

Abstract
Previously, we reported that (S)-3,5-dihydroxypenylglycine (DHPG), an agonist for group I metabotropic glutamate receptors (mGluRs), stimulates CK1 and Cdk5 kinase activities in neostriatal neurons, leading to enhanced phosphorylation, respectively, of Ser-137 and Thr-75 of DARPP-32 (dopamine and cAMP-regulated phosphoprotein, 32 kDa). We have now investigated the signaling pathway that leads from mGluRs to casein kinase 1 (CK1) activation. In mouse neostriatal slices, the effect of DHPG on phosphorylation of Ser-137 or Thr-75 of DARPP-32 was blocked by the phospholipase Cβ inhibitor U73122, the Ca2+ chelator 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA/AM), and the calcineurin inhibitor cyclosporin A. In neuroblastoma N2a cells, the effect of DHPG on the activity of transfected HA-tagged CK1ε was blocked by BAPTA/AM and cyclosporin A. In neostriatal slices, the effect of DHPG on Cdk5 activity was also abolished by BAPTA/AM and cyclosporin A, presumably through blocking activation of CK1. Metabolic labeling studies and phosphopeptide mapping revealed that a set of C-terminal sites in HA-CK1ε were transiently dephosphorylated in N2a cells upon treatment with DHPG, and this was blocked by cyclosporin A. A mutant CK1ε with a nonphosphorylatable C-terminal domain was not activated by DHPG. Together, these studies suggest that DHPG activates CK1ε via Ca2+-dependent stimulation of calcineurin and subsequent dephosphorylation of inhibitory C-terminal autophosphorylation sites.
Casein kinase 1 (CK1)1 was one of the first serine/threonine protein kinases to be isolated and characterized. There are at least seven mammalian CK1 isoforms (α, β, γ1, γ2, γ3, δ, and ε (1, 2)), and distinct CK1 family members are likely to have a variety of roles in eukaryotic cells. An increasing number of potential physiologic substrates for CK1 isoforms have been identified. CK1α phosphorylates M1 and M3 muscarinic receptors and rhodopsin in an agonist-dependent manner (3, 4). CK1ε and CK1δ phosphorylate N-terminal residues of p53 in vitro and in vivo, and DNA-damaging drugs enhance this activity (4–6). CKIε is an important regulator of β-catenin in the Wnt pathway; CKIε mimicked Wnt in inducing a secondary axis in Xenopus, stabilizing β-catenin, and stimulating β-catenin-dependent gene transcription (7–11). In Drosophila, the double-time gene product, a CK1ε homolog, has been found to interact with dPER and regulate circadian cycle length (12). CK1δ and CK1ε have also both been implicated in the regulation of the circadian clock in mammals (13–15).
CK1 family members contain a highly related, central kinase domain that is flanked by N- and C-terminal extensions of variable length. The amino acid sequences of the C-terminal extensions are in general not highly related. However, the 124-amino acid C-terminal domain of mammalian CK1ε is 50% identical to that of CK1δ. Notably, several in vitro studies have shown that the activities of CK1δ and CK1ε are regulated by autophosphorylation of their respective C-terminal domains (16, 17). Autophosphorylation of more than eight sites leads to inhibition of kinase activity. Moreover, it has been shown in vitro that treatment of CK1ε with several different serine/threonine phosphatases including PP1, PP2A, and PP2B (calcineurin) causes a marked increase in kinase activity (13, 17, 18). Dephosphorylation of CK1δ and CK1γ isoforms by the catalytic subunit of PP1 has also been found in vitro to result in enzyme activation (16).
Recently, we have reported that both CK1 and Cdk5 are regulated by activation of metabotropic glutamate receptors (mGluRs) in neostriatal neurons (19). DHPG, an agonist for group I mGluRs, increased CK1 and Cdk5 activities in neostriatal slices, leading to enhanced phosphorylation of Ser-137 and Thr-75 of DARPP-32, respectively. The effects of DHPG on both Ser-137 and Thr-75 were blocked by CK1–7 and IC261, specific inhibitors of CK1, suggesting that activation of Cdk5 by mGluRs required activation of CK1. In support of this possibility, the DHPG-induced increase in Cdk5 activity, subsequently measured in extracts of neostriatal slices, was abolished by treatment of slices with CK1–7 or IC261. Finally, treatment of acutely dissociated neurons with DHPG enhanced voltage-dependent Ca2+ currents. This enhancement was eliminated by either CK1–7 or butyrolactone (an inhibitor of Cdk5), indicating that CK1 and Cdk5 may be involved in the regulation by mGluR agonists of Ca2+ channels.
In the present study, we have investigated the processes that lead from mGluRs to CK1 activation and the mechanism that underlies CK1 activation in response to group I mGluR agonists. The results obtained support a signal transduction pathway in which group I mGluRs increase intracellular Ca2+ and stimulate calcineurin to dephosphorylate autoinhibitory phosphorylation sites in CK1ε. Transient dephosphorylation and subsequent autophosphorylation of CK1ε leads to transient activation and inactivation, respectively, of the enzyme.
MATERIALS AND METHODS
Antibodies, Plasmids, and Chemicals
Phosphospecific antibodies that recognize either phospho-Ser-137 DARPP-32 or phospho-Thr-75 DARPP-32 were developed as described (19, 20). The expression plasmids pCDP4HA-CKIε and pCS-Myc-MM2-CK1ε were prepared as described (18). Anti-HA (12CA5) was obtained from Roche Molecular Biochemicals and anti-Myc (9E10) from Upstate Biotechnology. Anti-Cdk5 (C-8) and anti-CKIε were obtained from Santa Cruz Biotechnology. U73122, BAPTA/AM, and cyclosporin A were obtained from Calbiochem; (S)-3,5-DHPG, ZM241385, and L-AP3 were obtained from Tocris. Protease inhibitor mixture tablets were obtained from Roche Molecular Biochemicals. Lambda protein phosphatase was obtained from Upstate Biotechnology.
Preparation and Treatment of Striatal Slices
Neostriatal slices were prepared from male C57/BL6 mice (6–8 weeks old) as described (21). Briefly, coronal (usually 3–4/mouse) slices (350 μm) were prepared using a vibratome. From each coronal slice, two neostriatal slices (left and right) were dissected. When slices were treated with drugs, one slice from a pair served as a control for the drug-treated slice. After drug treatment, slices were immediately frozen in liquid nitrogen and stored at −80 °C until assayed.
Immunoblotting
Frozen slices were sonicated in hot homogenization buffer containing 1% SDS and 50 mm NaF, and samples were boiled for 10 min. SDS-PAGE sample buffer was then added, and samples were boiled for 5 min. Samples (~120 μg protein) were separated by SDS-PAGE (10% polyacrylamide) and transferred to nitrocellulose. Immunoblots were first probed with anti-phospho-Ser-137 DARPP-32 antibody. The blots were stripped and probed with anti-phospho-Thr-75 DARPP-32 antibody. Blots were stripped again and probed with anti-total DARPP-32 antibody. Antibody binding was detected by enhance chemiluminescence (ECL) using x-ray film, and images were analyzed by laser scanning densitometry using NIH Image 1.52 software. Data were statistically analyzed by Student’s t test in Microsoft Excel software as indicated. For each neostriatal slice sample, the level of phospho-Ser-137 or phospho-Thr-75 was normalized to the total level of DARPP-32. In every individual experiment (i.e. for each mouse brain), a control without drug and a control with DHPG were always included. Results from slices from a single mouse brain were normalized to the control slice without drug (arbitrarily set as 1). The figures show ECL blots that were obtained in some cases from different mouse brains and from different experiments. The cumulative data shown in the bar graphs were obtained from at least three independent experiments.
Transfection, Immunoprecipitation, and Assay of CK1 and Cdk5
Neuroblastoma N2a cells were cultured to 50–60% confluence in Dulbecco’s modified Eagle’s medium containing 5% fetal bovine serum. Four μg of the expression plasmid for HA-CK1ε or Myc-MM2-CK1ε was transfected into N2a cells in 100-mm dishes using FuGENE™ 6. Twenty-four hours after transfection, cells were incubated at room temperature in phosphate-buffered Krebs-Henseleit solution (Sigma) for 10 min and then with or without inhibitors for 30 min before treatment with (S)-3,5-DHPG for 2 min. Cells were then lysed in 1 ml of radioimmune precipitation buffer containing 1% Nonidet P-40, 150 mm NaCl, 0.1% SDS, 50 mm Tris, pH 8.0, 5 mm Na3VO4, 20 mM NaF, 20 mm β-glycerol-phosphate, and protease inhibitors. Lysates were centrifuged at 10,000 × g, and supernatants were used for immunoprecipitation and kinase assay.
For immunoprecipitation of CK1ε from N2a cells, lysates (1 mg of total protein) were precleared with 5 μl of mouse IgG (ICN) and 50 μl of protein A-agarose for 30 min. Five μl (~2 μg) of anti-HA antibody was added, and samples were incubated for 1 h at 4 °C. Five μl of anti-mouse rabbit IgG and 50 μl of protein A-agarose were then added for 45 min. Immunocomplexes were washed three times in lysis buffer and two times in kinase buffer (30 mm Hepes, pH 7.5, 7 mm MgCl2, 0.5 mM dithiothreitol).
CK1 assays were performed in a 30-μl assay volume with 2 μg of purified DARPP-32, 500 μm ATP, and 5 μCi of [γ-32P]ATP. Samples were incubated at 30 °C for 10 min, and reactions were stopped by the addition of SDS sample buffer and boiled for 5 min. Samples were separated by SDS-PAGE (12% polyacrylamide). SDS-polyacrylamide gels were dried and exposed to Kodak film for autoradiography. Results were quantified using a PhosphorImager (Amersham Biosciences). The amount of HA-CK1ε in each immunoprecipitated sample was determined by immunoblotting using an anti-HA antibody. Kinase activity in each immunoprecipitated sample was normalized to total HA-CK1ε. Immunoprecipitation and assay of Cdk5 were performed as described (19).
Immunoprecipitated CKIε, from N2a cells treated with DHPG, was added to a mixture consisting of 50 mm Tris-HCl, 0.1 mm Na2EDTA, 5 mm dithiothreitol, 2 mm MnCl2, and 200 units of lambda phosphatase (Upstate Biotechnology, no. 14–405). Control reactions without lambda protein phosphatase were also performed. Dephosphorylation reactions were incubated at 37 °C for 15 min. To stop the reactions, beads with CKIε were washed three times with radioimmune precipitation buffer and two times with kinase buffer (30 mm Hepes, pH 7.5, 7 mm MgCl2, 0.5 mm dithiothreitol). Kinase activity was measured as described above.
Metabolic Labeling and Two-dimensional Phosphopeptide Mapping
Twenty-four hours after transfecting N2a cells with CK1ε expression plasmids, cells were incubated in 200 μCi/ml (PerkinElmer Life Sciences) of 32P-inorganic phosphate and phosphate-free, serum-free Dulbecco’s modified Eagle’s medium for 2 h. Cyclosporin A was added to the transiently transfected cultures at a final concentration at 1 μm during the last 30 min of metabolic labeling. Cells were treated with DHPG for various periods of time as indicated, harvested by lysis in radioimmune precipitation buffer, and clarified by centrifugation at 14,000 × g for 10 min. Soluble extracts containing HA- or Myc-tagged proteins were immunoprecipitated with 12CA5 or 9E10 monoclonal antibody and protein A-agarose. The immunoprecipitates were eluted from the protein A-agarose and separated by SDS-PAGE on 10% gel. Protein was stained briefly with Coomassie Brilliant Blue, the gels were dried, and the labeled protein was visualized by autoradiography. Radioactivity was determined using a PhosphorImager and ImageQuant software (Amersham Biosciences).
Radiolabeled protein bands were excised, rehydrated, destained, and dried in a Speedvac. The gel slices were minced and rehydrated in 75 μg/ml TPCK/trypsin in 50 mm NH4CO3H (1 ml final volume) for 24 h at 37 °C. The supernatant was removed from the gel slices and then lyophilized to dryness. Recovery of tryptic phosphopeptides was determined by Cerenkov counting. The two-dimensional peptide mapping method was used to separate phosphopeptides. Lyophilized tryptic peptides were suspended in 10 μl of electrophoresis buffer (10% acetic acid and 1% pyridine, pH 3.5) and spotted onto thin-layer cellulose plates (20 × 20 cm, Analtech). Electrophoresis was carried out at 400 V for 1.5 h. Following electrophoresis, cellulose plates were dried and then subjected to ascending chromatography in buffer containing 25% l-butanol, 7.5% acetic acid, and 37.5% pyridine. Phosphopeptides were visualized using a PhosphorImager and radioactivity in individual phosphopeptides was measured using ImageQuant software (Amersham Biosciences).
RESULTS
The Effect of DHPG on Ser-137 and Thr-75 phosphorylation of DARPP-32 Is Blocked by U73122, BAPTA, and Cyclosporin A
Previously we showed that DHPG, an agonist for group I mGluRs, increased CK1 and Cdk5 activities in neostriatal slices, leading to enhanced phosphorylation of Ser-137 and Thr-75 of DARPP-32, respectively. Activation of group I mGluRs results in the stimulation of phosphoinositide hydrolysis (22). Therefore, one possible mechanism for DHPG-dependent activation of CK1 might involve activation of PLCβ. However, it has also been reported that DHPG can potentiate the response of adenosine A2a receptors to agonist (23, 24), raising the possibility of an involvement of other signal transduction pathways. We tested the effect of the specific PLCβ inhibitor, U73122, in slices. Preincubation with 12.5 μm U73122 for 20 min did not change the basal phosphorylation of Ser-137 or Thr-75, but the effect of DHPG was abolished (Fig. 1). In contrast, the adenosine A2a receptor antagonist, ZM241385 (10 μm), did not affect the ability of DHPG to stimulate phosphorylation of Ser-137 or Thr-75. These results support a role for a signal transduction pathway involving PLCβ.
Fig. 1. The effect of DHPG on Ser-137 and Thr-75 phosphorylation of DARPP-32 is blocked by the PLCβ inhibitor U73122, the Ca2+ chelator BAPTA/AM, and the calcineurin inhibitor cyclosporin A.
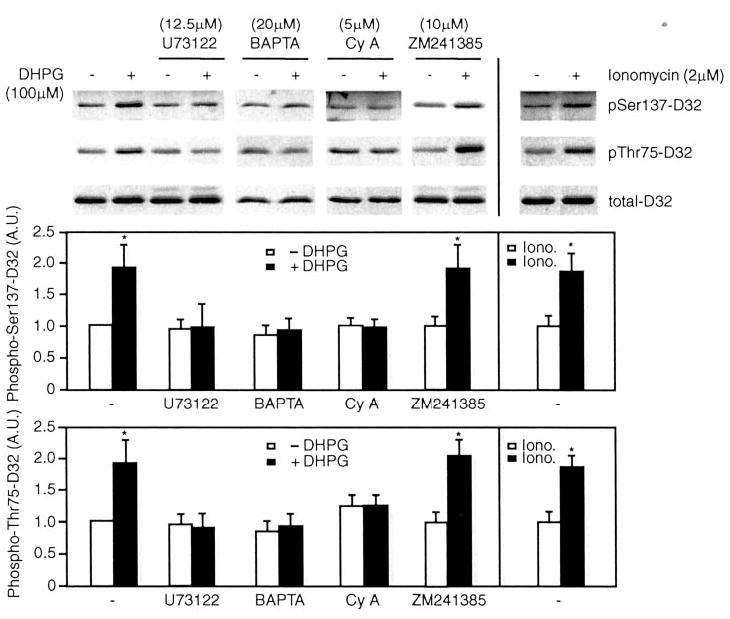
The effect of the mGluR group I agonist, DHPG, on phosphorylation of DARPP-32 at Ser-137 (CK1 site) and Thr-75 (Cdk5 site) was examined in mouse neostriatal slices using phosphorylation state-specific antibodies. Slices were treated without or with DHPG (100 μm) or ionomycin (Iono., 2 μm) for 2 min following preincubation with vehicle (U73122, 12.5 μm for 20 min), BAPTA/AM (BAPTA, 20 μm for 20 min), cyclosporin A (Cy A, 5 μm for 60 min), or the adenosine A2a receptor antagonist (ZM241385, 10 μm for 20 min). Slices were homogenized and analyzed by SDS-PAGE and immunoblotting using phospho-Ser-137, phospho-Thr-75, and total DARPP-32 antibodies. Immunoblots are shown in the top panel, and cumulative data (means ± S.E.) obtained from three experiments are shown in graphical format in the lower panels. Data for each sample were normalized to the total level of DARPP-32. Data were then normalized to the value obtained in the absence of any addition (−DHPG, set as 1). *, p < 0.05, Student’s t test, compared with untreated slices.
Activation of PLCβ leads to production of inositol 1,4,5-triphosphate (IP3) and release of Ca2+ from the endoplasmic reticulum. To examine the role of Ca2+, we used the Ca2+chelator, BAPTA/AM, in studies in slices. Preincubation with 20 μm BAPTA/AM did not change the basal phosphorylation of DARPP-32, but the effect of DHPG was abolished (Fig. 1). Moreover, treatment of slices with the Ca2+ ionophore, ionomycin (2 μm), resulted in increased phosphorylation of both Ser-137 and Thr-75 of DARPP-32 (Fig. 1). Based on previous studies of the regulation of CK1ε (13, 17, 18), we hypothesized that increased intracellular Ca2+ might activate a Ca2+-dependent protein phosphatase to dephosphorylate inhibitory autophosphorylation sites on CK1. The Ca2+-dependent phosphatase, calcineurin (PP2B), is expressed at high levels in striatum (25). Treatment of slices with cyclosporin A (5 μm), a specific calcineurin inhibitor, for 1 h attenuated the effect of DHPG on phosphorylation of Ser-137 and Thr-75 of DARPP-32 (Fig. 1).
The Effect of DHPG on CKIε Activity Is Blocked by U73122, BAPTA, and Cyclosporin A
To further characterize the effect of DHPG on CK1ε activity, we used a transfection system. An expression plasmid containing HA-tagged CK1ε was transiently transfected into N2a cells, and cells were treated with DHPG for 2 min. CK1ε was immunoprecipitated using anti-HA antibody, and CK1ε activity was assayed using DARPP-32 as substrate (Fig. 2). An initial screen for different subtypes of group I mGluRs indicated that mGluR1 is expressed in N2a cells. For example, treatment of cells with DHPG resulted in an increase in CK1ε activity; this effect was blocked by the group I mGluR antagonist L-AP3 (Fig. 2). Preincubation of cells with U73122 (10 μm), BAPTA/AM (20 μm), or cyclosporin A (1 μm) abolished the effect of DHPG on CK1ε activity, consistent with a role for Ca2+-dependent activation of calcineurin in the regulation of CK1ε.
Fig. 2. The effect of DHPG on CK1ε activity is blocked by BAPTA/AM and cyclosporin A.
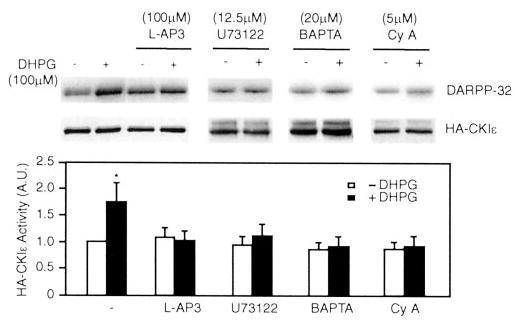
N2a cells were transiently transfected with HA-tagged CK1ε. Cells were preincubated without or with the group I mGluR antagonist L-AP3 (100 μm for 20 min), U73122 (12.5 μm for 20 min), BAPTA/AM (BAPTA, 20 μm for 20 min), or cyclosporin A (Cy A, 1 μm for 20 min) prior to treatment without or with DHPG (100 μm for 2 min). HA-CK1ε was immunoprecipitated, and CK1 was assayed using DARPP-32 as a substrate. Samples were analyzed by SDS-PAGE and autoradiography. Top panel, autoradiogram of DARPP-32 phosphorylation; middle panel, immunoblot using HA antibody; bottom panel, cumulative data obtained from five experiments (means ± S.E.). Data for each sample were normalized to the total level of HA-CK1ε. Data were then normalized to the value obtained in the absence of any addition (−DHPG, set as 1). Data were normalized to values for untreated cells. *, p < 0.05, Student’s t test, compared with untreated cells.
DHPG Regulates Cdk5 Kinase Activity through a PLCβ/Ca2+/Calcineurin Pathway
Previously we demonstrated by using specific CK1 inhibitors that group I mGluRs activate Cdk5 kinase activity via a pathway that involves CK1. To further examine whether Cdk5 activation by DHPG is through a PLCβ/Ca2+/calcineurin pathway, we analyzed Cdk5 kinase activity following its immunoprecipitation from mouse neostriatal slices. Preincubation of mouse neostriatal slices with U73122 (12.5 μm), BAPTA/AM (20 μm) for 30 min, or cyclosporin A (5 μm) for 60 min abolished the effect of DHPG on Cdk5 activity (Fig. 3). Treatment of slices with ionomycin (2 μm) for 2 min also resulted in an increase in Cdk5 kinase activity by 2-fold; the effect of ionomycin was blocked by cyclosporin A (5 μm).
Fig. 3. The effect of DHPG on Cdk5 activity is blocked by U73122, BAPTA/AM, and cyclosporin A.
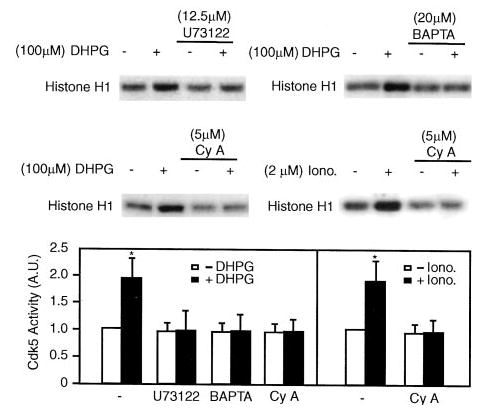
Mouse neostriatal slices were preincubated with U73122 (12.5 μm for 20 min), BAPTA/AM (BAPTA, 20 μm for 20 min), or cyclosporin A (Cy A, 5 μm for 60 min) and then without or with DHPG (100 μm for 2 min) or ionomycin (Iono., 2 μm for 2 min). Slices were homogenized, and Cdk5 was immunoprecipitated with anti-Cdk5 (C-8) antibody. Cdk5 activity was assayed using histone H-1 as substrate, and samples were analyzed by SDS-PAGE and autoradiography. The top and middle panels show autoradiograms indicating histone H-1 phosphorylation. The bottom panel shows cumulative data (means ± S.E.) from three experiments. Data for each sample were normalized to the total level of cdk5 (determined by immunoblotting, not shown). Data were then normalized to the value obtained in the absence of any addition (−DHPG, set as 1). *, p < 0.05, Student’s t test, compared with untreated slices.
DHPG Treatment Induces Transient Dephosphorylation of CKIε
The ability of cyclosporin A to block the effect of DHPG suggested that the regulation of CK1ε by DHPG might involve direct dephosphorylation of CK1ε by calcineurin. To examine the phosphorylation state of CK1ε in response to DHPG, N2a cells that expressed HA-CK1ε were metabolically labeled with 32P and then treated with DHPG for various periods of time. HA-CK1ε was immunoprecipitated, separated by SDS-PAGE (Fig. 4a), and then subjected to two-dimensional phosphopeptide mapping (Fig. 4b). There was little apparent change in the total level of phosphorylation of CK1ε after incubation of cells with DHPG (Fig. 4a). However, peptide mapping revealed that DHPG treatment resulted in rapid and transient dephosphorylation of a subset of phosphopeptides (Fig. 4b). At time 0 (in the absence of DHPG), wild-type CK1ε was found to be strongly phosphorylated at one site (basic peptide labeled “C ” in Fig. 4b, top left). In addition, 7–10 negatively charged peptides were phosphorylated (acidic sites, circled in Fig. 4b, top left). Treatment with DHPG for 2 or 4 min resulted in the dephosphorylation of the acidic peptides, whereas there was no dephosphorylation of the control (basic) peptide. Indeed, after calculating the relative radioactivity in the acidic peptides and in the C peptide, phosphorylation of the C peptide actually increased ~2-fold at 2 or 4 min. Ten minutes after DHPG treatment, the phosphorylation level of the acidic peptides, and also of the C peptide, returned close to the same levels observed in the “0 min” time point sample. Preincubation of cells with cyclosporin A (1 μm) for 30 min before the addition of DHPG prevented the transient dephosphorylation of the acidic peptides in CK1ε (Fig. 4c, measured at the 4 min time point). Together these results indicate that DHPG stimulates calcineurin and results in transient dephosphorylation of a subset of autophosphorylation sites in CK1ε, whereas phosphorylation of a separate site increases transiently.
Fig. 4. CK1ε is transiently dephosphorylated upon DHPG treatment.
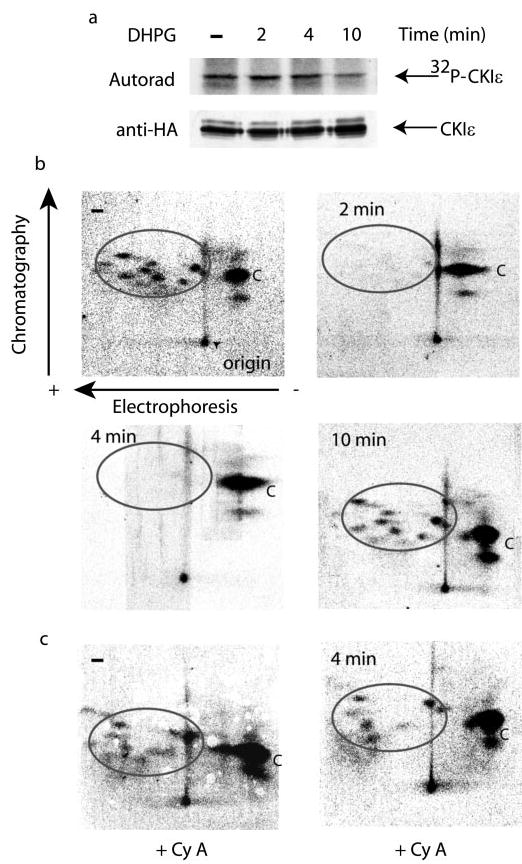
N2a cells were transiently transfected with HA-CK1ε. Cells were incubated with 200 μCi/ml H3 32PO4 in phosphate-free medium for 2 h. For the last 30 min of labeling, cells were treated without or with cyclosporin A (Cy A, 1 μm) and then without or with DHPG for various times as indicated. a, HA-CK1ε was immunoprecipitated, and samples were analyzed by SDS-PAGE and autoradiography (upper panel). The lower panel shows an immunoblot using HA antibody. Radioactivity was determined using a PhosphorImager and Image-Quant software. The values obtained were: − DHPG, 10522; 2 min DHPG, 12204; 4 min DHPG, 10606; 10 min DHPG, 9540. b, gel bands containing 32P-labeled HA-CK1ε were subjected to two-dimensional tryptic phosphopeptide mapping. Electrophoresis was in the horizontal direction (positive electrode at left, point of origin marked by arrowhead in top left), and chromatography was in the vertical direction. Radioactivity in the phosphopetides for each peptide map was determined using a PhosphorImager and ImageQuant software. To account for variations in the peptide mapping process, the ratios in the radioactivity of the circled peptides and the C peptide were used to calculate the absolute radioactivity from the values obtained for phosphorylation of HA-CK1ε (see above). The values obtained were: − DHPG, 4840 in the circled peptides, 5682 in the C peptide; 2 min DHPG, 366 in the circled peptides, 11900 in the C peptide; 4 min DHPG, 0 in the circled peptides, 10606 in the C peptide; 10 min DHPG, 3339 in the circled peptides, 6201 in the C peptide. c, cells transfected with HA-CK1ε were incubated with cyclosporin A (Cy A) and DHPG for 0 or 4 min, as indicated. Cell extracts were analyzed as described in a and b.
C-terminal Autophosphorylation of CKIε Is Involved in Regulation of Its Activity by DHPG
Eight phosphorylation sites in the C-terminal domain of CK1ε were identified as probable in vivo autophosphorylation sites (18). To further examine the details of CK1ε activation, we tested a mutant of CK1ε, MM2, that lacked the eight sites (S323A/T325A/T334A/T337A/S368A/S405A/T407A/S408A). Myc-MM2-CK1ε was transiently transfected into N2a cells, immunoprecipitated, and subjected to phosphopeptide mapping and kinase activity assay. Phosphopeptide mapping revealed that MM2-CK1ε was autophosphorylated only at the control (basic) peptide, and treatment with DHPG had no effect on phosphorylation of this site (Fig. 5a and data not shown). Treatment with DHPG had no effect on MM2-CK1ε activity assayed using DARPP-32 as substrate, and this was also unaffected by preincubation with cyclosporin A (Fig. 5b). The expression level of Myc-tagged MM2-CK1ε in N2a cells was about the same as the HA-tagged wild-type CK1ε, but the immunoprecipitated MM2-CK1ε was ~2-fold more active than HA-CK1ε (Fig. 5b).
Fig. 5. A CK1ε mutant lacking inhibitory autophosphorylation sites is not activated by DHPG.
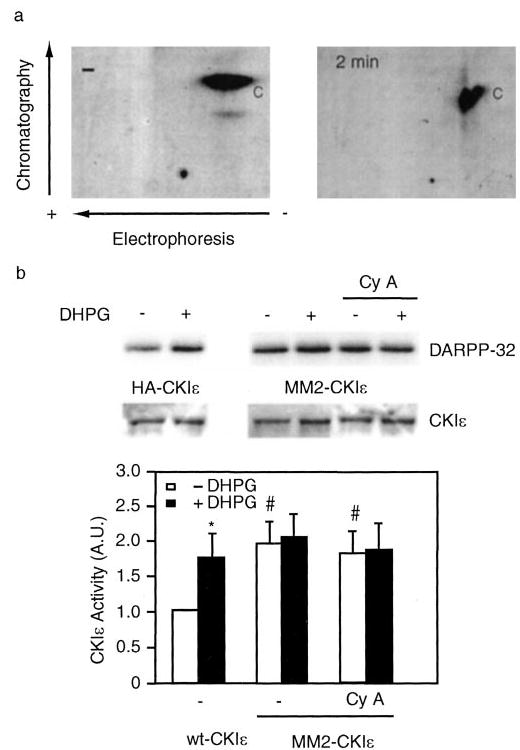
N2a cells were transiently transfected with either wild-type HA-tagged CK1ε or Myc-tagged MM2-CK1ε, a mutant enzyme in which Ser-323, Thr-325, Thr-334, Thr-337, Ser-368, Ser-405, Thr-407, and Ser-408 are mutated to alanine. a, cells were labeled with 200 μCi/ml H3 32PO4 in phosphate-free medium for 2 h and treated with DHPG for 2 min. HA- or Myc-tagged CKIε was immunoprecipitated, analyzed by SDS-PAGE and subjected to phosphopeptide mapping as described in the legend to Fig. 4. b, cells were pretreated without or with cyclosporin A (Cy A, 1 μm for 30 min) prior to incubation without or with DHPG (100 μm for 2 min). HA-CK1ε or MM2-CK1ε was immunoprecipitated, and CK1ε activity was assayed using DARPP-32 as a substrate. Samples were analyzed by SDS-PAGE and autoradiography. Top panel, autoradiogram of DARPP-32 phosphorylation; middle panel, immunoblot showing expression of HA- or Myc-tagged CK1ε using an anti-CK1 antibody that recognized both wild-type and MM2-CK1ε; bottom panel, cumulative kinase activity data obtained from five experiments (mean ± S.E.). Data for each sample were normalized to the total level of CK1ε. Data were then normalized to the value obtained in the absence of any addition (−DHPG, set as 1). *, p < 0.05, Student’s t test compared with untreated cells; #, p < 0.001, Student’s t test compared with untreated cells that were transfected with wild-type HA-CKIε.
Previous studies carried out in vitro had suggested that inhibitory autophosphorylation site(s) within the catalytic domain might also contribute to autoinhibition of CK1ε (17). The phosphopeptide maps revealed that the control (basic) phosphopeptide was present in both wild-type and MM2-CKIε, and that phosphorylation of this site increased transiently in wild-type CK1ε (but, notably, not in MM2-CK1ε) following treatment with DHPG (Figs. 4 and 5). The identity of the control site phosphorylated under basal conditions is not known. To examine whether phosphorylation of this site has any influence on CKIε activity, tagged CK1ε was immunoprecipitated from N2a cell lysates and incubated with nonspecific lambda protein phosphatase. The incubation with lambda phosphatase substantially reduced phosphorylation of either wild-type or MM2-CK1ε, as revealed by studies in which cells were prelabeled with 32P (and treated with DHPG) (Fig. 6a, upper panel). Other samples were prepared in parallel with unlabeled N2a cells and treated with lambda phosphatase, and CK1 activity was assayed using DARPP-32 as substrate. Phosphatase treatment did not apparently affect the activity of wild-type or MM2-CKIε (all cells were preincubated with DHPG) (Fig. 6a, lower panel, and Fig. 6b), suggesting that phosphorylation of the control site in intact cells did not regulate CK1ε.
Fig. 6. Constitutive phosphorylation of CK1ε does not regulate enzyme activity.
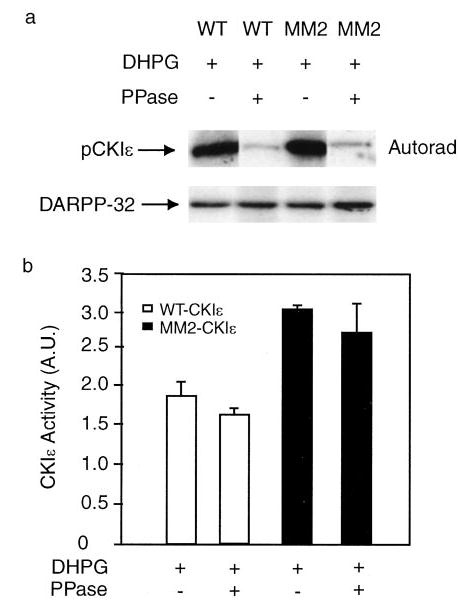
N2a cells were transfected with HA-CK1ε or MM2-CKIε. a, cells were labeled with H3 32PO4 in phosphate-free medium (200 μCi/ml for 2 h) and then treated with DHPG for 2 min. HA-CK1ε or MM2-CKIε were immunoprecipitated and incubated without or with lambda protein phosphatase for 15 min. 32P-labeled samples were analyzed by SDS-PAGE and autoradiography (upper panel). Other samples prepared in parallel were analyzed for CK1 activity using DARPP-32 as substrate (lower panel). b, cumulative kinase activity data obtained from three experiments (means ± S.E.). Data for each sample were normalized to the total level of CK1ε. Data were then normalized to the value obtained in the absence of any addition (−DHPG set as 1; not shown).
Comparison of our phosphopeptide maps with those obtained in a previous study of wild-type CK1ε and a kinase-dead mutant suggest that the control site might not be autophosphorylated by an intramolecular mechanism (cf. peptide f in Fig. 1B in Gietzen et al. (18)) and could possibly be phosphorylated by another protein kinase in intact cells. In the present study, we found that as the activity of CK1ε was stimulated ~2-fold by dephosphorylation of a subset of inhibitory sites, phosphorylation of the control site was increased ~2-fold (see Fig. 4, a and b). This observation supports the idea that the phosphorylation of the control site is linked directly to the activity of CK1ε and probably occurs, at least in part, via autophosphorylation.
DISCUSSION
In the present study we have examined the signal transduction pathway that links stimulation of group I mGluRs to CK1ε activation. The results obtained are consistent with the mechanism illustrated in Fig. 7. Activation of mGluR1 receptors stimulates G proteins that are coupled to PLCβ; Ca2+ released from IP3-sensitive stores actives the Ca2+/calmodulin-dependent phosphatase, calcineurin; and calcineurin dephosphorylates the inhibitory autophosphorylation sites on CKIε. Dephosphorylation of CK1ε results in an increase in kinase activity. However, this increase is transient because of the subsequent autophosphorylation and autoinhibition of the kinase.
Fig. 7. Model for regulation of CK1 activity by activation of group I mGluRs.
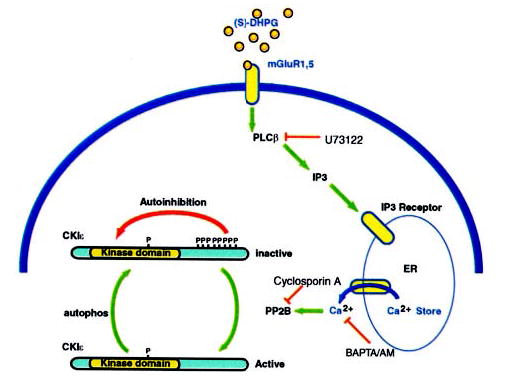
DHPG activates group I mGluRs that are coupled to PLCβ via Gq. Activation of PLCβ generates IP3, and IP3 binds to IP3 receptors on the endoplasmic reticulum and releases Ca2+ into the cytosol. Elevated intracellular Ca2+actives the Ca2+-dependent phosphatase calcineurin, which in turn dephosphorylates the regulatory autophosphorylation sites on CKIε. CKIε is transiently activated, but gradual autophosphorylation restores the inhibited level of kinase activity. A site that is basally phosphorylated is likely to be present within the kinase domain but does not appear to regulate enzyme activity.
Our previous studies had shown that in neostriatal slices, DHPG, an agonist for group I mGluRs, increased CK1 activity, leading to enhanced phosphorylation of Ser-137 of DARPP-32. In the present study, we found that the phosphorylation of Ser-137 of DARPP-32 in neostriatal neurons was sensitive to U73122, BAPTA, and cyclosporin A. The phosphorylation of Thr-75 was also sensitive to these inhibitors, supporting our previous results indicating that activation of CK1 leads to activation of Cdk5 (19). The present results, from studies carried out largely using transfected cell lines, support the conclusion that CK1ε is the likely target for regulation by type I mGluRs in neostriatal neurons. However, it is possible that other CK1 isoforms may be activated through a pathway similar to that shown in Fig. 7. The C-terminal 125 amino acids of CK1δ is ~50% identical to the corresponding domain of CK1ε. In addition, several in vitro studies have found that the activity of CK1δ, like CK1ε, is regulated by autophosphorylation (16, 26). In vitro studies have also indicated that all three CK1γ isoforms can be autophosphorylated (16), raising the possibility that these isoforms are regulated as well. CK1α, -δ, and -ε isoforms have all been found to be expressed in brain and are likely to be distributed widely in neurons (27–29). In addition, our preliminary results indicate that CK1α, -δ, and -ε isoforms are expressed in neostriatum (data not shown). Thus it is possible that activation of calcineurin could lead to transient activation of several CK1 isoforms in neostriatal slices (see also further discussion below).
The precise molecular mechanism by which autophosphorylation of CK1 isoforms regulates enzyme activity is not clear. Autophosphorylation of multiple C-terminal sites in CK1δ and CK1ε appear to be required, although the precise relationship between individual sites and enzyme activity remains to be clarified (16–18). Autophosphorylation is associated with inhibition of enzyme activity toward protein substrates but does not affect phosphorylation of some short synthetic peptides. This latter observation suggests that autophosphorylation serves to influence protein substrate binding negatively by a process that does not block access to the active site of the kinase. Ser-137, the site phosphorylated in DARPP-32 by CK1, is situated at the C-terminal end of a highly acidic region of the protein (23 of 30 residues are either glutamate or aspartate) (30). Possibly, the phosphorylated C-terminal domain of CK1ε (or other isoforms) could act to block binding of longer polypeptide substrates containing acidic domains but not shorter synthetic peptides. Dephosphorylation of the C-terminal domain would then lead to a loss of this inhibitory constraint. Alternatively, an unphosphorylated C-terminal domain (which in CK1ε contains a significant excess of basic amino acids) could serve a positive role in binding to polypeptide substrates that contain acidic domains.
The present study establishes that autophosphorylation of CK1ε at least is a regulated physiological event in intact cells. Although we did not investigate in detail the identity of the site(s) of autophosphorylation that are regulated by calcineurin, the results provide some further insight into the molecular events involved in regulation of CK1ε activity in intact cells. Previous studies of CK1ε have indicated that at least eight sites are autophosphorylated in vitro. However, in intact cells autophosphorylation of many of these sites is apparent only in the presence of okadaic acid or calyculin A, inhibitors of PP1 and PP2A (18, 26). Moreover, autophosphorylation of these sites in the presence of PP1/PP2A inhibitors is associated with a decrease in electrophoretic mobility, detected using SDS-PAGE. In the present study, a significant level of autophosphorylation of CK1ε was observed in intact cells under basal conditions. Moreover, treatment with DHPG or cyclosporin A had no effect on the electrophoretic mobility of the protein, despite the dephosphorylation of a subset of the sites phosphorylated. A reasonable explanation for these results is that there are a least two subsets of autophosphorylation sites. One set is subject to dephosphorylation by calcineurin, is not associated with any alteration of electrophoretic mobility, and is phosphorylated under basal conditions in intact cells as long as calcineurin is inactive. The second set is subject to dephosphorylation by PP1 or PP2A, is associated with a decrease in electrophoretic mobility, and is maintained in a dephosphorylated state in intact cells by active PP1 or PP2A. Additional mutagenesis will be required to identify the site(s) in CK1ε that are specifically dephosphorylated by either calcineurin or PP1/PP2A in intact cells.
The results from our present study indicate that one or more of the sites phosphorylated under basal conditions, and dephosphorylated by activated calcineurin, is associated with regulation of CK1ε activity. It also seems likely that autophosphorylation of one or more of the sites dephosphorylated in intact cells by PP1/PP2A may be associated with regulation of CK1ε activity. Although the sites that are sensitive to PP1/PP2A are maintained in a dephosphorylated state in cells in culture (and apparently in neostriatal neurons under basal conditions), it is possible that physiological inhibition of PP1 or PP2A would result in additional inhibition of CK1ε. For example, in neostriatal neurons, stimulation of phosphorylation of DARPP-32 by D1 dopamine receptors would lead to inhibition of PP1 and could influence CK1ε activity. Further studies will be required to determine whether autophosphorylation of these different sets of sites results in independent modes of regulation of CK1ε or whether there is some sort of interdependence between autophosphorylation of the different sites and regulation of CK1. Autophosphorylation of different sites could have additive or synergistic effects, could cause them to occlude one another, or perhaps could even modulate the ability of CK1 to interact with distinct substrates. Interestingly, three of the eight sites in CK1ε (Thr-325, Ser-368, and Ser-405) appear to be conserved in CK1δ. It is possible that these conserved sites might also confer regulation of CK1δ by calcineurin in intact cells. Alternatively, different autophosphorylation sites may be used to confer differential physiological regulation of CK1 isoforms by protein phosphatases.
The present studies were motivated by our original observations indicating that CK1 could phosphorylate Ser-137 of DARPP-32 (31, 32). Phosphorylation of Ser-137 impairs the ability of Thr-34 of DARPP-32 to be dephosphorylated by calcineurin, thereby modulating the DARPP-32/PP1 cascade. Our more recent studies support the conclusion that activation of CK1 by group I mGluRs results in phosphorylation of Ser-137 of DARPP-32 in neostriatal neurons and that this leads to regulation of voltage-dependent Ca2+ channels (19). A variety of other studies have provided strong support for a role of CK1 isoforms, particularly CK1ε and CK1δ, in diverse cellular processes such as regulation of Wnt signaling and of the circadian clock (7–11, 13–15). CK1ε and CK1δ have also been implicated in the pathophysiology of Alzheimer’s disease (28, 33, 34). Regulation of CK1ε (and possibly CK1δ) by autophosphorylation and transient dephosphorylation by calcineurin may play an important role in the regulation of these other processes that involve the enzyme. Activation of calcineurin may result from stimulation of mGluRs or via many alternative pathways that increase the concentration of intracellular Ca2+ in mammalian cells. Regulation of CK1ε also adds to the diversity of signal transduction pathways utilized by mGluRs and may be responsible for various actions of this increasingly important family of G protein-coupled receptors (35, 36). Finally, the results from these studies indicate that CK1ε represents an additional example of a group of protein kinases including Cdks, Src family members, and Raf-1 (37–39) that are inhibited by phosphorylation and that require dephosphorylation to allow signaling to occur.
Footnotes
The abbreviations used are: CK1, casein kinase 1; HA, hemagglutinin; mGluR, metabotropic glutamate receptor; PP, protein phosphatase; DHPG, (S)-3,5-dihydroxypenylglycine; DARPP-32, dopamine and cAMP-regulated phosphoprotein, 32 kDa; BAPTA, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid; TPCK, l-1-tosylamido-2-phenylethyl chloromethyl ketone; PLCβ, phospholipase Cβ; IP3, inositol 1,4,5-triphosphate; Cdk, cyclin-dependent kinase.
References
- 1.Gross SD, Anderson RA. Cell Signal. 1998;10:699–711. doi: 10.1016/s0898-6568(98)00042-4. [DOI] [PubMed] [Google Scholar]
- 2.Vielhaber E, Virshup DM. IUBMB Life. 2001;51:73–78. doi: 10.1080/15216540117461. [DOI] [PubMed] [Google Scholar]
- 3.Tobin AB, Totty NF, Sterlin AE, Nahorski SR. J Biol Chem. 1997;272:20844–20849. doi: 10.1074/jbc.272.33.20844. [DOI] [PubMed] [Google Scholar]
- 4.Budd DC, McDonald JE, Tobin AB. J Biol Chem. 2000;275:19667–19675. doi: 10.1074/jbc.M000492200. [DOI] [PubMed] [Google Scholar]
- 5.Knippschild U, Milne DM, Campbell LE, DeMaggio AJ, Christenson E, Hoekstra MF, Meek DW. Oncogene. 1997;15:1727–1736. doi: 10.1038/sj.onc.1201541. [DOI] [PubMed] [Google Scholar]
- 6.Sakaguchi K, Saito S, Higashimoto Y, Roy S, Anderson CW, Appella E. J Biol Chem. 2000;275:9278–9283. doi: 10.1074/jbc.275.13.9278. [DOI] [PubMed] [Google Scholar]
- 7.Sakanaka C, Leong P, Xu L, Harrison SD, Williams LT. Proc Natl Acad Sci U S A. 1999;96:12548–12552. doi: 10.1073/pnas.96.22.12548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peters JM, McKay RM, McKay JP, Graff JM. Nature. 1999;401:345–350. doi: 10.1038/43830. [DOI] [PubMed] [Google Scholar]
- 9.McKay RM, Peters JM, Graff JM. Dev Biol. 2001;235:388–396. doi: 10.1006/dbio.2001.0308. [DOI] [PubMed] [Google Scholar]
- 10.Gao ZH, Seeling JM, Hill V, Yochum A, Virshup DM. Proc Natl Acad Sci U S A. 2002;99:1182–1187. doi: 10.1073/pnas.032468199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. Cell. 2002;108:837–847. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- 12.Eide EJ, Virshup DM. Chronobiol Int. 2001;18:389–398. doi: 10.1081/cbi-100103963. [DOI] [PubMed] [Google Scholar]
- 13.Lowrey PL, Shimomura K, Antoch MP, Yamazaki S, Zemenides PD, Ralph MR, Menaker M, Takahashi JS. Science. 2000;288:483–492. doi: 10.1126/science.288.5465.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toh KL, Jones CR, He Y, Eide EJ, Hinz WA, Virshup DM, Ptacek LJ, Fu YH. Science. 2001;291:1040–1043. doi: 10.1126/science.1057499. [DOI] [PubMed] [Google Scholar]
- 15.Lee C, Etchegaray JP, Cagampang FR, Loudon AS, Reppert SM. Cell. 2001;107:855–867. doi: 10.1016/s0092-8674(01)00610-9. [DOI] [PubMed] [Google Scholar]
- 16.Graves PR, Roach PJ. J Biol Chem. 1995;270:21689–21694. doi: 10.1074/jbc.270.37.21689. [DOI] [PubMed] [Google Scholar]
- 17.Cegielska A, Gietzen KF, Rivers A, Virshup DM. J Biol Chem. 1998;273:1357–1364. doi: 10.1074/jbc.273.3.1357. [DOI] [PubMed] [Google Scholar]
- 18.Gietzen KF, Virshup DM. J Biol Chem. 1999;274:32063–32070. doi: 10.1074/jbc.274.45.32063. [DOI] [PubMed] [Google Scholar]
- 19.Liu F, Ma XH, Ule J, Bibb JA, Nishi A, DeMaggio AJ, Yan Z, Nairn AC, Greengard P. Proc Natl Acad Sci U S A. 2001;98:11062–11068. doi: 10.1073/pnas.191353898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bibb JA, Snyder GL, Nishi A, Yan Z, Meijer L, Fienberg AA, Tsai LH, Kwon YT, Girault JA, Czernik AJ, Huganir RL, Hemmings HC, Jr, Nairn AC, Greengard P. Nature. 1999;402:669–671. doi: 10.1038/45251. [DOI] [PubMed] [Google Scholar]
- 21.Nishi A, Snyder GL, Greengard P. J Neurosci. 1997;17:8147–8155. doi: 10.1523/JNEUROSCI.17-21-08147.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Conn PJ, Pin JP. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 23.Paolillo M, Montecucco A, Zanassi P, Schinelli S. Eur J Neurosci. 1998;10:1937–1945. doi: 10.1046/j.1460-9568.1998.00203.x. [DOI] [PubMed] [Google Scholar]
- 24.Cartmell J, Goepfert F, Knoflach F, Pink JR, Bleuel Z, Richards JG, Schaffhauser H, Kemp JA, Wichmann J, Mutel V. Brain Res. 1998;791:191–199. doi: 10.1016/s0006-8993(98)00094-8. [DOI] [PubMed] [Google Scholar]
- 25.Goto S, Matsukado Y, Mihara Y, Inoue N, Miyamoto E. Brain Res. 1986;397:161–172. doi: 10.1016/0006-8993(86)91381-8. [DOI] [PubMed] [Google Scholar]
- 26.Rivers A, Gietzen KF, Vielhaber E, Virshup DM. J Biol Chem. 1998;273:15980–15984. doi: 10.1074/jbc.273.26.15980. [DOI] [PubMed] [Google Scholar]
- 27.Graves PR, Haas DW, Hagedorn CH, DePaoli-Roach AA, Roach PJ. J Biol Chem. 1993;268:6394–6401. [PubMed] [Google Scholar]
- 28.Ghoshal N, Smiley JF, DeMaggio AJ, Hoekstra MF, Cochran EJ, Binder LI, Kuret J. Am J Pathol. 1999;155:1163–1172. doi: 10.1016/S0002-9440(10)65219-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takano A, Shimizu K, Kani S, Buijs RM, Okada M, Nagai K. FEBS Lett. 2000;477:106–112. doi: 10.1016/s0014-5793(00)01755-5. [DOI] [PubMed] [Google Scholar]
- 30.Hemmings HC, Jr, Nairn AC, Elliott JI, Greengard P. J Biol Chem. 1990;265:20369–20376. [PubMed] [Google Scholar]
- 31.Desdouits F, Cohen D, Nairn AC, Greengard P, Girault JA. J Biol Chem. 1995;270:8772–8778. doi: 10.1074/jbc.270.15.8772. [DOI] [PubMed] [Google Scholar]
- 32.Desdouits F, Siciliano JC, Greengard P, Girault JA. Proc Natl Acad Sci U S A. 1995;92:2682–2685. doi: 10.1073/pnas.92.7.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuret J, Johnson GS, Cha D, Christenson ER, DeMaggio AJ, Hoekstra MF. J Neurochem. 1997;69:2506–2515. doi: 10.1046/j.1471-4159.1997.69062506.x. [DOI] [PubMed] [Google Scholar]
- 34.Schwab C, DeMaggio AJ, Ghoshal N, Binder LI, Kuret J, McGeer PL. Neurobiol Aging. 2000;21:503–510. doi: 10.1016/s0197-4580(00)00110-x. [DOI] [PubMed] [Google Scholar]
- 35.Sallese M, Iacovelli L, Cumashi A, Capobianco L, Cuomo L, De Blasi A. Biochim Biophys Acta. 2000;1498(2–3):112–121. doi: 10.1016/s0167-4889(00)00088-4. [DOI] [PubMed] [Google Scholar]
- 36.Hermans E, Challiss RA. Biochem J. 2001;359:465–484. doi: 10.1042/0264-6021:3590465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Williams JC, Wierenga RK, Saraste M. Trends Biochem Sci. 1998;23:179–184. doi: 10.1016/s0968-0004(98)01202-x. [DOI] [PubMed] [Google Scholar]
- 38.Obaya AJ, Sedivy JM. Cell Mol Life Sci. 2002;59:126–142. doi: 10.1007/s00018-002-8410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dhillon AS, Meikle S, Yazici Z, Eulitz M, Kolch W. EMBO J. 2002;21:64–71. doi: 10.1093/emboj/21.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]