DOI] [PubMed] [Google Scholar]
- 67.Orr, G., D. Hu, S. Ozcelik, L. K. Opresko, H. S. Wiley, and S. D. Colson. 2005. Cholesterol dictates the freedom of EGF receptors and HER2 in the plane of the membrane. Biophys. J. 89:1362–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Streb, J. W., and J. M. Miano. 2005. Cross-species sequence analysis reveals multiple charged residue-rich domains that regulate nuclear/cytoplasmic partitioning and membrane localization of a kinase anchoring protein 12 (SSeCKS/gravin). J. Biol. Chem. 280:28007–28014. [DOI] [PubMed] [Google Scholar]
- 69.McIntosh, T. J., and S. A. Simon. 2006. Roles of bilayer material properties in function and distribution of membrane proteins. Annu. Rev. Biophys. Biomol. Struct. 35:177–198. [DOI] [PubMed] [Google Scholar]
- 70.Lee, A. G. 2004. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta. 1666:62–87. [DOI] [PubMed] [Google Scholar]
- 71.Pike, L. J. 2004. Lipid rafts: heterogeneity on the high seas. Biochem. J. 378:281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ebihara, L., J. E. Hall, R. C. MacDonald, T. J. McIntosh, and S. A. Simon. 1979. Effect of benzyl alcohol on lipid bilayers. A comparisons of bilayer systems. Biophys. J. 28:185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Frey, S., and L. K. Tamm. 1990. Membrane insertion and lateral diffusion of fluorescence-labelled cytochrome c oxidase subunit IV signal peptide in charged and uncharged phospholipid bilayers. Biochem. J. 272:713–719. [DOI] [PMC free article] [PubMed] [Google Scholar]