

Abstract
Objective
The objective of the study was to evaluate the role of endothelin-1 and platelet-activating factor in ischemia/reperfusion-induced fetal growth restriction in the rat.
Study design
On day 17 of gestation, the right uterine and ovarian arteries were occluded for 30 minutes in experimental but not sham-operated rats. All rats received endothelin receptor A antagonist, A-127722 (10 mg/kg per day), platelet-activating factor antagonist, WEB-2086 (1 mg/kg), or vehicle. On gestational day 21, litter size, fetal viability, and fetal and placental weights were recorded. Reverse transcription–polymerase chain reaction for phospholipase A2-IIA and preproendothelin-1 messenger ribonucleic acid was performed on uterus and placentas from each uterine horn. Groups were compared statistically by analysis of variance.
Results
Ischemia/reperfusion reduced fetal weights, in both the ischemic horn and the nonischemic horn (P < .001). Antagonism of either endothelin receptor A or platelet-activating factor normalized fetal growth in both horns. Neither placental weight nor the incidence of fetal demise was affected by ischemia/reperfusion. Phospholipase A2-IIA and preproendothelin-1 messenger ribonucleic acid expression did not differ between right and left uterine horns in any group. Uterine and placental tissues in the ischemia/reperfusion group exhibited increased phospholipase A2-IIA (P < .01) but not preproendothelin-1.
Conclusion
Endothelin-1 and platelet-activating factor are both important mediators in the pathophysiology of ischemia/reperfusion-induced fetal growth restriction in the rat, contributing to the fetal growth restriction observed in both the ischemic and nonischemic horns. Antagonism of either mediator produces normal fetal growth in this model of fetal growth restriction.
Keywords: Endothelin, Platelet-activating factor, Ischemia/reperfusion, Fetal growth restriction, Rat
Fetal growth restriction (FGR) remains a common and important cause of perinatal morbidity and mortality.1–3 This is particularly true when growth restriction complicates a pregnancy remote from term.4 Suboptimal uteroplacental perfusion is the most common identifiable cause of FGR. Histologic evaluation of placentas from pregnancies complicated by FGR commonly demonstrates evidence of placental ischemia and/or infarction. The severity of fetal growth restriction correlates with number and severity of ischemic lesions observed.5 The distribution of these ischemic findings implicates the limitation of blood flow through the maternal spiral arteries.6 Because the redundancy of uteroplacental vessels allows for collateral blood flow to placental units, the presence of an infarct suggests not only the obstruction of a uteroplacental vessel but also compromise of collateral blood flow.7 Evidence for redistribution of blood flow within the placental lobules is commonly observed, suggesting that the dynamics of ischemia/reperfusion (IR) are at work within the substance of the placenta.8 The mechanisms regulating IR in the placenta have not been elucidated.
Animal models have been developed to evaluate the role of uterine ischemia in FGR. Wigglesworth9 originally demonstrated that ligation of the uterine artery in the rat results in fetal growth restriction. More recently temporary uterine ischemia by unilateral occlusion of uterine perfusion followed by reperfusion of the ischemic horn10 has been shown to result in FGR in both uterine horns, not just the one subjected to ischemic insult.11 This model is more analogous to the pathophysiology occurring in human FGR than the previous uterine artery ligation model, particularly because it reveals the impact of IR on regional blood flow in the placenta. The occurrence of FGR even after relatively brief occlusion of uterine blood flow suggests that it is not only the restriction of blood flow per se that results in a compromise of fetal growth but the long-term consequences of IR that is at play. Furthermore, the restriction of fetal growth in the contralateral uterine horn suggests that circulating factors or mediators are involved.
Endothelin-1 (ET-1) is a vasoactive mediator produced primarily by endothelial cells. ET-1 is the most potent endogenous vasoconstrictor known and, along with its receptors, is expressed in the placenta. ET-1 infusion in maternal rats results in FGR independent of maternal hypertension.12 ET-1 also plays a primary role in the pathophysiology of both nitric oxide synthase inhibition as well as hypoxia-induced FGR.13,14 Ischemia is a potent stimulus to ET-1 production. ET-1 has previously been shown to play an important role in the pathophysiology of IR injury in the lung.15,16 Consequently, we hypothesize that ET-1 may also play a role in the pathophysiology of IR-induced FGR.
Platelet-activating factor (PAF) is a proinflammatory mediator that also has vasoactive properties by virtue of its effect on vascular smooth muscle. We have previously demonstrated that maternal PAF infusion in the rat results in FGR.17 PAF has previously been shown to act synergistically with ET-1 to decrease local perfusion in other models, including bacteremia-induced pulmonary hypertension in the lung,18 acute pancreatitis,19 and IR in transplanted lung.16 Ishimoto et al20 demonstrated that PAF plays a role in the pathophysiology of IR-induced FGR, ameliorating FGR by administration of a PAF receptor antagonist. The relative contributions of ET-1 and PAF in this model have not been evaluated. We hypothesize that PAF, acting synergistically with ET-1, contributes to the pathophysiology of IR-induced FGR. The purpose of this study was to evaluate the significance of ET-1 and PAF in IR-induced FGR.
Material and methods
Endothelin receptor and PAF receptor antagonists
A-127722 (provided by Abbott Laboratories, Abbott Park, IL) is a nonpeptide compound with a high affinity for ETA receptors (Ki = 0.069 nM) and a low affinity for ETB receptors (Ki = 139 nM). It has a 35% bio-availability in rats and a plasma half-life of 3.5 hours.21 WEB-2086 (provided by Boehringer-Ingelheim, Ingelheim, Germany) is a thieno-triazolodiazepine PAF receptor antagonist with a median inhibitory concentration of 170 nM and a plasma half-life of 1 hour.22–24
Animals
Nonpregnant Sprague-Dawley female and male rats were purchased from Harlan Laboratories (Madison, WI), housed in the Evanston Northwestern Healthcare Research Institute Center for Comparative Medicine, maintained in 12-hour light/12-hour dark cycles, and allowed free access to a standard laboratory rodent diet and water. Animal care and the conduct of all experiments were in accord with guidelines approved by the Evanston Northwestern Healthcare Research Institute Institutional Animal Care and Use Committee. Rats were bred at an age of 11 to 20 weeks and a weight of 225 to 250 g. Thirty-six maternal rats were used for this study, 6 in each experimental group.
IR
On day 17 of gestation, a laparotomy was performed under general anesthesia consisting of a single intraperitoneal injection of xylazine, ketamine, and acepromazine in combination (8, 40, and 1.3 mg/kg, respectively). A midline abdominal incision was utilized to expose both uterine horns. Vascular clamps were then applied to both the uterine and ovarian arteries of the right uterine horn to totally occlude perfusion. The uterine horns were replaced into the abdominal cavity, with the occlusion clamps in place. After 30 minutes, the vascular clamps were removed. The abdominal incision was closed. Rats in the sham-operated group had the uterine horns exposed and returned to the abdomen, and then, after 30 minutes, the abdomen was closed.
ET and PAF receptor antagonism
The ETA antagonist, A-127722 (10 mg/kg per day), was administered intraperitoneally on days 17 to 21 of gestation via an Alzet osmotic pump (5.1 cm long and 1.4 cm in diameter, Durect Corp, Cupertino, CA) placed into the abdomen during IR surgery (sham-ETA antagonist and IR-ETA antagonist groups). The PAF receptor antagonist, WEB-2086 (1 mg/kg), was administered intravenously into the femoral vein just prior to the IR surgery (sham-PAF antagonist and IR-PAF antagonist groups). Control rats received only vehicle (20% ethyl alcohol, 40% propylene glycol, and 0.04 M NaOH in H2O, intraperitoneally by osmotic pump and saline intravenously).
Pregnancy outcome
On gestational day 21, hysterotomies were performed, litter size was noted, fetal viability was determined for each pup, and fetal and placental weights were obtained for each live pup.
Prepro-ET-1 and phospholipase A2 (PLA2) expression analysisreverse transcription–polymerase chain reaction (PCR)
Placental and uterine tissues were frozen in liquid nitrogen and stored at −80°C. Total ribonucleic acid (RNA) was extracted using RNA STAT-60 (TEL-TEST, Friendswood, TX), according to the manufacturer’s instructions. The concentration of RNA was determined by absorbance at 260 nm, and the purity was checked by the 260:280 nm ratio (greater than 1.8). RNA integrity was verified by electrophoresis in a 1% agarose gel. For each sample, 3 μg of total RNA were reverse transcribed at 37°C for 1 hour in a total of 20 μL of reaction mixture (50 mM Tris-HCl, 75 mM potassium chloride, 2 mM magnesium chloride, 10 mM dithiothreitol, 1.25 mM of each deoxynucleotide-triphosphate, 7.5 pM random hexamer, 1 U/μL RNasin [RNase inhibitor], and 10 U/μL Moloney-murine leukemia virus reverse transcriptase [Invitrogen, Carlsbad, CA]).
Complementary deoxyribonucleic acid (DNA) was amplified by PCR with 0.1 U/μL AmpliTaq DNA polymerase on a GeneAmp 5700 real-time sequence detector (Applied Biosystems, Foster City, CA) in a total volume of 50 μL consisting of 1.0 μL reverse transcription product, 10 mM Tris-HCl, 50 mM potassium chloride, 2 mM magnesium chloride, 0.1 mM of each deoxynucleotide-triphosphate, and 1.0 μM of each primer.
The primer sequences were, for rat prepro-ET-1, sense 5′-GACCAGCGTCCTTGTTCCAA-3′ and antisense 5′-TTGCTACCAGCGGATGCAA-3′ for rat PL A2-IIA, sense 5′-CCGTCTGGAGAAACGTGGAT-3′ and antisense 5′-GTTCCGGGCAAAACATTCAG-3′. TaqMan MGB probes were, for rat prepro-ET-1, 6FAM-TCCAAGAGAGGTTGAGGTGT-MGBNFQ and for rat PLA2-IIA, 6FAM-TGGCACAAAGTTTC-MGBNFQ. The reaction mixtures were heated at 50°C for 2 minutes to allow UNG to degrade dUTP-containing double-stranded DNA and then heated at 95°C for 10 minutes and immediately carried through 60 cycles of PCR with 15 seconds denaturation at 95°C, 20 seconds annealing at 60°C, and 40 seconds extension at 72°C. Quantification was on the basis of standard curves generated from copy number standards. Control ribosomal RNA reagents (Applied Biosystems) were used to normalized all results, which are expressed as the ratio of a specific messenger RNA (mRNA)/ribosomal RNA.
Statistical analysis
Results are presented as mean ± SEM. Statistical comparisons among groups were made using an analysis of variance (ANOVA) followed by a post hoc Newman-Keuls test or a Kruskal-Wallis nonparametric ANOVA with post hoc Dunn’s test, as appropriate. All statistical tests were 2-tailed and results were considered statistically significant at P < .05.
Results
Fetal weights were reduced in response to IR, in both the ischemic uterine horn and the contralateral, nonischemic horn (P < .001, compared with all other groups). With IR, treatment with either ETA antagonist or PAF receptor antagonist resulted in normalization of fetal growth in both uterine horns (Figure 1). In the absence of IR, neither ETA antagonism alone nor PAF receptor antagonism alone had any impact on fetal weight.
Figure 1.
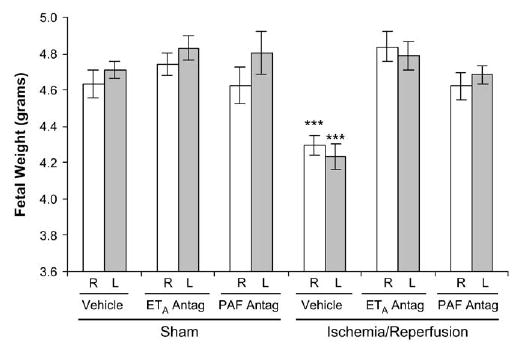
Fetal weights on gestational day 21 in the right (R, white bars) and left (L, gray bars) uterine horns 4 days after maternal sham operation or 30-minute IR of the right uterine horn. Fetal weights in both uterine horns of IR rats were significantly lower than those in all other groups. ETA antagonism and PAF receptor antagonism each prevented fetal growth restriction. Data are mean ± SEM. ***, P < .001 versus all other groups.
Placental weight was not affected by IR. Placental weights in rats treated only with the ETA antagonist were not significantly different from the control (sham operated, vehicle treated) rats but were significantly lower than those in the IR group (P < .05; Figure 2). This reduced placental size in the ETA antagonist-treated rats had no impact on fetal growth in this group (Figures 1 and 2).
Figure 2.
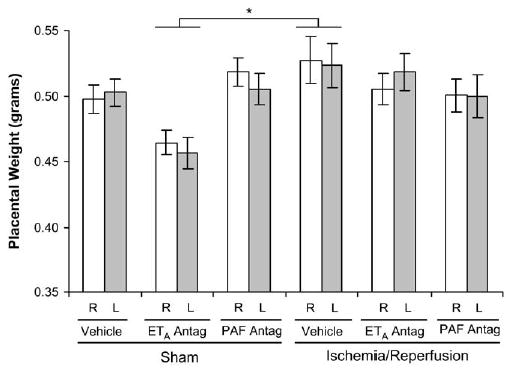
Placental weights on gestational day 21 in the right (R, white bars) and left (L, gray bars) uterine horns 4 days after maternal sham operation or 30-minute IR of the right uterine horn. Placental weights in ETA antagonist-treated rats were significantly reduced, compared with those in vehicle-treated, IR rats. None of the placental weights were significantly different from those in the sham/vehicle-treated rats. Data are mean ± SEM. *, P < .05 for sham/ETA antagonist versus IR/vehicle groups.
Litter size and numbers of live pups in each litter did not differ among the groups or between right and left uterine horns in any individual group (Table). Fetal demise occurred with similar frequencies in each of the groups in the study.
Table.
Litter size and fetal viability on gestational day 21, 4 days after maternal sham operation or 30-minute IR of the right uterine horn
| Sham
|
IR
|
|||||
|---|---|---|---|---|---|---|
| Vehicle | A-127722 | WEB-2086 | Vehicle | A-127722 | WEB-2086 | |
| Right uterine horn | ||||||
| Litter size | 7.0 ± 1.2 | 8.0 ± 0.7 | 7.2 ± 0.6 | 6.4 ± 0.8 | 5.5 ± 0.9 | 5.4 ± 0.6 |
| Live births | 6.5 ± 1.1 | 7.5 ± 0.7 | 6.3 ± 0.8 | 5.4 ± 0.6 | 5.5 ± 0.9 | 5.1 ± 0.5 |
| Fetal deaths | 0.5 ± 0.2 | 0.5 ± 0.3 | 0.8 ± 0.5 | 1.1 ± 0.7 | 0.5 ± 0.5 | 0.3 ± 0.2 |
| Left uterine horn | ||||||
| Litter size | 6.7 ± 1.0 | 6.8 ± 0.7 | 5.7 ± 1.1 | 6.6 ± 0.8 | 7.2 ± 1.2 | 6.1 ± 0.8 |
| Live births | 6.2 ± 1.1 | 6.1 ± 0.9 | 5.0 ± 1.0 | 5.9 ± 1.2 | 7.2 ± 1.2 | 6.0 ± 0.8 |
| Fetal deaths | 0.5 ± 0.2 | 0.6 ± 0.3 | 0.7 ± 0.7 | 0.9 ± 0.4 | 0.3 ± 0.3 | 0.1 ± 0.1 |
Data are mean ± SEM, n = 6 litters/group. There were no statistically significant differences among these groups by ANOVA.
The transcription of both PLA2-IIA and prepro-ET-1 mRNA exhibited considerable variability within each group at 4 days after ischemia. There was no difference in the transcription of either PLA2-IIA or prepro-ET-1 mRNA between the right and left uterine horn in any of the experimental groups; therefore, the results are presented as single values for each tissue and group. Uterine PLA2-IIA expression was increased in the IR group (P < .001), compared with all other groups (Figure 3). There was a trend toward increased prepro-ET-1 mRNA expression in the IR group, compared with the sham-operated, vehicle-treated control group (P < .09; Figure 4). Placental PLA2-IIA expression increased in the IR group, compared with all other groups (P < .01), but prepro-ET-1 expression was not significantly different among any of the groups (Figures 5 and 6).
Figure 3.
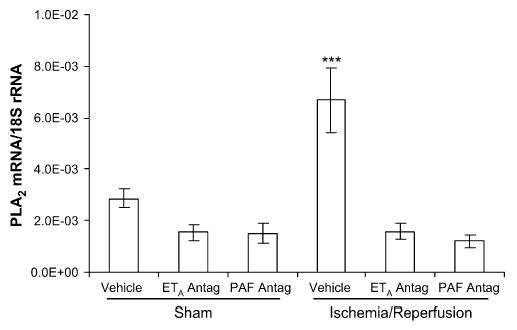
Uterine PLA2-IIA mRNA expression on gestational day 21, 4 days after maternal sham operation or 30-minute IR of the right uterine horn. PLA2-IIA expression was significantly increased in the IR group, compared with all other groups. ETA antagonism and PAF receptor antagonism each prevented the increase in PLA2-IIA mRNA. Data are mean ± SEM. ***, P < .001 versus all other groups.
Figure 4.
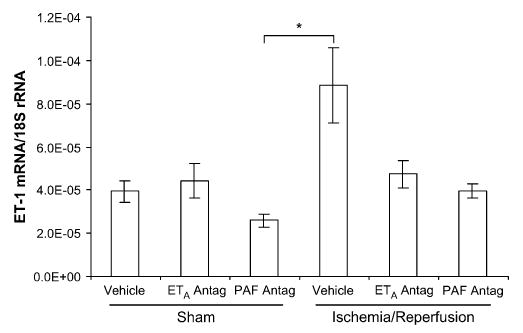
Uterine prepro-ET-1 mRNA expression on gestational day 21, 4 days after maternal sham operation or 30-minute IR of the right uterine horn. Prepro-ET-1 expression was significantly different only between the sham-operated/PAF antagonist-treated rats and the IR/vehicle-treated rats. Data are mean ± SEM. *, P < .05.
Figure 5.
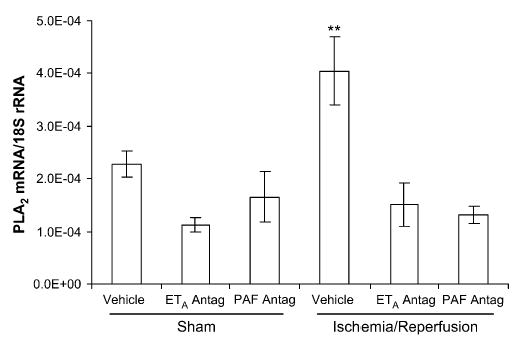
Placental PLA2-IIA mRNA expression on gestational day 21, 4 days after maternal sham operation or 30-minute IR of the right uterine horn. PLA2-IIA expression was significantly increased in the IR group, compared with all other groups. ETA antagonism and PAF receptor antagonism each prevented the increase in PLA2-IIA mRNA. Data are mean ± SEM. **, P < .01 versus all other groups.
Figure 6.
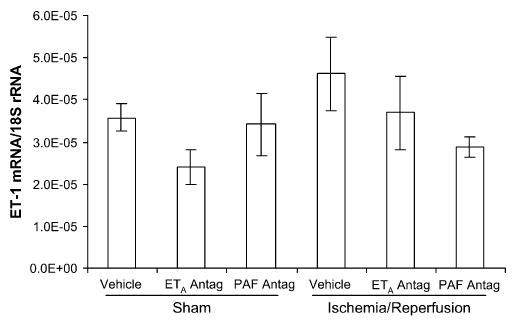
Placental prepro-ET-1 mRNA expression on gestational day 21, 4 days after maternal sham operation or 30-minute IR of the right uterine horn. Prepro-ET-1 expression was not significantly different among any of the groups. Data are mean ± SEM.
Comment
IR injury in the maternal rat leads to fetal growth restriction. The temporary interruption of uterine blood flow to one horn of the rat uterus leads to reduced fetal growth in not only the temporarily ischemic horn but also the contralateral horn. This suggests that the reduction of fetal growth is caused by not only the temporary interruption of blood flow to the uteroplacental unit(s) but also circulating mediators that are produced in response to this ischemic insult. In this study we have demonstrated that both ET-1 and PAF contribute to the pathophysiology of IR-induced FGR.
Antagonism of either ET-1 or PAF results in normalization of fetal growth in this animal model of FGR. PAF receptor antagonism has been shown previously to ameliorate IR-induced FGR.20 Using a different PAF receptor antagonist, we have observed the same result. ETA receptor antagonism ameliorates FGR equally well. There are several possible reasons for the complete efficacy of either ETA or PAF receptor antagonists. Prevention of the vasoconstrictive actions of either of these mediators could result in improved placental perfusion and thereby function. Furthermore, decreased activity of the inflammatory effects of these mediators may also improve placental function. Whether by improving placental perfusion or decreasing inflammation, either of these antagonists improves placental function in the setting of IR sufficiently to normalize fetal growth. Our observation that IR did not have an impact on placental weight is consistent with that of Lewis et al.11 Thus, IR appears to have its primary impact on placental function rather than on placental size, and administration of either antagonist normalizes this function.
ET-1 and PAF may act synergistically to decrease local perfusion and augment the inflammatory response. Stimulation of PLA2 expression and PAF synthesis by ET-1 via the ETA receptor has been shown in a variety of cells and tissues.25–31 PAF and ET have been shown to contribute synergistically to posttransplant lung IR injury in the rat.16 Studies in rat models of bacteremia-induced lung injury have demonstrated rapid induction of pulmonary hypertension by PAF but mediated in part by ET-1.18 Similarly, PAF and ET-1 both affect capillary blood flow and vascular permeability in rat models of acute pancreatitis.19 With respect to IR-induced FGR in the rat, if ET-1 and PAF are functioning synergistically, the expectation would be additional improvement in fetal growth with addition of a second antagonist rather than complete normalization of growth with either antagonist. However, if these antagonists improve placental perfusion and/or function sufficiently to meet the demands of the developing fetus, this additive effect may not be observed. The lack of observation of an additive effect does not rule out synergism between these mediators.
In this study, we evaluated transcription of PLA2-IIA and prepro-ET-1 mRNA in the uterus and placenta 4 days after the IR insult. We demonstrated that transcription of both of these mediators was responsive to the IR insult. Uterine and placental PLA2-IIA was significantly increased by IR, but uterine prepro-ET-1 mRNA, even though it appeared elevated in the IR group, did not achieve statistical significance because of high variability. The high degree of variation may in part be due to the timing of our evaluation. After 4 days, compensatory mechanisms may be actively moderating the molecular response. Furthermore, the response of these mediators to the initial insult may be waning. The expression of these mediators may best be studied at a time closer to the ischemic insult to capture the early molecular responses. This is the subject of ongoing investigation in our laboratory.
The fact that expression of these mediators was similar in both horns of the uterus in spite of the ischemic insult occurring in only 1 horn supports the concept that the observed growth restriction is secondary to circulating mediators rather than to the ischemic insult itself. This study demonstrates that both ET-1 and PAF are prominent mediators in the pathophysiology of this model of FGR.
Other mediators may also contribute to the pathophysiology of IR-induced FGR. Tanaka et al32 demonstrated that thromboxane plays a salient role in the pathophysiology of this model. Leukocytes are known to accumulate in response to tissue reperfusion and to limit postischemic perfusion and fetal growth.33 Furthermore, other proinflammatory proteins, free radical oxygen species, or nitric oxide (either directly or indirectly) may all contribute to the pathophysiology of this model, as they have in other models of IR.34 Their role in this model of FGR, however, has not been evaluated. IR is likely to set in motion a cascade of events. The prominence of any individual component in these events likely varies in proportion to its contribution to the overall process. However, factors having the greatest impact on perfusion are likely to have the greatest significance in the pathophysiologic process as a whole.
The uterine IR model of FGR in the rat is uniquely suited to study the contribution of vascular mediators to decreased uteroplacental perfusion in FGR. This model may provide valuable insights into the mechanisms operative in uteroplacental dysfunction in humans as well. Inadequate uteroplacental perfusion ultimately leads to ischemia and/or infarction of the placenta, resulting in a compromised capacity for maternal-fetal gas and nutrient exchange. Histologic evidence of placental ischemia is commonly observed in cases of human FGR. Salafia et al5 evaluated placentas from nonanomalous preterm singleton live-born fetuses for evidence of uteroplacental ischemia. They found that growth restriction is primarily related to the cumulative number and severity of placental ischemic lesions.
Vascular pathology associated with obliteration of small muscular arteries in the tertiary stem villi,35 and resulting in reduced vascularization of the intermediate and terminal villi,36,37 implies that IR may occur in relatively localized areas of the placenta. The mediators produced in response to IR in these localized areas may affect perfusion and function of not only these but also surrounding areas of the placenta. The uterine IR model in the rat is then an attractive model to investigate the pathophysiology of FGR caused by suboptimal uteroplacental perfusion (the most common identifiable cause of FGR in humans) and specifically to study the role of mediators affecting regional perfusion and function and ultimately fetal growth.
Ischemic insults to the placenta result in compromised nutrient and gas exchange in the affected area but also result in the expression of mediators, which may further compromise the function of surrounding areas of the placenta. ET-1 and PAF, possibly acting synergistically, both play a prominent role in this pathophysiologic process. Antagonists to these mediators have been shown to improve pregnancy outcome in several animal models of FGR and deserve consideration as therapeutic modalities in human FGR as well.
Acknowledgments
The authors thank Ms. Sylvia Gontar for her technical assistance.
Footnotes
Supported in part by National Institutes of Health Grant HD042581.
References
- 1.McIntire DD, Bloom SL, Casey BM, Leveno KJ. Birth weight in relation to morbidity and mortality among newborn infants. N Engl J Med. 1999;340:1234–8. doi: 10.1056/NEJM199904223401603. [DOI] [PubMed] [Google Scholar]
- 2.Lackman F, Capewell V, Richardson B, daSilva O, Gagnon R. The risks of spontaneous preterm delivery and perinatal mortality in relation to size at birth according to fetal versus neonatal growth standards. Am J Obstet Gynecol. 2001;184:946–53. doi: 10.1067/mob.2001.111719. [DOI] [PubMed] [Google Scholar]
- 3.Bernstein IM, Horbar JD, Badger GJ, Ohlsson A, Golan A. Morbidity and mortality among very-low-birth-weight neonates with intrauterine growth restriction. Am J Obstet Gynecol. 2000;182:198–206. doi: 10.1016/s0002-9378(00)70513-8. [DOI] [PubMed] [Google Scholar]
- 4.Garite TJ, Clark R, Thorp JA. Intrauterine growth restriction increases morbidity and mortality among premature neonates. Am J Obstet Gynecol. 2004;191:481–7. doi: 10.1016/j.ajog.2004.01.036. [DOI] [PubMed] [Google Scholar]
- 5.Salafia CM, Minior VK, Pezzullo JC, Popek EJ, Rosenkrantz TS, Vintzileos AM. Intrauterine growth restriction in infants of less than thirty-two weeks’ gestation: associated placental pathologic features. Am J Obstet Gynecol. 1995;173:1049–57. doi: 10.1016/0002-9378(95)91325-4. [DOI] [PubMed] [Google Scholar]
- 6.De Wolf F, Brosens I, Renaer M. Fetal growth retardation and the maternal arterial supply of the human placenta in the absence of sustained hypertension. Br J Obstet Gynaecol. 1980;87:678–85. doi: 10.1111/j.1471-0528.1980.tb04601.x. [DOI] [PubMed] [Google Scholar]
- 7.Salafia CM. Placental pathology of fetal growth restriction. Clin Obstet Gynecol. 1997;40:740–9. doi: 10.1097/00003081-199712000-00008. [DOI] [PubMed] [Google Scholar]
- 8.Shen-Schwarz S, Macpherson TA, Mueller-Heubach E. The clinical significance of hemorrhagic endovasculitis of the placenta. Am J Obstet Gynecol. 1988;159:48–51. doi: 10.1016/0002-9378(88)90492-9. [DOI] [PubMed] [Google Scholar]
- 9.Wigglesworth JS. Experimental growth retardation in the foetal rat. J Pathol Bacteriol. 1964;88:1–13. [PubMed] [Google Scholar]
- 10.Tanaka M, Natori M, Ishimoto H, Miyazaki T, Kobayashi T, Nozawa S. Experimental growth retardation produced by transient period of uteroplacental ischemia in pregnant Sprague-Dawley rats. Am J Obstet Gynecol. 1994;171:1231–4. doi: 10.1016/0002-9378(94)90138-4. [DOI] [PubMed] [Google Scholar]
- 11.Lewis R, Ahokas R, Sibai B. Unilateral uterine ischemia/reperfusion induces oxidative stress with fetal growth restriction in both horns in the rat. Am J Obstet Gynecol. 2000;182:S13. (abstract #6). [Google Scholar]
- 12.Neerhof MG, Silver RK, Caplan MS, Thaete LG. Endothelin-1-induced placental and fetal growth restriction in the rat. J Matern Fetal Med. 1997;6:125–8. doi: 10.1002/(SICI)1520-6661(199705/06)6:3<125::AID-MFM1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 13.Thaete LG, Neerhof MG, Caplan MS. Endothelin receptor A antagonism prevents hypoxia-induced intrauterine growth restriction in the rat. Am J Obstet Gynecol. 1997;176:73–6. doi: 10.1016/s0002-9378(97)80014-2. [DOI] [PubMed] [Google Scholar]
- 14.Thaete LG, Neerhof MG, Silver RK. Differential effects of endothelin A and B receptor antagonism on fetal growth in normal and nitric oxide-deficient rats. J Soc Gynecol Invest. 2001;8:18–23. [PubMed] [Google Scholar]
- 15.Ogata M, Iwamoto T, Tazawa N, Nishikawa M, Yamashita J, Takaoka M, et al. A novel and selective Na+/Ca2+ exchange inhibitor, SEA0400, improves ischemia/reperfusion-induced renal injury. Eur J Pharmacol. 2003;478:187–98. doi: 10.1016/j.ejphar.2003.08.082. [DOI] [PubMed] [Google Scholar]
- 16.Stammberger U, Carboni GL, Hillinger S, Schneiter D, Weder W, Schmid RA. Combined treatment with endothelin- and PAF-antagonists reduces posttransplant lung ischemia/reperfusion injury. J Heart Lung Transplant. 1999;18:862–8. doi: 10.1016/s1053-2498(99)00039-x. [DOI] [PubMed] [Google Scholar]
- 17.Thaete LG, Neerhof MG, Jilling T, Caplan MS. Infusion of exogenous platelet-activating factor produces intrauterine growth restriction in the rat. J Soc Gynecol Invest. 2003;10:145–50. doi: 10.1016/s1071-5576(03)00005-4. [DOI] [PubMed] [Google Scholar]
- 18.Clavijo LC, Carter MB, Matheson PJ, Wills-Frank LA, Wilson MA, Wead WB, et al. Platelet-activating factor and bacteremia-induced pulmonary hypertension. J Surg Res. 2000;88:173–80. doi: 10.1006/jsre.1999.5748. [DOI] [PubMed] [Google Scholar]
- 19.Foitzik T, Hotz HG, Eibl G, Hotz B, Kirchengast M, Buhr HJ. Therapy for microcirculatory disorders in severe acute pancreatitis: effectiveness of platelet-activating factor receptor blockade vs. endothelin receptor blockade. J Gastrointest Surg. 1999;3:244–51. doi: 10.1016/s1091-255x(99)80066-3. [DOI] [PubMed] [Google Scholar]
- 20.Ishimoto H, Minegishi K, Miyakoshi K, Tanigaki S, Nishimura O, Miyazaki T, et al. Platelet-activating factor receptor antagonism prevents ischemia-reperfusion-induced intrauterine growth restriction in the rat. J Soc Gynecol Invest. 2000;7:258A. (abstract #766). [Google Scholar]
- 21.Opgenorth TJ, Adler AL, Calzadilla SV, Chiou WJ, Dayton BD, Dixon DB, et al. Pharmacological characterization of A-127722: an orally active and highly potent ETA-selective receptor antagonist. J Pharmacol Exp Ther. 1996;276:473–81. [PubMed] [Google Scholar]
- 22.Casals-Stenzel J, Muacevic G, Weber K-H. Pharmacological actions of WEB 2086, a new specific antagonist of platelet activating factor. J Pharmacol Exp Ther. 1987;241:974–81. [PubMed] [Google Scholar]
- 23.Summers JB, Albert DH. Platelet activating factor antagonists. In: August JT, Anders MW, Murad F, Coyle J, eds. Advances in pharmacology. vol 32. San Diego, CA: Academic Press; 1995. p. 67–168. [DOI] [PubMed]
- 24.Heuer HO. Pharmacology of hetrazepines as PAF-antagonists. In: Braquet P, editor. CRC handbook of PAF and PAF antagonists. Boca Raton (FL): CRC Press; 1991. p. 171–202.
- 25.Zouki C, Baron C, Fournier A, Filep JG. Endothelin-1 enhances neutrophil adhesion to human coronary artery endothelial cells: role of ET(A) receptors and platelet-activating factor. Br J Pharmacol. 1999;127:969–79. doi: 10.1038/sj.bjp.0702593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Resink TJ, Scott-Burden T, Bühler FR. Activation of phospholipase A2 by endothelin in cultured vascular smooth muscle cells. Biochem Biophys Res Commun. 1989;158:279–86. doi: 10.1016/s0006-291x(89)80209-8. [DOI] [PubMed] [Google Scholar]
- 27.Abdel-Latif AA, Zhang Y, Yousufzai SYK. Endothelin-1 stimulates the release of arachidonic acid and prostaglandins in rabbitiris sphincter smooth muscle: activation of phospholipase A2. Curr Eye Res. 1991;10:259–65. doi: 10.3109/02713689109003448. [DOI] [PubMed] [Google Scholar]
- 28.Montero A, Rodriguez-Barbero A, López-Novoa JM. A role for platelet-activating factor in endothelin-1-induced rat mesangial cell proliferation. Eur J Pharmacol. 1993;243:235–40. doi: 10.1016/0014-2999(93)90180-p. [DOI] [PubMed] [Google Scholar]
- 29.Schramek H, Wang Y, Konieczkowski M, Simonson MS, Dunn MJ. Endothelin-1 stimulates cytosolic phospholipase A2 activity and gene expression in rat glomerular mesangial cells. Kidney Int. 1994;46:1644–52. doi: 10.1038/ki.1994.464. [DOI] [PubMed] [Google Scholar]
- 30.Filep JG, Fournier A, Földes-Filep É. Endothelin-1-induced myocardial ischaemia and oedema in the rat: involvement of the ETA receptor, platelet-activating factor and thromboxane A2. Br J Pharmacol. 1994;112:963–71. doi: 10.1111/j.1476-5381.1994.tb13175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mustafa SB, Gandhi CR, Harvey SAK, Olson MS. Endothelin stimulates platelet-activating factor synthesis by cultured rat Kupffer cells. Hepatology. 1995;21:545–53. [PubMed] [Google Scholar]
- 32.Tanaka M, Natori M, Ishimoto H, Miyakoshi K, Miyazaki T, Kobayashi T, et al. Effects of a thromboxane synthetase inhibitor (OKY-046) in an ischemia-reperfusion model of intrauterine growth retardation in Sprague-Dawley rats. Biol Neonate. 1997;72:181–6. doi: 10.1159/000244482. [DOI] [PubMed] [Google Scholar]
- 33.Miyakoshi K, Ishimoto H, Nishimura O, Tanigaki S, Tanaka M, Miyazaki T, et al. Role of leukocytes in uterine hypoperfusion and fetal growth retardation induced by ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2001;280:H1215–21. doi: 10.1152/ajpheart.2001.280.3.H1215. [DOI] [PubMed] [Google Scholar]
- 34.Anaya-Prado R, Toledo-Pereyra LH. The molecular events underlying ischemia/reperfusion injury. Transplant Proc. 2002;34:2518–9. doi: 10.1016/s0041-1345(02)03471-1. [DOI] [PubMed] [Google Scholar]
- 35.Giles WB, Trudinger BJ, Baird PJ. Fetal umbilical artery flow velocity waveforms and placental resistance: pathological correlation. Br J Obstet Gynaecol. 1985;92:31–8. doi: 10.1111/j.1471-0528.1985.tb01045.x. [DOI] [PubMed] [Google Scholar]
- 36.Bracero LA, Beneck D, Kirshenbaum N, Peiffer M, Stalter P, Schulman H. Doppler velocimetry and placental disease. Am J Obstet Gynecol. 1989;161:388–93. doi: 10.1016/0002-9378(89)90528-0. [DOI] [PubMed] [Google Scholar]
- 37.Hitschold TP. Doppler flow velocity waveforms of the umbilical arteries correlate with intravillous blood volume. Am J Obstet Gynecol. 1998;179:540–3. doi: 10.1016/s0002-9378(98)70392-8. [DOI] [PubMed] [Google Scholar]