

Abstract
Inositol 1,4,5-trisphosphate (IP3) receptors form tetrameric, IP3-gated channels in endoplasmic reticulum membranes that govern the release of Ca2+ from this organelle. In response to activation of certain G protein-coupled receptors that persistently elevate IP3 concentration, IP3 receptors are ubiquitinated and degraded by the ubiquitin-proteasome pathway. IP3 receptor ubiquitination is mediated by the ubiquitin-conjugating enzyme, mamUbc7, a component of the endoplasmic reticulum-associated degradation pathway. However, the mechanism by which ubiquitinated IP3 receptors are transferred to the proteasome is not known. Here, we examine this process and show in several mammalian cell types that the ATPase p97 associates with IP3 receptors in response to hormonal stimuli that induce IP3 receptor ubiquitination. To examine the functional relevance of the p97 interaction with IP3 receptors, we stably and specifically reduced p97 protein levels by 62 ± 3% in Rat-1 fibroblasts using RNA interference. In these cells, endothelin-1-induced IP3 receptor degradation was markedly retarded and the accumulation of ubiquitinated IP3 receptors was markedly enhanced. These effects were reversed by expression of exogenous p97. In addition, Ufd1 and Npl4, which complex with p97, also associated with IP3 receptors upon hormonal stimulation. We conclude that the p97-Ufd1-Npl4 complex couples ubiquitinated IP3 receptors to proteasomal degradation and, thus, plays a key role in IP3 receptor processing. These data also establish that the p97-Ufd1-Npl4 complex mediates endoplasmic reticulum-associated degradation in mammalian cells.
Activation of various G protein-coupled receptors (GPCRs)2 increases phospholipase C activity and generates the second messengers inositol 1,4,5-trisphosphate (IP3) and diacylglycerol. IP3 diffuses into the cytosol and mobilizes Ca2+ by binding to IP3 receptors (IP3Rs) present in membranes of the endoplasmic reticulum (ER). Hence, IP3Rs play a pivotal role in converting cues conveyed by extracellular stimuli into intracellular Ca2+ signals (1, 2). There are three different IP3R types (IP3R1, IP3R2, and IP3R3) expressed at various levels in different tissues (3, 4). Each IP3R type is ~2700 amino acids in length and has six membrane-spanning regions, a long cytosolic amino-terminal region, and a short cytosolic carboxyl-terminal tail (5–7).
In response to stimulation of GPCRs that persistently elevate IP3 concentration, the cellular levels of IP3R1–3 are rapidly reduced, a phenomenon termed IP3R down-regulation. This has been demonstrated in many mammalian cell lines and in native tissues (3, 8–15), with the consequence that the frequency, amplitude, and duration of elementary Ca2+ puffs are reduced (16), and global Ca2+ signaling is restrained (8, 9, 13–16). IP3R down-regulation appears to be an adaptation to GPCR stimulation that protects cells against the deleterious effects of chronic elevation of cytosolic Ca2+ (17).
IP3R down-regulation is mediated by the ubiquitin-proteasome pathway (UPP) (10–15). Protein degradation via the UPP has two distinct steps: first, polyubiquitin chains are covalently attached to the targeted protein, and second, the polyubiquitinated protein is degraded by the 26 S proteasome (18, 19). The process by which ER resident proteins or proteins traversing the ER are degraded is termed endoplasmic reticulum-associated degradation or ERAD (20–23). Two forms of ERAD appear to exist: quality control ERAD and regulated ERAD (21). Quality control ERAD ensures that misfolded proteins, such as mutant rhodopsin (24) and cystic fibrosis transmembrane conductance regulator (25), or unassembled subunits of multiprotein complexes, such as T-cell receptor subunits TCRα (26) and CD3-δ (26, 27), do not accumulate in the ER and do not transit the secretory pathway. Regulated ERAD, on the other hand, accounts for the degradation of native ER proteins in response to various stimuli. For example, in yeast and mammalian cells, 3-hydroxy 3-methylglutaryl-CoA reductase, an ER membrane protein, is polyubiquitinated and degraded in response to sterols (28, 29). In the case of IP3Rs, putative conformational changes associated with receptor activation (5–7) appear to almost instantaneously render IP3Rs susceptible to ubiquitination and degradation (30, 31). This process is mediated by mamUbc7 (32), which catalyzes the ubiquitination of many ERAD substrates (27, 32). Furthermore, as IP3Rs are unique in being rapidly convertible from their native form into ERAD substrates, they provide a valuable system for studying the mechanism of ERAD in mammalian cells.
An intriguing feature of ERAD substrates is that they have to be translocated from the ER to reach the proteasome. While the mechanism involved is poorly understood, several recent studies have implicated a cytosolic ATPase, termed p97 or VCP in mammals and Cdc48p in yeast, in this retrotranslocation process (33–39). p97 is a member of AAA ATPase family and is involved in a wide variety of cellular activities, including vesicular transport, homotypic membrane fusion, dissociation of a ubiquitinated membrane-tethered transcription factor from its non-ubiquitinated binding partner, and proteasome-mediated protein degradation (40–42). p97 is coupled to these various cellular activities by a set of cofactors or adaptor proteins (40, 42). For example, VCIP135 and p47 associate with p97 to recruit it to membrane fusion events (43–45). Likewise, heterodimers of Ufd1 and Npl4 form a complex with p97 and help recruit it to ubiquitinated substrates (46–48).
In the present study, we examined whether p97 and its cofactors are involved in IP3R down-regulation. We report that p97, Ufd1, and Npl4 all rapidly associate with IP3Rs under conditions that lead to IP3R ubiquitination and that this association correlates with the extent to which IP3Rs are ubiquitinated. Moreover, suppression of p97 expression by RNA interference (RNAi) impaired the regulated ERAD of IP3Rs. Our results indicate that the p97-Ufd1-Npl4 complex is required for the processing of ubiquitinated IP3Rs and, thus, that this complex plays a key role in ERAD in mammalian cells.
EXPERIMENTAL PROCEDURES
Materials
αT3-1 mouse anterior pituitary gonadotrope cells were obtained and cultured as described (11). Rat-1 rat fibroblasts were a kind gift from Dr. A. Cox, University of North Carolina, and were grown as monolayers in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. Cells were fed every other day and subcultured every 3–5 days using 0.25% trypsin, 1 mm EDTA. Antibodies used were: rabbit polyclonal anti-IP3R1 (11), mouse monoclonal anti-p97 (Research Diagnostics Inc.), rabbit polyclonal anti-Npl4 (a kind gift from Dr. Akira Kakizuka, Kyoto University, Kyoto, Japan), rabbit polyclonal anti-Sec61β (a kind gift from Dr. T. Rapoport, Harvard Medical School, Boston, MA), rabbit polyclonal anti-calnexin (Stressgen Biotechnologies Corp.), mouse monoclonal anti-p27 (Santa Cruz Biotechnology Inc.), mouse monoclonal anti-p53 (Calbiochem), mouse monoclonal anti-ubiquitin (Zymed Laboratories Inc.), mouse monoclonal anti-HSP90, anti-IP3R3, anti-Ufd1 and anti-Ufd2 (BD Transduction Laboratories), mouse monoclonal anti-hemagglutinin (HA) epitope (Covance), and horseradish peroxidase-conjugated secondary antibodies (Sigma). SDS, Triton X-100, Igepal CA-630, protease inhibitors, gonadotropin-releasing hormone (GnRH), cycloheximide, and polybrene (hexamethrine bromide) were purchased from Sigma; endothelin-1 (ET1) was from Calbiochem; T4 ligase, BglII, and HindIII were obtained from New England Biolabs; Precision Plus Protein™ Standards and dithiothreitol were from Bio-Rad; G418 was from Cellgro; Protein A-Sepharose CL-4B was from Amersham Biosciences; N-acetyl-Leu-Leu-norleucinal (ALLN) was from Alexis; bortezomib (PS-341) was obtained from Millennium Pharmaceuticals Inc. (Cambridge, MA); and puromycin was from Clontech.
Electrophoresis, Immunoblotting, and Quantitation
Samples were resolved by SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose essentially as described (10), probed with relevant primary antibodies, followed by horseradish peroxidase-conjugated secondary antibodies, and immunoreactivity was visualized with Pierce chemiluminescence reagents. Immunoreactivity was quantitated using a Genegnome Imager (Syngene Bio Imaging). Data shown are mean ± S.E. or representative of ≥3 independent experiments.
IP3R1 Immunoprecipitation
For αT3-1 gonadotropes, cells were grown to near confluence in 15-cm diameter dishes and were incubated with or without GnRH. Cells were then harvested by vigorous scraping and pipetting in culture medium, pelleted by centrifugation (1000 × g for 6 min at 4 °C), and solubilized in Igepal CA-630 lysis buffer (50 mm Tris-HCl, 120 mm NaCl, 0.5% Igepal CA-630 (v/v), 1 mm EDTA, 0.2 mm phenylmethylsulfonyl fluoride, 10 μm leupeptin, 10 μm pepstatin, 0.2 μm soybean trypsin inhibitor, 1 mm dithiothreitol, pH 8) for 30 min at 4 °C. Lysates were clarified by centrifugation (16,000 × g for 10 min at 4 °C), and IP3R1 was immunoprecipitated by incubating with anti-IP3R1 for 1 h followed by Protein A-Sepharose CL-4B for 3 h. Immunocomplexes were washed thoroughly with Igepal CA-630 lysis buffer, resuspended in gel loading buffer (10), electrophoresed, and immunoblotted. For Rat-1 fibroblasts, cells were grown to near confluence in 15-cm diameter dishes and were serum-starved for about 15 h. Cells were then incubated with or without ET1 and after removing culture medium were lysed by adding Triton X-100 lysis buffer (50 mm Tris base, 150 mm NaCl, 1% Triton X-100, 1 mm EDTA, 0.2 mm phenylmethylsulfonyl flouride, 10 μm leupeptin, 10 μm pepstatin, 0.2 μm soybean trypsin inhibitor, 1 mm dithiothreitol, pH 8) directly to cell monolayers followed by vigorous scraping. After 30 min at 4 °C, lysates were clarified by centrifugation (16,000 × g for 10 min at 4 °C), and IP3R1 was immunoprecipitated and processed in immunoblots as described for αT3-1 cells.
Levels of IP3Rs and Other Proteins
For measurement of IP3R down-regulation, serum-starved Rat-1 cells were incubated with or without ET1 and after removing culture medium were harvested by adding Triton X-100 lysis buffer directly to cell monolayers followed by vigorous scraping. After 30 min at 4 °C, lysates were cleared by centrifugation (16,000 × g for 10 min at 4 °C), supernatants were collected, estimated for protein content, and equal amounts of protein were immunoblotted with anti-IP3R1 and anti-IP3R3. Essentially, identical methods were used to monitor the levels of ubiquitin conjugates, p27 and p53, in cell lysates.
Subcellular Fractionation
Rat-1 cells were harvested by scraping into 155 mm NaCl, 10 mm HEPES, 2 mm EDTA, pH 7.4, pelleted by centrifugation (1000 × g for 6 min at 4 °C), resuspended in homogenization buffer (10 mm Tris base, 1 mm EGTA, 0.2 mm phenylmethylsulfonyl flouride, 10 μm leupeptin, 10 μm pepstatin, 0.2 μm soybean trypsin inhibitor, 1 mm dithiothreitol, pH 7.4), and disrupted with 40 strokes of a Dounce homogenizer. The homogenate was centrifuged (1100 × g for 6 min at 4 °C) to pellet nuclei, and the supernatants were re-centrifuged (100,000 × g for 1h at 4 °C). The supernatants from this step were designated as cytosolic fractions, while pellets and nuclei were then solubilized with Triton X-100 lysis buffer and re-centrifuged (16,000 × g for 10 min at 4 °C) to obtain membrane and nuclear fractions. Supernatants were estimated for protein content and equivalent amounts of each fraction were then immunoblotted with anti-ubiquitin.
RNAi Design and Generation of Stable Cell Lines
The pSUPER.retro vector (49, 50) was used to introduce short interfering RNA (siRNA) targeting p97 into Rat-1 fibroblasts. Two 64-base complementary oligonucleotides (forward, 5′-gatccccgtaggctatgatgacatcgttcaagagacgat-gtcatcatagcctactttttggaaa-3′and reverse, 5′-agcttttccaaaaagtaggctatgatgacatcgtc-tcttgaacgatgtcatcatagcctacggg-3′) were synthesized to contain a 19-nucleotide sequence (bold) corresponding to nucleotides 812–830 of Rattus norvegicus p97 mRNA (GenBank™ accession number NM_053864), separated from the reverse complement of the same 19 nucleotides by a 9-nucleotide spacer. The annealed product contains 5′ and 3′ overhangs compatible with BglII and HindIII restriction sites, respectively, and was ligated into pSUPER.retro digested with BglII and HindIII, generating pSUPER.retro.p97. In parallel, three different control vectors encoding random siRNA (pSUPER.retro.ran1–3) were constructed using a 19-nucleotide sequence with no known homology to any of the known rat mRNAs. Correct ligation into pSUPER.retro was confirmed by restriction digestion and sequencing. To generate infectious retroviral stock, pSUPER.retro.p97 or pSUPER.retro.ran1–3 were transfected together with pVPack-Eco, which encodes viral envelope, and pVPack-GP, which encodes Gag-pol (Stratagene), into 293T cells using the calcium-phosphate method (51). 72 h posttransfection, culture medium was removed and passed through a 0.45-μm filter (Nalgene). Virus-containing media were aliquoted and frozen at −80 °C until use. To transduce Rat-1 cells, cells were seeded in a 12-well plate, and after 24 h, viruses were added along with 8 μg/ml polybrene, and cells were incubated for 8 h at 37 °C, followed by a change of medium. After 48 h, transduced cells were selected with 2.5 μg/ml puromycin and after several passages were maintained in 1 μg/ml puromycin.
Expression of Exogenous p97
cDNA encoding full-length, amino-terminal HA-tagged mouse p97 (HA-p97) was a kind gift from Dr. Masaki Matsumoto, Kyushu University, Fukuoka, Japan. To render the mRNA encoded by this plasmid refractory to the siRNA targeting endogenous p97, five silent mutations were introduced into the targeted region using the QuikChange™ kit (Stratagene) and were confirmed by sequencing. p97 cells (~80% confluent) were transfected with 1 μg of either pcDNA3 (vector) or HA-p97 plasmid using 8 μl of CytoPure-™huv (Qbiogene) according to the manufacturer’s instructions. After 48 h, cells were subcultured and grown in the presence of 500 μg/ml G418. Sterile cloning cylinders were used to select G418-resistant clones, which were then screened for HA-p97 expression. Five clones expressing high levels of HA-p97 and four vector-transfected clones were used to analyze IP3R ubiquitination and down-regulation.
Measurement of Ca2+ Mobilization
Serum-starved Rat-1 cells were harvested in and washed with 155 mm NaCl, 10 mm HEPES, 2 mm EDTA, pH 7.4, resuspended in Krebs-HEPES buffer (30) and then incubated with 10 μm Fura2-AM at 37 °C for 1 h. Cells were centrifuged at 500 × g for 2 min, washed twice, incubated again at 37 °C for 30 min, again washed twice, resuspended in 2 ml of Krebs-HEPES buffer, placed in a cuvette, and excited at 340 and 380 nm. Fluorescence emission was recorded using a computerized LS-50B flourimeter (PerkinElmer Life Sciences). 0.1% Triton X-100 and 10 mm EGTA were used to determine the maximum and minimum fluorescence values, respectively. 340/380 nm emission ratios were used to calculate intracellular Ca2+ as described (32).
RESULTS
Dynamic Association of the p97-Ufd1-Npl4 Complex with IP3Rs
Initially, we used αT3-1 mouse anterior pituitary gonadotropes to investigate mechanisms involved in GPCR-initiated IP3R down-regulation. These cells express phospholipase C-linked GnRH receptors, and GnRH induces rapid and robust ubiquitination and down-regulation of IP3R1, the predominant IP3R in this cell type (11). Fig. 1A shows that GnRH-induced IP3R1 ubiquitination peaked at ~5 min and thereafter declined as IP3R1 was degraded (lanes 1–4). As is typical for UPP substrates, inclusion of proteasome inhibitor (bortezomib) caused the accumulation of ubiquitinated IP3R1 and blocked IP3R1 down-regulation (lane 5). Probing for co-immunoprecipitating proteins revealed that p97, Ufd1, and Npl4 all associated with IP3R1 after GnRH stimulation in a manner that correlated with the levels of ubiquitinated species (Fig. 1A, lanes 2–4), indicating that the p97-Ufd1-Npl4 complex is recruited to participate in the processing of ubiquitinated IP3R1. Additionally, Fig. 1A shows that the U-box protein Ufd2 (52–54) associated with ubiquitinated IP3R1. Intriguingly, the amount of p97-Ufd1-Npl4 and Ufd2 that co-precipitated with IP3R1 in the presence of bortezomib, when the accumulation of ubiquitinated IP3R1 was greatly enhanced (Fig. 1A, lane 5), was less than that seen maximally in its absence (Fig. 1A, lane 2). This likely reflects the fact that bortezomib causes generalized accumulation of ubiquitin-protein conjugates in αT3-1 cells and other cells (55) and that under such circumstances, the p97-Ufd1-Npl4 complex is deployed to process these conjugates rather than ubiquitinated IP3R1.
FIGURE 1. The p97-Ufd1-Npl4 complex associates with IP3R1 in stimulated cells.
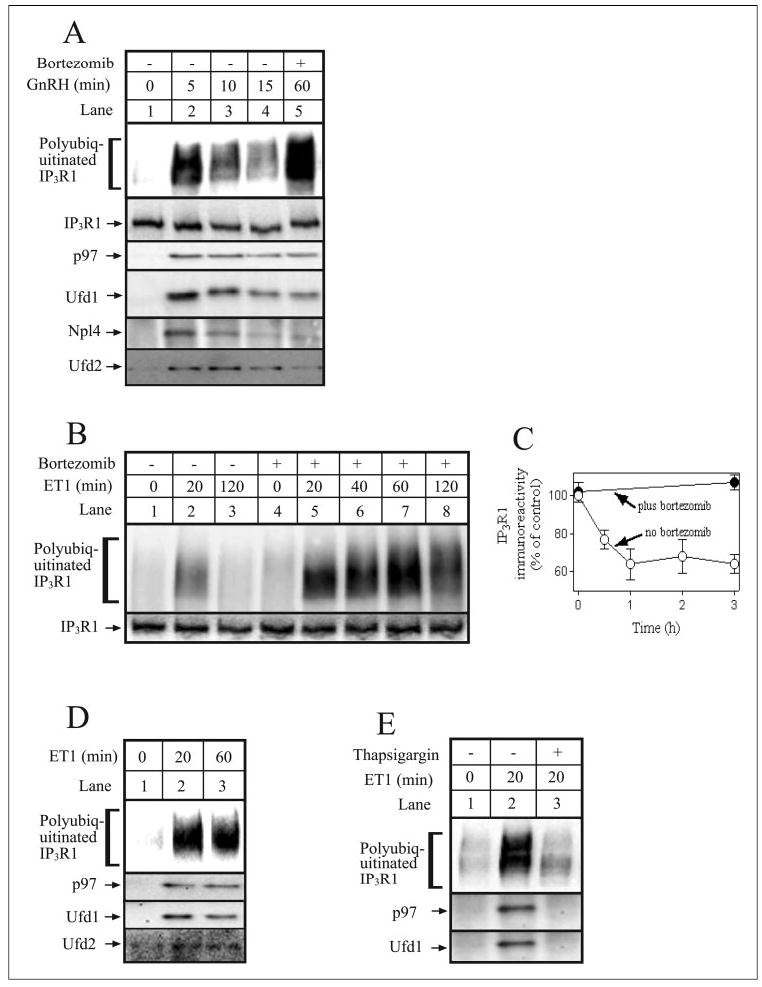
A, αT3-1 cells were stimulated with GnRH (0.1 μm) for the times indicated without (lanes 1–4) or with 1-h preincubation with 1 μm bortezomib (lane 5). Cells were then harvested and lysed, and IP3R1 was immunoprecipitated with anti-IP3R1. Samples were then electrophoresed and immunoblotted with anti-ubiquitin, anti-IP3R1, anti-p97, anti-Ufd1, anti-Npl4, and anti-Ufd2. B, Rat-1 cells were incubated for times indicated with 10 nm ET1 without (lanes 1–3) or with 1h preincubation with 1 μm bortezomib (lanes 4 – 8). Cells were then harvested and lysed, and IP3R1 was immunoprecipitated with anti-IP3R1. Samples were then electrophoresed and immunoblotted with anti-ubiquitin and anti-IP3R1. C, Rat-1 cells were incubated with 100 nm ET1 for the times indicated without or with 1h preincubation with 1 μm bortezomib. Cells were then harvested, lysates were prepared, and IP3R1 immunoreactivity was assessed, quantitated, and expressed as percentage of immunoreactivity at time 0. D and E, Rat-1 cells were incubated with 10 nm ET1 for the times indicated without or with 1 μm thapsigargin. Cells were then lysed, IP3R1 was immunoprecipitated with anti-IP3R1, and samples were immunoblotted as described for A. Proteins migrated as follows: polyubiquitinated IP3R1 at 275–380 kDa, IP3R1 at 260 kDa, p97 at 97 kDa, Ufd1 at 40 kDa, Npl4 at 60 kDa, and Ufd2 at 146 kDa.
To extend our analysis to other cell types, we examined Rat-1 fibroblasts, since our preliminary experiments indicated that RNAi was feasible and efficient in this cell type. Rat-1 cells were found to express approximately equal amounts of IP3R1 and IP3R3 and very little IP3R2, and ET1, a potent IP3-forming ligand (56), caused IP3R1 ubiquitination (Fig. 1B) and down-regulation (Fig. 1C). In the absence of bortezomib, IP3R1 ubiquitination peaked at ~20 min and thereafter declined as IP3Rs were degraded (Fig. 1B, lanes 2 and 3), whereas in the presence of bortezomib, ubiquitinated IP3R1 accumulated (Fig. 1B, lanes 5–8), and IP3R1 down-regulation was blocked (Fig. 1C). Stimulation with ET1 also caused the association of p97, Ufd1, and Ufd2 with IP3R1 (Fig. 1D), indicating that the same interactions that occur in GnRH-stimulated αT3-1 cells also occur in ET1-stimulated Rat-1 cells. However, we were unable to detect Npl4 co-immunoprecipitation, most likely because of the insensitivity of the Npl4 antibody. Fig. 1E shows that depletion of ER Ca2+ with the Ca2+ -ATPase inhibitor thapsigargin (11) blocked both IP3R1 ubiquitination and co-precipitation of p97 and Ufd1 in Rat-1 cells. Taken together, these data indicate that in αT3-1 and Rat-1 cells the p97-Ufd1-Npl4 complex and Ufd2 are recruited to and participate in the processing of ubiquitinated IP3Rs.
Specific Inhibition of p97 Expression by RNAi
To investigate the role of p97 in IP3R1 processing, we used the pSUPER.retro vector (49, 50) to stably express siRNA targeting p97 mRNA in Rat-1 cells. The p97 siRNA chosen concurred with that used in a recent study to inhibit p97 expression in HeLa cells using chemically synthesized siRNA (57). pSUPER.retro directs the synthesis of short hairpin RNAs (Fig. 2A) that can be processed by the Dicer enzymatic machinery into active siRNAs (49, 50, 58). We established three cell lines harboring p97 siRNA (p97–1, p97–2 and p97–3, henceforth referred to as p97 cells) and three control cell lines harboring random siRNAs (ran-1, ran-2, and ran-3, henceforth referred to as ran cells). Expression of p97 was reduced by 62 ± 3% in p97 cells (Fig. 2B, lanes 4–6) as compared with ran cells (Fig. 2B, lanes 1–3), or unmodified Rat-1 cells,3 and this “knockdown” was specific, since the levels of Ufd1, Ufd2, and HSP90 (cytosolic proteins), and Sec61β and calnexin (ER membrane proteins) were unaffected (Fig. 2B). In addition, p97 knockdown did not alter IP3R levels (Fig. 4, A and E). The reduction in p97 expression was maintained for >8 passages, and p97 cell growth and morphology were identical to that of ran or unmodified Rat-1 cells.3 p97 expression was not reduced further by re-transduction of p97 cells,3 indicating that ~62% knockdown was the maximum possible using this technique. Since we planned to examine ET1-induced IP3R ubiquitination and down-regulation in these cells, we also examined whether signaling down-stream of the ET1 receptor was affected by p97 knockdown. Fig. 2C shows this was not the case, since ET1-induced Ca2+ release was not significantly different in ran and p97 cells. Taken together, these data show that stable and specific inhibition of p97 expression is possible in Rat-1 cells and that this does not affect overall cell function. Furthermore, the three p97 cell lines behaved identically, as did the three ran cell lines, and thus, we used them interchangeably for subsequent experiments.
FIGURE 2. RNAi of p97 in Rat-1 fibroblasts.
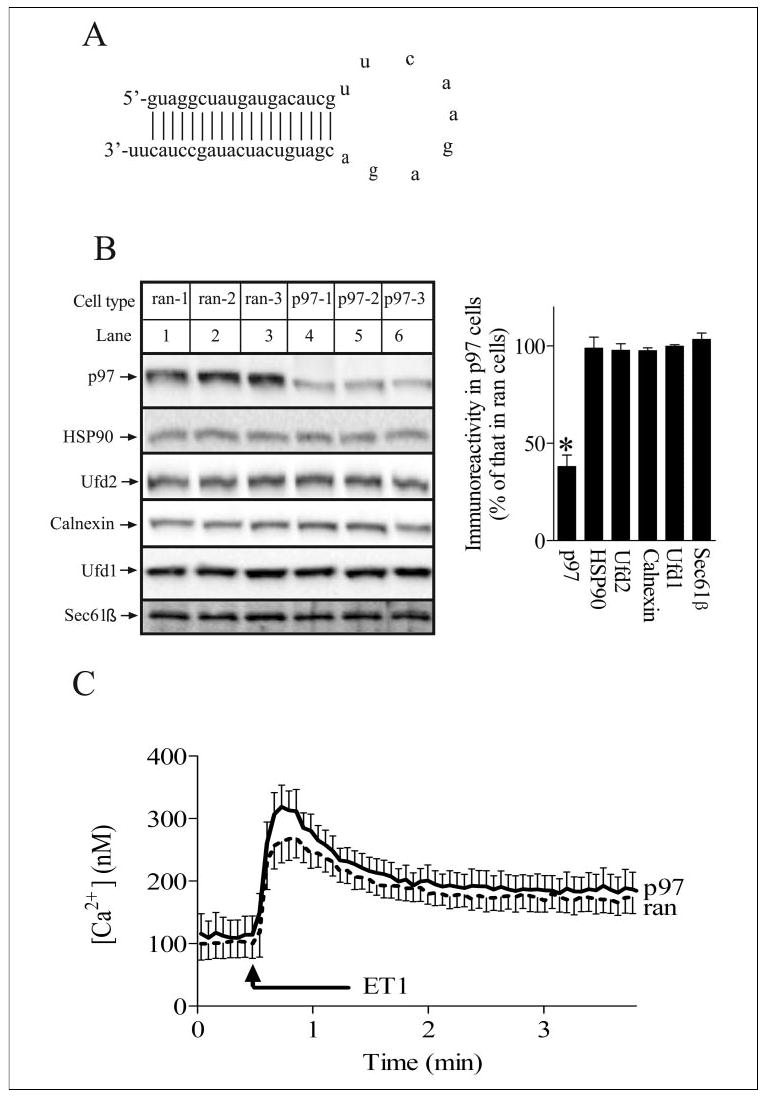
A, sequence and predicted secondary structure of the short hairpin RNA that yields the siRNA that targets p97 mRNA. B, Rat-1 cells expressing three different control siRNA sequences (lanes 1–3) or p97 siRNA (lanes 4 – 6) were harvested and lysed, and equal amounts of cell protein were electrophoresed and immunoblotted with anti-p97, anti-HSP90, anti-Ufd2, anti-calnexin, anti-Ufd1, and anti-Sec61β. Proteins detected migrated as follows: p97 at 97 kDa, HSP90 at 90 kDa, Ufd2 at 146 kDa, calnexin at 90kDa, Ufd1 at 40 kDa, and Sec61β at 14 kDa. The histogram shows combined quantitated immunoreactivity from three independent experiments with * indicating significant differences (p < 0.05) from ran cell values by Welch’s unpaired t test. C, ran and p97 cells were stimulated with 100 nm ET1, and cytosolic free Ca2+ concentration was recorded. Data shown are mean ± S.E. of seven independent experiments; any differences between ran and p97 cells were not significant (p > 0.2 by unpaired t test).
FIGURE 4. p97 is required for IP3R processing.
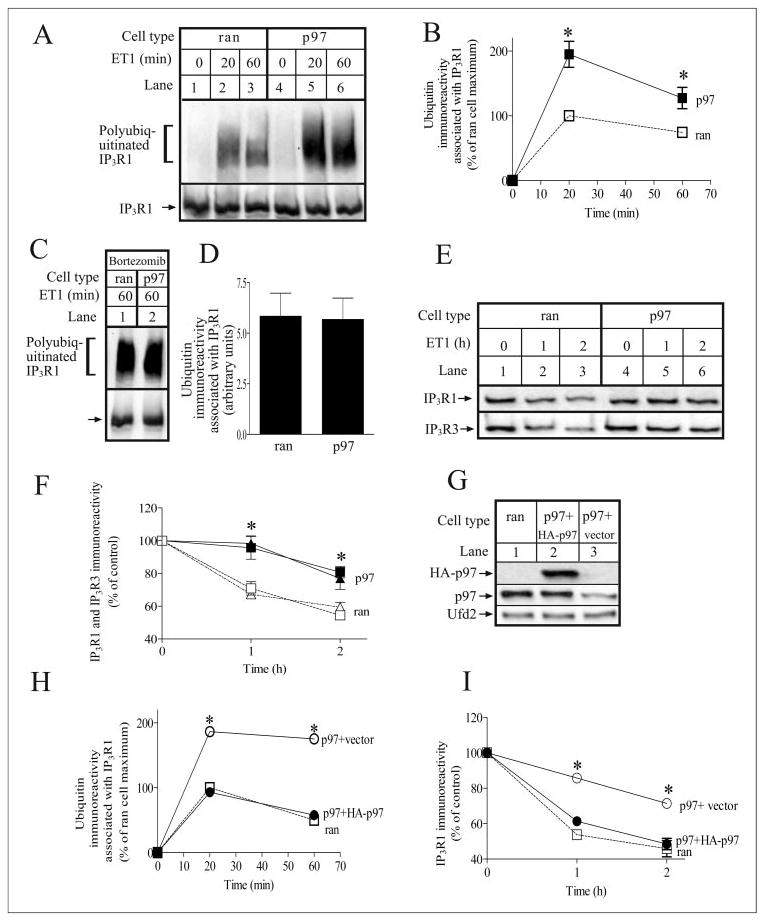
A, ran and p97 cells were incubated with 10 nm ET1 for the times indicated. IP3R1 was then immunoprecipitated with anti-IP3R1 and was electrophoresed and immunoblotted with anti-ubiquitin and anti-IP3R1. B, quantitated ubiquitin immunoreactivity associated with IP3R1 expressed as a percentage of that associated with IP3R1 in ET1-stimulated ran cells at 20 min; * denotes p < 0.05, when comparing ran and p97 cells at each time point, by Welch’s unpaired t test. C and D, ran and p97 cells were stimulated with 10 nm ET1 for 60 min in the presence of 1 μm bortezomib. IP3R1 was then immunoprecipitated, processed, and quantitated as described for A and B. E and F, ran and p97 cells were incubated with 10 nm ET1 for the times indicated. Cells were then harvested and lysed, and equal amounts of cell protein were electrophoresed and immunoblotted with anti-IP3R1 and anti-IP3R3. Quantitated immunoreactivity of IP3R1 (squares) and IP3R3 (triangles) are expressed as percentage of immunoreactivity at time 0; * denotes p < 0.05, when comparing ran and p97 cells at each time point, by unpaired t test. G, p97 cells stably transfected with either empty vector or HA-p97 and ran cells were harvested and lysed, and equal amounts of cell protein were electrophoresed and immunoblotted with anti-HA (upper panel), anti-p97 (middle panel), or anti-Ufd2 (lower panel). H and I, p97 cells stably transfected with either empty vector (open circles) or HA-p97 (filled circles) and ran cells (open squares) were stimulated and analyzed as in B and F, respectively; * denotes significant differences (p < 0.05) from ran cell values.
We also examined whether p97 knockdown had general effects on the UPP. ran and p97 cells were disrupted and the steady state level of ubiquitin-protein conjugates in different subcellular fractions was examined (Fig. 3A). However, no differences were detected between ran and p97 cells, indicating that the UPP was not perturbed. Likewise, the rate of degradation of ubiquitin-protein conjugates, measured in the presence of cycloheximide, a protein synthesis inhibitor, was the same in ran and p97 cells (Fig. 3B), showing that the bulk degradation of ubiquitinated proteins was unaffected by p97 knockdown. Furthermore, analysis of p27, a cyclin-dependent kinase inhibitor (59), and p53, a tumor suppressor (60), both of which are degraded by the UPP (61–63), showed that their accumulation in the presence of ALLN, a proteasome inhibitor, and their degradation following ALLN withdrawal were the same in both ran and p97 cells (Fig. 3C). Overall, these data show that ~62% knockdown of p97 does not affect general protein turnover by the UPP.
FIGURE 3. p97 knockdown does not perturb the UPP.
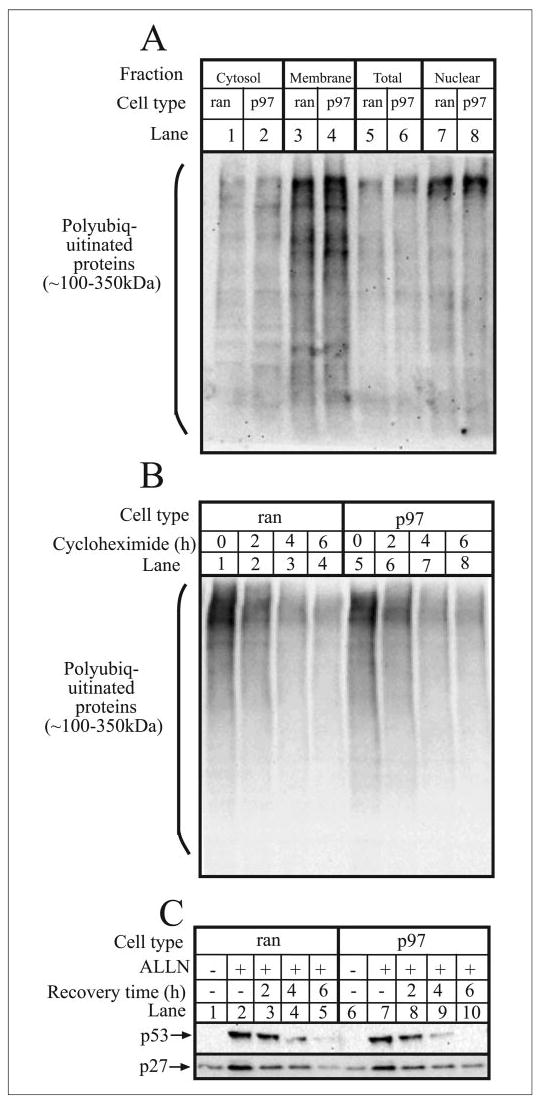
A, ran and p97 cells were harvested, homogenized, and fractionated to prepare cytosol, membrane, and nuclear fractions, as well as total cell lysates. Equal amounts of protein from each fraction were then electrophoresed and immunoblotted with anti-ubiquitin. B, ran and p97 cells were incubated with 25 μg/ml cycloheximide for the times indicated, and equal amounts of cell lysates were electrophoresed and immunoblotted with anti-ubiquitin. C, ran and p97 cells were incubated without or with 20 μg/ml ALLN for 4 h, after which ALLN was removed, and cells were washed and allowed to recover for the times indicated. Equal amounts of cell protein were then electrophoresed and immunoblotted with anti-p53 and anti-p27.
Effects of p97 Knockdown on IP3R1 Processing
Fig. 4A, lanes 1–3, shows that ET1-induced IP3R1 ubiquitination in ran cells was kinetically and quantitatively similar to that seen in unmodified Rat-1 cells (Fig. 1B), indicating that the expression of control siRNA does not affect IP3R processing. In contrast, in p97 cells (Fig. 4A, lanes 4–6), ET1-induced accumulation of ubiquitinated IP3R1s was significantly enhanced such that at both 20 and 60 min, the amount of ubiquitinated IP3R1s in p97 cells was approximately twice that seen in ran cells (Fig. 4B). To determine whether this effect was due to an increase in the rate of IP3R1 ubiquitination or a reduction in the rate of ubiquitinated IP3R1 processing, ran and p97 cells were incubated with ET1 in the presence of bortezomib to block degradation of ubiquitinated IP3R1. Under these conditions, IP3R1 was ubiquitinated equally in ran and p97 cells (Fig. 3, C and D), indicating that the rate of IP3R1 ubiquitination was not enhanced in p97 cells. Thus, the marked accumulation of ubiquitinated IP3R1 in p97 cells was due to reduced processing of ubiquitinated IP3R1.
Fig. 4, E and F, show the effect of p97 knockdown on ET1-induced IP3R down-regulation. Both IP3R1 and IP3R3 were down-regulated in ran cells (Fig. 4, E, lanes 1–3, and F) similarly to that seen in unmodified Rat-1 cells (Fig. 1C), showing that this process was unaffected by stable expression of control siRNA. However, down-regulation of IP3R1 and IP3R3 was significantly inhibited in p97 cells (Fig. 4, E, lanes 4–6, and F). Together with the enhanced accumulation of ubiquitinated IP3R1 in p97 cells, these data indicate that p97 plays a key role in processing IP3R1 once it has been ubiquitinated.
Exogenous p97 Reverses the Effects of Endogenous p97 Knockdown
To confirm that the defect in IP3R processing in p97 cells was truly due to p97 knockdown, we expressed exogenous HA-p97 in p97 cells. Expression of HA-p97 was confirmed in immunoblots with anti-HA (Fig. 4G, upper panel) and anti-p97 (Fig. 4G, middle panel) and raised total p97 immunoreactivity in the five cell lines chosen for analysis to 88 ± 5% of that seen in ran cells. The growth and morphology of HA-p97-expressing cells was identical to ran cells, and the expression level of Ufd2, a representative control protein, was not affected by HA-p97 expression (Fig.4G, lowerpanel). Importantly, the expression of exogenous p97 restored IP3R1 ubiquitination and down-regulation to a level not significantly different from that seen in ran cells (Fig. 4, H and I). These data demonstrate that expression of exogenous p97 can overcome the effects of endogenous p97 knockdown and confirm that p97 plays a role in IP3R processing.
DISCUSSION
The ATPase p97 forms a complex with heterodimers of Ufd1 and Npl4 (46, 47) and plays a role in the UPP (34, 36–39). Each member of the p97-Ufd1-Npl4 complex can bind ubiquitin (35, 46, 47), and with regard to ERAD, it has been proposed that p97 initially binds a non-ubiquitinated segment of ERAD substrates en route from the ER and then, in concert with Ufd1 and Npl4, binds polyubiquitin chains as they are attached to the substrate (35). Additionally, it is thought that p97 uses ATP hydrolysis to translocate ubiquitinated proteins from the ER membrane to the proteasome (34–39). However, these conclusions about p97 function are based on data from a limited repertoire of ERAD substrates often expressed in model systems, and it remains unclear what role p97 and its cofactor play in the ERAD of endogenously expressed proteins in mammalian cells.
In the present study, we provide two lines of evidence that implicate the p97-Ufd1-Npl4 complex in the regulated ERAD of endogenously expressed IP3Rs. First, it interacted with IP3R1 in cells stimulated with GPCR agonists that induced IP3R1 ubiquitination. This interaction was widespread, since it was detected in a variety of cell types, including αT3-1 mouse gonadotropes stimulated with GnRH (Fig. 1A), Rat-1 fibroblasts stimulated with ET1 (Fig. 1D), and SH-SY5Y human neuroblastoma cells stimulated with carbachol,3 a muscarinic agonist. Furthermore, the amount of co-precipitating p97-Ufd1-Npl4 complex correlated closely with the amount of ubiquitinated IP3R1. Interestingly, our data also showed that Ufd2 interacted with ubiquitinated IP3R1 in parallel with the pattern seen for the p97-Ufd1-Npl4 complex. Ufd2 is a U-box-containing protein that has been described as a multiubiquitin chain assembly factor, E4 (52), or as a ubiquitin-protein ligase, E3 (53, 54). Ufd2 and its homologues interact with ubiquitin-protein conjugates via the U-box domain, a stretch of ~70 amino acids that is present in U-box proteins from yeast to man (53). It is also known to interact with p97 (53). We are currently investigating the functional relevance of the interaction of Ufd2 with IP3Rs.
The second line of evidence is that p97 knockdown in Rat-1 fibroblasts caused a marked increase in the accumulation of ubiquitinated IP3R1 in ET1-stimulated cells, with concomitant inhibition of IP3R down-regulation. These effects could be attributed to inhibition of the degradation of ubiquitinated IP3Rs and were not due to perturbation of signaling induced by ET1 receptor activation, an increase in basal IP3R levels or alteration of other cell processes. Furthermore, the specificity of the effects of p97 knockdown was confirmed by experiments showing that expression of exogenous HA-p97 in p97 cells restored the degradation of ubiquitinated IP3Rs. Thus, it appears that p97 (and by extension, the p97-Ufd1-Npl4 complex) plays a key role in IP3R processing after ubiquitination has occurred. The role of the complex is most likely in the recognition and translocation of ubiquitinated IP3Rs from the ER membrane.
It seems somewhat surprising that p97 knockdown inhibited IP3R processing, while all other indices of UPP function were unchanged. p97 cells were indistinguishable from ran cells in terms of growth and morphology, the amounts and subcellular distribution of ubiquitin-protein conjugates, the rate of degradation of the total cellular pool of ubiquitin-protein conjugates, and the rate of degradation of specific UPP substrates (p27 and p53). Taken together, these data show that Rat-1 cells can withstand a 62 ± 3% reduction in p97 expression without deleterious effects, indicating that the remaining p97 is sufficient to maintain cell function. Why then is IP3R processing inhibited? The most likely reason is that IP3Rs are relatively abundant proteins (3, 4), and their activation after cell stimulation will almost instantaneously generate relatively large amounts of ubiquitinated IP3Rs that have to be processed. This processing may require a large proportion of the available p97-Ufd1-Npl4 complexes and knocking down p97 will reduce their abundance and thus slow IP3R processing.
It is intriguing that the maximum p97 knockdown we could obtain was only ~62%. This limit may result from a reduction in the viability of cells in which p97 knockdown was >62%; these cells would not proliferate well and would not be well represented in the cell lines we generated. In this regard, other studies in which endogenous p97 has been interfered with have noted that cell function was perturbed. In neuronal PC12 cells overexpressing dominant negative p97, there was generalized accumulation of ubiquitin-protein conjugates in nuclear and membrane fractions, cytoplasmic vacuolization, ER stress and expansion, and cell death (64). In another study, transient transfection of HeLa cells with synthetic siRNAs to knockdown p97 mRNA by ~80% caused accumulation of ubiquitin-protein conjugates, extensive cytoplasmic vacuolization, ER expansion, impaired cell proliferation, and apoptosis (57).
It is important to note that the studies described herein are the first in mammalian cells in which inhibition of endogenous p97 activity has been demonstrated to affect the processing of an ERAD substrate, in the absence of possible confounding effects from UPP perturbation. Furthermore, our studies addressed the regulation of endogenous IP3Rs by endogenous signaling pathways, as opposed to overexpressed proteins activated by non-physiological means. This focus on proteins expressed and activated normally, and the lack of UPP perturbation resulting from p97 knockdown means that the results obtained should accurately reflect the role of p97 under physiological conditions.
If p97 participates in driving ubiquitinated IP3Rs from the ER membrane, what else mediates this retrotranslocation? It has recently been shown that the ER membrane protein derlin-1 interacts with ERAD substrates (65) and might constitute part of the retrotranslocation complex (65). It has also been shown that derlin-1 interacts with VIMP, an ER membrane protein that serves as receptor for p97 (66). Thus, VIMP may recruit p97 to ER membranes and bring it to the vicinity of the retrotranslocation complex. Therefore, based on our findings and those of others, we propose the following model. In response to GPCR stimulation, IP3 is liberated and binds to IP3Rs, inducing conformational changes that render them susceptible to ubiquitination by mamUbc7. Simultaneously, the Ufd1-Npl4 heterodimer and p97, which is tethered to the ER membrane by VIMP, interact with ubiquitinated IP3Rs and transport them through the retrotranslocation complex to the proteasome. Support for this model comes from preliminary experiments showing that derlin-1 co-precipitates with IP3R1 in GnRH-stimulated αT3-1 cells.4
In summary, the current study provides evidence that the p97-Ufd1-Npl4 complex plays a key role in post-ubiquitination but pre-proteasomal processing of IP3Rs in response to GPCR activation. These findings both further our understanding of the mechanism IP3R down-regulation and how ERAD substrates are processed in mammalian cells.
Acknowledgments
We thank Matt Soulsby, Qun Xu, and Sarah Reks for valuable discussions.
Footnotes
This work was supported by National Institutes of Health Grants DK49194 (to R. J. H. W.) and DK56294 (to G. G. K.).
The abbreviations used are: GPCR, G protein-coupled receptor; IP3, inositol 1,4,5-trisphosphate; IP3R, inositol 1,4,5-trisphosphate receptor; ER, endoplasmic reticulum; UPP, ubiquitin-proteasome pathway; ERAD, endoplasmic reticulum-associated degradation; RNAi, RNA interference; siRNA, short interfering RNA; GnRH, gonadotropin-releasing hormone; ET1, endothelin-1; ALLN, N-acetyl-Leu-Leu-norleucinal; HA, hemagglutinin.
K. J. Alzayady and R. J. H. Wojcikiewicz, unpublished data.
M. M. Panning and R. J. H. Wojcikiewicz, unpublished data.
References
- 1.Patterson RL, Boehning D, Snyder SH. Annu Rev Biochem. 2004;73:437–465. doi: 10.1146/annurev.biochem.73.071403.161303. [DOI] [PubMed] [Google Scholar]
- 2.Berridge MJ, Lipp P, Bootman MD. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 3.Wojcikiewicz RJH. J Biol Chem. 1995;270:11678–11683. doi: 10.1074/jbc.270.19.11678. [DOI] [PubMed] [Google Scholar]
- 4.Taylor CW, Genazzani AA, Morris SA. Cell Calcium. 1999;26:237–251. doi: 10.1054/ceca.1999.0090. [DOI] [PubMed] [Google Scholar]
- 5.Taylor CW, da Fonseca PCA, Morris EP. Trends Biochem Sci. 2004;29:210–219. doi: 10.1016/j.tibs.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 6.Vermassen E, Parys JB, Mauger JP. Biol Cell. 2004;96:3–17. doi: 10.1016/j.biolcel.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 7.Bosanac I, Michikawa T, Mikishiba K, Ikura M. Biochim Biophys Acta. 2004;1742:89–102. doi: 10.1016/j.bbamcr.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 8.Wojcikiewicz RJH, Nahorski SR. J Biol Chem. 1991;266:22234–22241. [PubMed] [Google Scholar]
- 9.Wojcikiewicz RJH, Furuichi T, Nakade S, Mikoshiba K, Nahorski SR. J Biol Chem. 1994;269:7963–7969. [PubMed] [Google Scholar]
- 10.Oberdorf J, Webster JM, Zhu CC, Luo SG, Wojcikiewicz RJH. Biochem J. 1999;339:453–461. [PMC free article] [PubMed] [Google Scholar]
- 11.Wojcikiewicz RJH, Xu Q, Webster JM, Alzayady K, Gao C. J Biol Chem. 2003;278:940–947. doi: 10.1074/jbc.M206607200. [DOI] [PubMed] [Google Scholar]
- 12.Wojcikiewicz RJH, Ernst SA, Yule DI. Gastroenterology. 1999;116:1194–1201. doi: 10.1016/s0016-5085(99)70023-5. [DOI] [PubMed] [Google Scholar]
- 13.Jellerette T, He CL, Wu H, Parys JB, Fissore RA. Dev Biol. 2000;223:238–250. doi: 10.1006/dbio.2000.9675. [DOI] [PubMed] [Google Scholar]
- 14.Sipma H, Deelman L, De Smedt H, Missiaen L, Parys JB, Vanlingen S, Henning RH, Casteels R. Cell Calcium. 1998;23:11–21. doi: 10.1016/s0143-4160(98)90070-7. [DOI] [PubMed] [Google Scholar]
- 15.Bokkala S, Joseph SK. J Biol Chem. 1997;272:12454–12461. doi: 10.1074/jbc.272.19.12454. [DOI] [PubMed] [Google Scholar]
- 16.Tovey SC, de Smet P, Lipp P, Thomas D, Young KW, Missiaen L, De Smedt H, Parys JB, Berridge MJ, Thuring J, Holmes A, Bootman MD. J Cell Sci. 2001;114:3979–3989. doi: 10.1242/jcs.114.22.3979. [DOI] [PubMed] [Google Scholar]
- 17.Wojcikiewicz RJH. Trends Pharmacol Sci. 2004;25:35–41. doi: 10.1016/j.tips.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 18.Glickman MH, Ciechanover A. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 19.Hatakeyama S, Nakayama KI. J Biochem (Tokyo) 2003;134:1–8. doi: 10.1093/jb/mvg106. [DOI] [PubMed] [Google Scholar]
- 20.Kostova Z, Wolf DH. EMBO J. 2003;22:2309–2317. doi: 10.1093/emboj/cdg227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hampton RY. Curr Opin Cell Biol. 2002;14:476–482. doi: 10.1016/s0955-0674(02)00358-7. [DOI] [PubMed] [Google Scholar]
- 22.McCracken AA, Brodsky JL. BioEssays. 2003;25:868–877. doi: 10.1002/bies.10320. [DOI] [PubMed] [Google Scholar]
- 23.Jarosch E, Lenk U, Sommer T. Int Rev Cytol. 2003;223:39–81. doi: 10.1016/s0074-7696(05)23002-4. [DOI] [PubMed] [Google Scholar]
- 24.Saliba RS, Munro PMG, Luthert PL, Cheetham ME. J Cell Sci. 2002;115:2907–2918. doi: 10.1242/jcs.115.14.2907. [DOI] [PubMed] [Google Scholar]
- 25.Ward CL, Omura S, Kopito RR. Cell. 1995;83:121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- 26.Yang M, Omura S, Bonifacino JS, Weissman AM. J Exp Med. 1998;187:835–846. doi: 10.1084/jem.187.6.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tiwari S, Weissman AM. J Biol Chem. 2001;276:16193–16200. doi: 10.1074/jbc.M007640200. [DOI] [PubMed] [Google Scholar]
- 28.Hampton RY, Gardner RG, Rine J. Mol Biol Cell. 1996;7:2029–2044. doi: 10.1091/mbc.7.12.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ravid T, Doolman R, Avner R, Harats D, Roitelman J. J Biol Chem. 2000;275:35840–35847. doi: 10.1074/jbc.M004793200. [DOI] [PubMed] [Google Scholar]
- 30.Zhu CC, Furuichi T, Mikoshiba K, Wojcikiewicz RJH. J Biol Chem. 1999;274:3476–3484. doi: 10.1074/jbc.274.6.3476. [DOI] [PubMed] [Google Scholar]
- 31.Zhu CC, Wojcikiewicz RJH. Biochem J. 2000;348:551–556. [PMC free article] [PubMed] [Google Scholar]
- 32.Webster JM, Tiwari S, Weissman AM, Wojcikiewicz RJH. J Biol Chem. 2003;278:38238–38246. doi: 10.1074/jbc.M305600200. [DOI] [PubMed] [Google Scholar]
- 33.Hirsch C, Jarosch E, Sommer T, Wolf DH. Biochim Biophys Acta. 2004;1695:208–216. doi: 10.1016/j.bbamcr.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 34.Ye Y, Meyer HH, Rapoport TA. Nature. 2001;414:652–656. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- 35.Ye Y, Meyer HH, Rapoport TA. J Cell Biol. 2003;162:71–84. doi: 10.1083/jcb.200302169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elkabetz Y, Shapira I, Rabinovich E, Bar-Nun S. J Biol Chem. 2004;279:3980–3989. doi: 10.1074/jbc.M309938200. [DOI] [PubMed] [Google Scholar]
- 37.Braun S, Matuschewski K, Rape M, Thoms S, Jentsch S. EMBO J. 2002;21:615–621. doi: 10.1093/emboj/21.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rabinovich E, Kerem A, Frohlich KU, Diamant N, Bar-Nun S. Mol Cell Biol. 2002;22:626–634. doi: 10.1128/MCB.22.2.626-634.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bays NW, Wilhovsky SK, Goradia A, Hodgkiss-Harlow K, Hampton RY. Mol Biol Cell. 2001;12:4114–4128. doi: 10.1091/mbc.12.12.4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dreveny I, Pye VE, Beuron F, Briggs LC, Isaacson RL, Matthews SJ, McKeown C, Xuan X, Zhang X, Freemont PS. Biochem Soc Trans. 2004;32:715–720. doi: 10.1042/BST0320715. [DOI] [PubMed] [Google Scholar]
- 41.Wang Q, Song C, Li CCH. J Struct Biol. 2004;146:44–57. doi: 10.1016/j.jsb.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 42.Woodman PG. J Cell Sci. 2003;116:4283–4290. doi: 10.1242/jcs.00817. [DOI] [PubMed] [Google Scholar]
- 43.Wang Y, Satoh A, Warren G, Meyer HH. J Cell Biol. 2004;164:973–978. doi: 10.1083/jcb.200401010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uchiyama K, Jokitalo E, Kano F, Murata M, Zhang X, Canas B, Newman R, Rabouille C, Pappin D, Freemont P, Kondo H. J Cell Biol. 2002;159:855–866. doi: 10.1083/jcb.200208112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kondo H, Rabouille C, Newman R, Levine TP, Pappin D, Freemont P, Warren G. Nature. 1997;388:75–78. doi: 10.1038/40411. [DOI] [PubMed] [Google Scholar]
- 46.Bruderer BM, Brasseur C, Meyer HH. J Biol Chem. 2004;279:49609–49616. doi: 10.1074/jbc.M408695200. [DOI] [PubMed] [Google Scholar]
- 47.Meyer HH, Wang Y, Warren G. EMBO J. 2002;21:5645–5652. doi: 10.1093/emboj/cdf579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bays NW, Hampton RY. Curr Biol. 2002;12:R366–R371. doi: 10.1016/s0960-9822(02)00862-x. [DOI] [PubMed] [Google Scholar]
- 49.Brummelkamp TR, Bernards R, Agami R. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 50.Brummelkamp TR, Bernards R, Agami R. Cancer Cell. 2002;2:243–247. doi: 10.1016/s1535-6108(02)00122-8. [DOI] [PubMed] [Google Scholar]
- 51.Sambrook, J., and Russel, D. W. (2001) Molecular Cloning: A Laboratory Manual, 3rd Ed., pp. 16.14–16.20, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY
- 52.Koegl M, Hoppe T, Schlenker S, Ulrich HD, Mayer TU, Jentsch S. Cell. 1999;96:635–644. doi: 10.1016/s0092-8674(00)80574-7. [DOI] [PubMed] [Google Scholar]
- 53.Hatakeyama S, Yada M, Matsumoto M, Ishida N, Nakayama KI. J Biol Chem. 2001;276:33111–33210. doi: 10.1074/jbc.M102755200. [DOI] [PubMed] [Google Scholar]
- 54.Kaneko C, Hatakeyama S, Matsumoto M, Yada M, Nakayama K, Nakayama KI. Biochem Biophys Res Commun. 2003;300:297–304. doi: 10.1016/s0006-291x(02)02834-6. [DOI] [PubMed] [Google Scholar]
- 55.Xu Q, Farah M, Webster JM, Wojcikiewicz RJH. Mol Cancer Ther. 2004;3:1263–1269. [PubMed] [Google Scholar]
- 56.Muldoon LL, Rodland KD, Forsythe ML, Magun BE. J Biol Chem. 1989;264:8529–8536. [PubMed] [Google Scholar]
- 57.Wojcik C, Yano M, DeMartino GN. J Cell Sci. 2004;117:281–292. doi: 10.1242/jcs.00841. [DOI] [PubMed] [Google Scholar]
- 58.Medema RH. Biochem J. 2004;380:593–603. doi: 10.1042/BJ20040260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sgambato A, Cittadini A, Faraglia B, Weinstein IB. J Cell Physiol. 2000;183:18–27. doi: 10.1002/(SICI)1097-4652(200004)183:1<18::AID-JCP3>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 60.Sutcliffe JE, Brehm A. FEBS Lett. 2004;567:86–91. doi: 10.1016/j.febslet.2004.03.122. [DOI] [PubMed] [Google Scholar]
- 61.Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, Yew PR, Draetta GF, Rolfe M. Science. 1995;269:682–685. doi: 10.1126/science.7624798. [DOI] [PubMed] [Google Scholar]
- 62.Vlach J, Hennecke S, Amati B. EMBO J. 1997;16:5334–5344. doi: 10.1093/emboj/16.17.5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maki CG, Huibregtse JM, Howley PM. Cancer Res. 1996;56:2649–2654. [PubMed] [Google Scholar]
- 64.Kobayashi T, Tanaka K, Inoue K, Kakizuka A. J Biol Chem. 2002;277:47358–47365. doi: 10.1074/jbc.M207783200. [DOI] [PubMed] [Google Scholar]
- 65.Lilley BN, Ploegh HL. Nature. 2004;429:834–840. doi: 10.1038/nature02592. [DOI] [PubMed] [Google Scholar]
- 66.Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA. Nature. 2004;429:841–847. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]