

Abstract
AMP-activated protein kinase (AMPK) is an energy sensor that regulates cellular metabolism. When activated by a deficit in nutrient status, AMPK stimulates glucose uptake and lipid oxidation to produce energy, while turning off energy-consuming processes including glucose and lipid production to restore energy balance. AMPK controls whole-body glucose homeostasis by regulating metabolism in multiple peripheral tissues, such as skeletal muscle, liver, adipose tissues, and pancreatic β cells — key tissues in the pathogenesis of type 2 diabetes. By responding to diverse hormonal signals including leptin and adiponectin, AMPK serves as an intertissue signal integrator among peripheral tissues, as well as the hypothalamus, in the control of whole-body energy balance.
Introduction
Living organisms are constantly challenged with irregular food supply; therefore the ability to maintain energy balance during food deprivation is critical for survival. Such selection pressure has driven organisms to evolve complex systems to store fuel substrates during food supply and to reduce energy expenditure during food shortage. Insulin is the primary anabolic hormone that stimulates uptake and storage of fuel substrates in skeletal muscle, liver, and fat cells, while inhibiting substrate production. Disruption of energy balance caused by overeating and a sedentary lifestyle has led to an increased prevalence of type 2 diabetes (T2D) (1, 2), a metabolic disorder associated with insulin resistance in peripheral tissues. Since energy balance is regulated by multiple processes, investigation of the cellular targets that regulate substrate intake and energy expenditure could enhance our understanding of the metabolic syndrome and conceivably lead to the development of novel preventive and pharmaceutical intervention strategies.
In multiple mammalian tissues, the AMP-activated protein kinase (AMPK) controls glucose and lipid metabolism (3–5) (Figure 1). Furthermore, AMPK integrates signaling circuits between peripheral tissues and the hypothalamus to regulate food intake and whole-body energy expenditure. The critical role of AMPK as an evolutionarily conserved energy sensor and master regulator of metabolism is further supported by the role of its ortholog in glucose metabolism of the unicellular eukaryote Saccharomyces cerevisiae (3–5). This Review will highlight the role of AMPK in the regulation of fuel substrate metabolism in peripheral tissues, intertissue communication, and food intake.
Figure 1. Structure and regulation of AMPK.
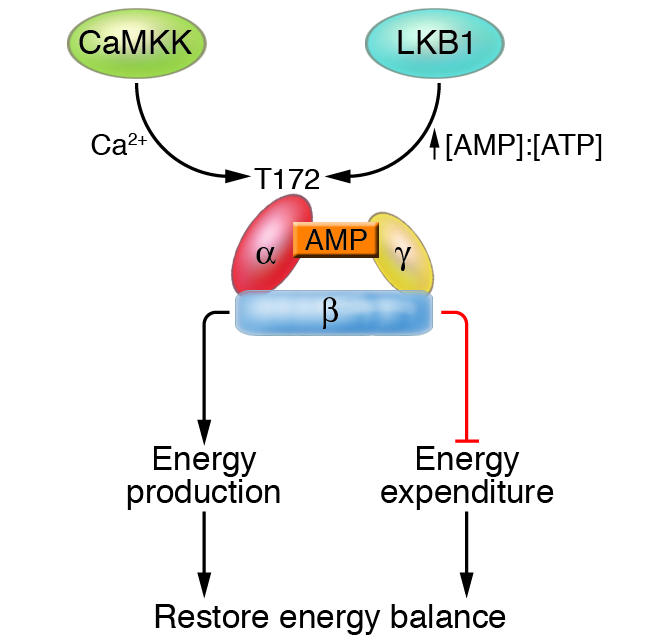
AMPK is a heterotrimeric complex consisting of an α, a β, and a γ subunit and is activated by the upstream kinases CaMKK and LKB1 via phosphorylation of threonine residue 172. Activation of AMPK by CaMKK and LKB1 is dependent on the intracellular calcium and AMP/ATP ratio, respectively. Generally, AMPK restores energy balance by activating processes that produce energy (e.g., lipid oxidation and glucose uptake) while inhibiting those that consume energy (e.g., protein synthesis).
Structure and regulation of AMPK
One of the most profound features of AMPK as a metabolic sensor is its sensitivity to the cellular energy status, which results from its unique biochemical properties.AMPK is a heterotrimeric protein consisting of a catalytic α and regulatory β and γ subunits (3–5) (Figure 1). Each α and β subunit is encoded by 2 genes (α1 and α2 or β1 and β2), whereas the γ subunit is encoded by 3 genes (γ1, γ2, and γ3). The protein is activated in response to an increase in the ratio of AMP to ATP within the cell and therefore acts as an efficient sensor for cellular energy state. Binding of AMP activates AMPK allosterically and induces phosphorylation of a threonine residue (Thr-172) within the activation domain of the α subunit by an upstream kinase, the tumor suppressor LKB1 (3, 4, 6). Furthermore, binding of AMP inhibits the dephosphorylation of Thr-172 by protein phosphatase, whereas a high concentration of ATP inhibits the activation of AMPK. Recent studies identified calmodulin-dependent protein kinase kinase (CaMKK) as an additional upstream kinase of AMPK (7–9). Activation of AMPK by CaMKK is triggered by a rise in intracellular calcium ions, without detectable changes in the AMP/ATP ratio (9). CaMKK is highly expressed in the CNS, and lower levels are detected in other tissues such as liver and skeletal muscle, suggesting that the AMPK pathway is regulated by multiple mechanisms that are likely to be tissue specific (10).
AMPK activity is activated by a wide array of metabolic stresses, including hypoxia, ischemia, and oxidative and hyperosmotic stresses (3, 4, 6, 11). Furthermore, exercise and glucose deprivation also activate AMPK, which suggests a role in exercise adaptations and β cell function. In general, activation of AMPK triggers catabolic pathways that produce ATP, while turning off anabolic pathways that consume ATP, to maintain cellular energy stores (4, 11). Metformin and thiazolidinedione, 2 widely prescribed drugs for the treatment of T2D, are also reported to increase AMPK activity (12), underscoring the potential role of the AMPK pathway in the treatment of T2D.
Pharmacological activation of AMPK can be achieved by treatment of cells with an artificial activator, 5-aminoimidazole-4-carboxamide riboside (AICAR). AICAR is a cell-permeable adenosine analog that is taken up by the cells and phosphorylated to form 5-aminoimidazole-4-carboxamide-1-d-ribofuranosyl-5′-monophosphate (ZMP), an AMP mimetic, and confers the activating effects of AMP on the AMPK pathway (13). However, ZMP is reported to affect other AMP-regulated enzymes (14, 15), and therefore caution has to be exercised in interpretation of data that involves the use of AICAR. Although elevations in the AMP/ATP ratio constitute a classical pathway of the activation of AMPK,activators of AMPK such as hyperosmotic stress do not appear to alter this ratio (12), suggesting that other mechanisms are involved in the regulation of the AMPK pathway.
AMPK regulates metabolism in peripheral tissues
Role of AMPK in exercise-induced glucose uptake in skeletal muscle.
Skeletal muscle is the major site of insulin-stimulated glucose disposal (16), and insulin resistance in this target tissue has long been viewed as a contributing factor in the pathogenesis of T2D. Therefore, alternative pathways that stimulate skeletal muscle glucose uptake independently of insulin signaling could potentially improve glycemic control in T2D subjects. Although exercise stimulates glucose uptake in skeletal muscle independently of the insulin pathway, the underlying molecular mechanism remains largely elusive.
The finding that glucose transport can be increased in isolated skeletal muscle in response to contraction in vitro suggests that the signaling pathways elicited by internal cellular energy deficiency are partly accountable for contraction-induced glucose uptake. AMPK is considered as an attractive candidate for contraction-induced skeletal muscle glucose uptake because it is activated in parallel with elevation in AMP and a reduction in creatine phosphate energy stores (17). Furthermore, AICAR-induced activation of AMPK increases glucose uptake (18), concomitantly with glucose transporter 4 (GLUT4)fusion with plasma membrane (19). The AICAR effect on glucose uptake is not affected by inhibition of the insulin-dependent PI3K pathway and is additive to insulin-stimulated glucose uptake, reminiscent of the exercise effect. However, in rat slow-twitch (oxidative) soleus muscle, AICAR does not induce glucose uptake, although phosphorylation of AMPK is increased (20). Furthermore, in slow-twitch muscle, contraction induces glucose uptake without any apparent increase in AMPK activity when there is a high glycogen content. (21). Given that AICAR induces AMPK phosphorylation and glucose uptake in rat fast-twitch (glycolytic) epitrochlearis muscle, the role of AMPK in mediating muscle glucose uptake is likely to be dependent on fiber type.
Earlier studies linking AMPK signaling with glucose metabolism were correlative in nature. The introduction of transgenic and genetic knockout mouse models to study AMPK action has enhanced our understanding of AMPK signaling in contraction-induced glucose uptake (Table 1). Overexpression of an α2 kinase–dead subunit in skeletal muscle abolishes AICAR, but only partially impairs contraction-stimulated glucose uptake (22). Furthermore, ablation of skeletal muscle AMPK α2 and γ3 subunit expression does not affect glucose uptake stimulated by contraction, although the AICAR effect is blunted (23–25). In the skeletal muscle–specific LKB1 knockout, however, glucose uptake in response to AICAR and contraction is impaired (26). These findings suggest that additional pathways mediate contraction-induced glucose uptake.
Table 1 .
In vivo validation of the AMPK pathway in mouse models

Despite extensive study on upstream stimuli that activate AMPK, investigation on the downstream substrate(s) of AMPK-mediated glucose uptake is lacking. Recent reports revealed that Akt substrate of 160 kDa (AS160) is an important substrate downstream of Akt that is involved in insulin-stimulated glucose uptake. In cultured adipocytes, insulin-stimulated GLUT4 translocation requires phosphorylation of AS160 (27). Phosphorylation of AS160 by Akt inhibits its GTPase-activating protein (GAP) activity, which leads to an elevation in the GTP form of a Rab protein, which in turn increases GLUT4 vesicle translocation to the plasma membrane (27–29). In addition to insulin, contraction and activation of AMPK by AICAR is associated with increased phosphorylation of AS160 in rodent skeletal muscle. Likewise, in human skeletal muscle, insulin (30) and exercise (31) stimulate phosphorylation of AS160. Phosphorylation of AS160 is impaired or abolished in skeletal muscle from AMPK α2 knockout, γ3 knockout, and α2 kinase–dead mice in response to AICAR treatment (32). This corroborates findings ofimpaired AICAR-stimulated glucose uptake in skeletal muscle of these mice (22, 24, 25). Therefore, AS160 appears to be a downstream target of AMPK in mediating glucose uptake in skeletal muscle. Moreover, AMPK complexes containing the α2 and γ3 subunits are required for AS160 phosphorylation. However, AMPK heterotrimers containing the AMPK α2 subunit are essential for AS160 phosphorylation in response to muscle contraction, whereas complexes containing the γ3 subunit are dispensable. Despite abolished contraction-stimulated AS160 phosphorylation in skeletal muscle of AMPK α2 knockout and α2 kinase–dead mice, contraction-mediated glucose uptake is unaltered or slightly impaired in these mice (22, 24), which again challenges the role of AMPK in exercise-induced glucose uptake.
Although it is apparent that AMPK mediates the effects of AICAR on glucose uptake, its role in contraction-induced glucose uptake remains unclear. Nonetheless, the therapeutic potential of AMPK activationin skeletal muscle to achieve whole-body glycemic control is apparent, given that genetic ablation of AMPK α2 activity abolishes the acute glucose-lowering effect of AICAR in mice (22–24). Further, activation of AMPK with metformin is associated with increased glucose uptake in rat skeletal muscle (33). Metformin also acts as an insulin sensitizer in isolated skeletal muscle from insulin-resistant humans (34). Therefore, the metabolic effects of metformin in T2D patients may be partly mediated by the activation of skeletal muscle AMPK (Figure 2).
Figure 2. Role of AMPK in the regulation of whole-body glucose homeostasis.

Activation of AMPK turns on ATP-generating processes, while switching off ATP-consuming processes. In skeletal muscle, acute activation of AMPK increases glucose uptake and lipid oxidation, while chronic activation of AMPK is associated with mitochondrial biogenesis. Activation of AMPK inhibits glucose and lipid synthesis in the liver but increases lipid oxidation. Lipolysis and lipogenesis in adipose tissue are also reduced by AMPK activation. Collectively, activation of AMPK in skeletal muscle, liver, and adipose tissue results in a favorable metabolic milieu for the prevention or treatment of T2D, i.e., decreased circulating glucose, reduced plasma lipid, and ectopic fat accumulation, as well as enhanced insulin sensitivity. Activation of pancreatic AMPK is associated with decreased insulin secretion, likely a protective measure to prevent hypoglycemia during food deprivation, although this effect needs to be considered in pharmaceutical targeting of AMPK for the treatment of T2D.
Regulation of fatty acid oxidation by AMPK.
The AMPK pathway has profound effects on the regulation of lipid metabolism. Fatty acid oxidation in skeletal muscle involves a rate-controlling step that is regulated by carnitine palmitoyltransferase 1 (CPT1). CPT1 transfers long-chain acyl-CoA into the mitochondria, and this process is inhibited allosterically by malonyl-CoA (35), synthesized by acetyl-CoA carboxylase (ACC) (36). The activity of ACC is regulated by reversible phosphorylation, and AMPK directly phosphorylates and inactivates this downstream target (37, 38). During exercise (37) and skeletal muscle contraction (17), activated AMPK inhibits ACC to reduce malonyl-CoA concentration, thereby driving the entry of long-chain acyl-CoA into the mitochondria for β-oxidation to restore energy balance (18, 39). The ability of AMPK to induce lipid oxidation and thus lower skeletal muscle (40) and liver (33) lipid deposition is considered an important feature for the insulin-sensitizing effect of AMPK activation. Indeed, when an activating form of AMPK γ3(R225Q) subunit is expressed in skeletal muscle via genetic manipulation, the transgenic mice are protected against the development of diet-induced skeletal muscle insulin resistance (25). This effect is associated with lower skeletal muscle triglyceride stores as a result of increased fatty acid oxidation (25).
AMPK activation, mitochondrial biogenesis, and insulin sensitivity in skeletal muscle.
Exercise has been long recognized as an essential element of clinical management of T2D. Long-term exercise training enhances insulin sensitivity and glucose uptake in skeletal muscle (41, 42) and promotes mitochondrial biogenesis, which leads to improved oxidative metabolism (43). Decreased mitochondrial density and activity have been proposed to partly account for the development of skeletal muscle insulin resistance (44–46). Given that activation of AMPK recapitulates some of the exercise-induced adaptations, it is likely to mediate some beneficial effects of exercise on glucose homeostasis (41, 42).
Activation of AMPK by hypoxia and AICAR mimics contraction-enhanced insulin sensitivity of glucose transport in isolated skeletal muscle (47). In long-term treatment studies, in vivoAICAR administration increases GLUT4 and hexokinase II expression (48, 49) and enhances insulin-stimulated muscle glucose transport (50, 51) and GLUT4 translocation (51). In genetic mouse models, an activating mutant AMPK γ3(R225Q) subunit enhanced expression of genes essential for skeletal muscle lipid and oxidative metabolism, whereas ablation of the AMPK γ3 subunit impaired the expression of these genes (52, 53). In response to exercise, mutant γ3(R225Q) mice demonstrated enhanced skeletal muscle expression of GLUT4 and hexokinase II, whereas expression of these genes during recovery was blunted in γ3 knockout mice (54). The mechanisms for such effects are unclear. Nonetheless, several studies revealed candidate downstream players; AMPK activation is associated with increased expression of myocyte enhancer factor 2A (MEF2A) and MEF2D (55), transcription factors that regulates the GLUT4 gene promoter. Furthermore, diet-induced chronic energy deprivation in rodents activates AMPK and increases skeletal muscle mitochondrial density (56, 57), concomitantly with an elevation in nuclear respiratory factor 1 (NRF-1) activity (57) and PPARγ coactivator 1 (PGC-1) content (56), which are key regulators of mitochondrial gene expression (58). These effects, however, are essentially abolished in the AMPK α2 kinase–dead mice (56). Nevertheless, activation of AMPK by chronic AICAR treatment increases some, but not all, mitochondrial enzyme activity in rat skeletal muscle (59), and exercise-induced gene expression is unaltered in α1 and α2 knockout mice (60). Therefore, AMPK may mediate some, but not all, exercise-induced gene expression, although chronic exercise effects in the AMPK mouse genetic models remain uncharacterized.
Regulation of liver metabolism by AMPK.
Glucose homeostasis is maintained by a balance between hepatic glucose production and glucose uptake by peripheral tissues. Elevated glucose production by the liver is a major cause of fasting hyperglycemia in T2D (61, 62). Gluconeogenesis in the liver is regulated by multiple enzymes such as phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase) (63). Activation of AMPK suppresses the transcription of these genes in hepatoma cells (64), providing clues for the role of AMPK in gluconeogenesis. Furthermore, AMPK α2 liver-specific knockout mice are glucose intolerant and display fasting hyperglycemia, presumably because of elevated gluconeogenesis associated with increased PEPCK and G6Pase activity (65). Recent data have provided evidence that inhibition of the gluconeogenic program by AMPK involves a transcriptional coactivator, transducer of regulated CREB activity 2 (TORC2) (66). Glucagon and fasting conditions induce hepatic TORC2 nuclear translocation, which enhances CREB-dependent transcription of the gluconeogenic program, including PGC-1α. Conversely, AMPK activation causes TORC2 phosphorylation and sequesters the coactivator in the cytoplasm, thus blunting the expression of the gluconeogenic program. Deletion of hepatic LKB1 (an upstream kinase of AMPK) abolishes AMPK activation and leads to nuclear accumulation of TORC2, which in turn drives gluconeogenesis (67). Consistently, liver LKB1 knockout mice on a high-fat diet exhibit fasting hyperglycemia and glucose intolerance and are unresponsive to metformin treatment (67).
In addition to gluconeogenesis, AMPK action has been implicated in regulation of liver lipogenesis, lipid oxidation, and cholesterol synthesis (4, 6). AMPK suppresses glucose-induced expression of lipogenesis-associated genes such as fatty acid synthase (33, 68), ACC, and pyruvate kinase (69). In rat primary hepatocytes, activation of AMPK by AICAR or metformin increases fatty acid oxidation via inhibition of ACC (33). Consistently, in rats treated with metformin, triglyceride levels are reduced, concomitantly with an increase in β-hydroxybutyrate, suggesting elevated hepatic lipid oxidation (33). These enhanced metabolic profiles are accompanied by downregulation of lipogenic genes such as SREBP-1, fatty acid synthase, and S14 (33). Moreover, the lipid-lowering effects of metformin in cultured hepatocytes require AMPK activity (70). Taken together, the studies provide evidence that AMPK suppresses liver gluconeogenesis and lipid production, while decreasing hepatic lipid deposition via increased lipid oxidation, thus improving the glucose and lipid profile in T2D.
AMPK regulates lipid metabolism in adipocytes.
A common characteristic of T2D is high circulating lipid levels of lipids, partly accounted for by impaired insulin-mediated suppression of lipolysis in adipose tissue (62, 71). Elevated circulating FFAs released by adipocytes are associated with ectopic fat accumulation and can cause insulin resistance in skeletal muscle and liver, as well as impaired insulin secretion by β cells (72). Insulin controls the release of FFAs from adipose tissue by inhibition of hormone-sensitive lipase, a rate-limiting enzyme of lipolysis. However, AMPK has also been implicated in the regulation of lipolysis through direct phosphorylation of hormone-sensitive lipase, which leads to inhibition of subsequent activation by protein kinase A (73). AMPK is thought to inhibit lipolysis, since treatment of isolated rat adipocytes with AICAR antagonizes isoprenaline-induced lipolysis (13, 74). Furthermore, expression of a constitutively active form of AMPK reduces isoproterenol-stimulated lipolysis, whereas reduced AMPK activity increases lipolysis in adipocytes (75). AMPK inhibits lipogenesis in isolated adipocytes via increased ACC phosphorylation in response to AICAR stimulation (74). AMPK also appears to control whole-body adiposity; when subjected to a high-fat diet, AMPK α2 knockout mice developed increased body weight and fat mass as compared with the wild-type mice (76). Although an in vivo antilipolytic effect of AICAR has been demonstrated (77), AMPK activity in adipocytes is paradoxically augmented in response to fasting (75), a condition when lipolysis is elevated. Furthermore, there is also evidence that activation of AMPK promotes lipolysis (78). Therefore, the physiological relevance and role of AMPK in the regulation of lipolysis in vivo remain to be addressed.
AMPK and insulin release by pancreatic β cells.
The SNF1 complex (a yeast ortholog of AMPK) in S. cerevisiae responds to changes in the glucose concentration in the medium and is essential for transcriptional activation of glucose-repressed genes (3–5). This glucose-sensing property is also found in mammalian pancreatic β cells. Glucose increases the intracellular ATP/ADP ratio in β cells (79, 80), which induces closure of ATP-sensitive potassium channels (81), and influx of calcium ions (82), a critical step in exocytosis of insulin (83). Moreover, AMP and ADP concentrations in β cells decrease in response to elevations in glucose concentration (84, 85), and this suggests that AMPK could play a role in insulin release by acting as a fuel sensor. An increase in glucose levels represses AMPK activity in β cell lines (85–88), whereas AICAR-induced activation of AMPK markedly reduced glucose-stimulated insulin release from primary pancreatic islets (85, 87) and β cell lines (87, 89). Furthermore, overexpression of a constitutively active form of AMPK reduces calcium influx in response to depolarizing agents and results in repressed glucose-induced insulin release from β cell lines (87, 90). Conversely, overexpression of a dominant-negative form of AMPK leads to increases in insulin release without apparent changes in glucose metabolism and calcium influx (87). Despite its profound effect on insulin release, the role of AMPK in β cell vesicle trafficking is unclear, and downstream targets of AMPK that mediate these physiological processes remain to be identified.
Given that antidiabetic drugs such as metformin activate AMPK, AMPK activators have been proposed to counter effects on insulin release in T2D patients (91). Incubation of either human islets or cultured β cells with metformin activates AMPK and inhibits glucose-stimulated insulin secretion (88). Although seemingly undesirable in the treatment of T2D, AMPK-mediated suppression of insulin release may be physiologically relevant for maintaining glucose homeostasis through inhibition of insulin secretion during glucose deficiency. The role of AMPK in the regulation of β cell function is clearly an unresolved question that requires further investigation.
Integration of intertissue signaling by AMPK
Leptin.
One of the central roles of AMPK in the regulation of whole-body glucose homeostasis is to integrate hormonal and nutrient signals in multiple tissues (Figure 3). Leptin is an adipocyte-derived hormone that diminishes adiposity by reducing food intake (92) and improves insulin sensitivity (93), at least in part by depleting triglyceride stores in peripheral tissues (94, 95). In peripheral tissues, leptin induces fatty acid oxidation (96) and glucose uptake (97, 98), 2 metabolic responses that are also triggered by AMPK activation; this implicates AMPK kinase as a candidate in the mediation of leptin responses. Intravenous injection of leptin was found to induce fatty acid oxidation via inhibition of ACC and activate AMPK α2 in skeletal muscle in a biphasic manner (99). An early and transient activation of AMPK in skeletal muscle occurred as a direct response to leptin treatment via an increase in the AMP concentration. A later, more sustained activation was mediated by hypothalamic–sympathetic nervous system and skeletal muscle α-adrenergic receptors without any apparent change in AMP level (99).
Figure 3. Integration of intertissue signaling by AMPK.

Adipocyte-derived leptin reduces food intake though inhibition of hypothalamic AMPK. Leptin increases skeletal muscle fatty acid oxidation via direct activation of AMPK and the hypothalamus–sympathetic nervous system axis. The involvement of hypothalamic AMPK in leptin-induced skeletal muscle lipid oxidation and glucose uptake is unknown. Adiponectin is secreted by adipose tissue and increases skeletal muscle lipid oxidation and glucose uptake, as well as suppressing liver glucose production by activating AMPK.
Adiponectin.
Adiponectin is another adipokine that lowers plasma glucose and FFAs in mice challenged with a high-fat diet (100). These effects are partly accounted for by adiponectin-induced AMPK activation, which in turn stimulates skeletal muscle fatty acid oxidation and glucose uptake (101, 102). Furthermore, activation of AMPK by adiponectin suppresses endogenous glucose production, concomitantly with inhibition of PEPCK and G6Pase expression (102). The AMPK pathway mediates metabolic effects of adipocyte-derived adiponectin in skeletal muscle and liver, thereby integrating communication among the 3 organs.
Role of AMPK in energy homeostasis.
The hypothalamus plays a key role in whole-body energy homeostasis by regulating peripheral metabolism (103, 104) and food intake (103–105) in response to hormones and nutrients. AMPK acts as a fuel sensor in the hypothalamus and responds to a variety of metabolic and nutrient factors. Injection of leptin and insulin (anorexigen) (106, 107) into rodents inhibits AMPK activity in the hypothalamus; conversely, administration of orexigenic peptides including ghrelin (106) and agouti-related protein (107) increases AMPK activity. The modulation of hypothalamic AMPK in rodents alters food intake: activation of AMPK by AICAR and α-lipoic acid in the hypothalamus increases food intake (106, 108), whereas anorexigenic and weight-reducing effects of leptin are attenuated when constitutively active AMPK is expressed in the hypothalamus (107). Apart from signals elicited by biological peptides, a decrease in fuel availability such as under fasting conditions (107, 108) activates hypothalamic AMPK activity. In contrast, hyperglycemia and refeeding inhibit AMPK activity in the hypothalamus (107).
Apart from the effects on food intake, additional roles of hypothalamic AMPK in the central control of peripheral metabolism are likely. The hypothalamic–sympathetic nervous system axis partly mediates leptin-induced skeletal muscle fatty acid oxidation (99). Intracerebroventricular administration of AICAR activates hypothalamic AMPK and triggers increased glycogen synthesis in skeletal muscle (109). Furthermore, total AMPK α2 knockout mice display impaired insulin-stimulated whole-body glucose utilization and skeletal muscle glycogen synthesis, although underlying mechanisms are unknown (99, 110). Therefore, further studies are critical for understanding the role of hypothalamic AMPK in the regulation of whole-body metabolism.
Conclusion
AMPK represents a diverse, yet versatile energy sensor and metabolic regulator that exerts a regulatory effect in the hypothalamus and multiple peripheral tissues. Activation of AMPK in skeletal muscle, liver, and adipose tissue enhances metabolism, insulin sensitivity, and gene expression to collectively promote a favorable metabolic milieu for the prevention or treatment of T2D. However, activated AMPK can induce food intake through its action in the hypothalamus. This effect is probably a physiological response for glucose homeostasis but will likely be undesirable for pharmaceutical targeting of AMPK for the treatment of T2D and obesity. Therefore, tissue-specific pharmacological activation of AMPK is essential and could potentially be achieved through isoform-specific activation or targeting of downstream substrates of AMPK. Given that many AMPK-regulated metabolic pathways are also controlled by other hormonal and metabolic signals, elucidation of the cross-talk between these pathways is also essential. Moreover, research into the AMPK system provides compelling molecular support for adopting exercise in the prevention and treatment of T2D. This of course does not disparage the need for pharmaceutical targeting of the AMPK pathway for a multimodal approach in the prevention and treatment of T2D.
Acknowledgments
The authors are grateful to current and former members of the laboratory for helpful discussion. Support has been obtained from the Swedish Research Council, the Swedish Diabetes Association, the Foundation for Scientific Studies of Diabetology, the Strategic Research Foundation (INGVAR II), and the Commission of the European Communities (contract no. LSHM-CT-2004-005272 EXGENESIS and contract no. LSHM-CT-2004-512013 EUGENE2). Y.C. Long is supported by scholarship from the Karolinska Institutet.
Footnotes
Nonstandard abbreviations used: ACC, acetyl-CoA carboxylase; AICAR, 5-aminoimidazole-4-carboxamide riboside; AMPK, AMP-activated protein kinase; AS160, Akt substrate of 160 kDa; CaMKK, calmodulin-dependent protein kinase kinase; CREB, cAMP response element–binding protein; G6Pase, glucose-6-phosphatase; GLUT4, glucose transporter 4; PEPCK, phosphoenolpyruvate carboxykinase; PGC-1, PPARγ coactivator 1; T2D, type 2 diabetes; TORC2, transducer of regulated CREB activity 2.
Conflict of interest: The laboratory of J.R. Zierath receives research funding from Arexis AB, Sweden, which investigates pharmaceutical targeting of AMPK.
Citation for this article: J. Clin. Invest. 116:1776–1783 (2006). doi:10.1172/JCI29044.
References
- 1.Klein S., et al. Weight management through lifestyle modification for the prevention and management of type 2 diabetes: rationale and strategies. A statement of the American Diabetes Association, the North American Association for the Study of Obesity, and the American Society for Clinical Nutrition. Diabetes Care. 2004;27:2067–2073. doi: 10.2337/diacare.27.8.2067. [DOI] [PubMed] [Google Scholar]
- 2.Wing R.R., et al. Behavioral science research in diabetes: lifestyle changes related to obesity, eating behavior, and physical activity. Diabetes Care. 2001;24:117–123. doi: 10.2337/diacare.24.1.117. [DOI] [PubMed] [Google Scholar]
- 3.Hardie D.G. The AMP-activated protein kinase pathway: new players upstream and downstream. J. Cell Sci. 2004;117:5479–5487. doi: 10.1242/jcs.01540. [DOI] [PubMed] [Google Scholar]
- 4.Carling D. The AMP-activated protein kinase cascade: a unifying system for energy control. . Trends Biochem. Sci. 2004;29:18–24. doi: 10.1016/j.tibs.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 5.Kemp B.E., et al. AMP-activated protein kinase, super metabolic regulator. Biochem. Soc. Trans. 2003;31:162–168. doi: 10.1042/bst0310162. [DOI] [PubMed] [Google Scholar]
- 6.Kahn B.B., Alquier T., Carling D., Hardie D.G. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 7.Woods A., et al. Ca2+/calmodulin-dependent protein kinase kinase-β acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 8.Hurley R.L., et al. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J. Biol. Chem. 2005;280:29060–29066. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- 9.Hawley S.A., et al. Calmodulin-dependent protein kinase kinase-β is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 10.Birnbaum M.J. Activating AMP-activated protein kinase without AMP. Mol. Cell. 2005;19:289–290. doi: 10.1016/j.molcel.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 11.Hardie D.G. Minireview. The AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology. 2003;144:5179–5183. doi: 10.1210/en.2003-0982. [DOI] [PubMed] [Google Scholar]
- 12.Fryer L.G.D., Parbu-Patel A., Carling D. The anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J. Biol. Chem. 2002;277:25226–25232. doi: 10.1074/jbc.M202489200. [DOI] [PubMed] [Google Scholar]
- 13.Corton J.M., Gillespie J.G., Hawley S.A., Hardie D.G. 5-Aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 1995;229:558–565. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- 14.Vincent M.F., Marangos P.J., Gruber H.E., Van den Berghe G. Inhibition by AICA riboside of gluconeogenesis in isolated rat hepatocytes. Diabetes. 1991;40:1259–1266. doi: 10.2337/diab.40.10.1259. [DOI] [PubMed] [Google Scholar]
- 15.Longnus S.L., Wambolt R.B., Parsons H.L., Brownsey R.W., Allard M.F. 5-Aminoimidazole-4-carboxamide 1-beta-D-ribofuranoside (AICAR) stimulates myocardial glycogenolysis by allosteric mechanisms. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003;284:R936–R944. doi: 10.1152/ajpregu.00319.2002. [DOI] [PubMed] [Google Scholar]
- 16.DeFronzo R.A., Gunnarsson R., Bjorkman O., Olsson M., Wahren J. Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin-dependent (type II) diabetes mellitus. . J. Clin. Invest. 1985;76:149–155. doi: 10.1172/JCI111938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hutber C.A., Hardie D.G., Winder W.W. Electrical stimulation inactivates muscle acetyl-CoA carboxylase and increases AMP-activated protein kinase. Am. J. Physiol. Endocrinol. Metab. 1997;272:E262–E266. doi: 10.1152/ajpendo.1997.272.2.E262. [DOI] [PubMed] [Google Scholar]
- 18.Merrill G.F., Kurth E.J., Hardie D.G., Winder W.W. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am. J. Physiol. Endocrinol. Metab. 1997;273:E1107–E1112. doi: 10.1152/ajpendo.1997.273.6.E1107. [DOI] [PubMed] [Google Scholar]
- 19.Kurth-Kraczek E., Hirshman M., Goodyear L., Winder W. 5′ AMP-activated protein kinase activation causes GLUT4 translocation in skeletal muscle. Diabetes. 1999;48:1667–1671. doi: 10.2337/diabetes.48.8.1667. [DOI] [PubMed] [Google Scholar]
- 20.Wright D.C., Geiger P.C., Holloszy J.O., Han D.H. Contraction- and hypoxia-stimulated glucose transport is mediated by a Ca2+-dependent mechanism in slow-twitch rat soleus muscle. Am. J. Physiol. Endocrinol. Metab. 2005;288:E1062–E1066. doi: 10.1152/ajpendo.00561.2004. [DOI] [PubMed] [Google Scholar]
- 21.Derave W., et al. Dissociation of AMP-activated protein kinase activation and glucose transport in contracting slow-twitch muscle. Diabetes. 2000;49:1281–1287. doi: 10.2337/diabetes.49.8.1281. [DOI] [PubMed] [Google Scholar]
- 22.Mu J., Brozinick J.T., Valladares O., Bucan M., Birnbaum M.J. A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol. Cell. 2001;7:1085–1094. doi: 10.1016/s1097-2765(01)00251-9. [DOI] [PubMed] [Google Scholar]
- 23.Fujii N., et al. AMP-activated protein kinase α2 activity is not essential for contraction- and hyperosmolarity-induced glucose transport in skeletal muscle. J. Biol. Chem. 2005;280:39033–39041. doi: 10.1074/jbc.M504208200. [DOI] [PubMed] [Google Scholar]
- 24.Jorgensen S.B., et al. Knockout of the α2 but not α1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-β-4-ribofuranoside but not contraction-induced glucose uptake in skeletal muscle. J. Biol. Chem. 2004;279:1070–1079. doi: 10.1074/jbc.M306205200. [DOI] [PubMed] [Google Scholar]
- 25.Barnes B.R., et al. The 5′-AMP-activated protein kinase γ3 isoform has a key role in carbohydrate and lipid metabolism in glycolytic skeletal muscle. J. Biol. Chem. 2004;279:38441–38447. doi: 10.1074/jbc.M405533200. [DOI] [PubMed] [Google Scholar]
- 26.Sakamoto K., et al. Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction. Embo J. 2005;24:1810–1820. doi: 10.1038/sj.emboj.7600667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sano H., et al. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J. Biol. Chem. 2003;278:14599–14602. doi: 10.1074/jbc.C300063200. [DOI] [PubMed] [Google Scholar]
- 28.Larance M., et al. Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J. Biol. Chem. 2005;280:37803–37813. doi: 10.1074/jbc.M503897200. [DOI] [PubMed] [Google Scholar]
- 29.Zeigerer A., McBrayer M.K., McGraw T.E. Insulin stimulation of GLUT4 exocytosis, but not its inhibition of endocytosis, is dependent on RabGAP AS160. Mol. Biol. Cell. 2004;15:4406–4415. doi: 10.1091/mbc.E04-04-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karlsson H.K.R., et al. Insulin-stimulated phosphorylation of the Akt substrate AS160 is impaired in skeletal muscle of type 2 diabetic subjects. . Diabetes. 2005;54:1692–1697. doi: 10.2337/diabetes.54.6.1692. [DOI] [PubMed] [Google Scholar]
- 31.Deshmukh A., et al. Exercise-induced phosphorylation of the novel Akt substrates AS160 and filamin A in human skeletal muscle. Diabetes. 2006 doi: 10.2337/db05-1419. [DOI] [PubMed] [Google Scholar]
- 32.Treebak J.T., et al. AMPK-mediated AS160 phosphorylation in skeletal muscle is dependent on AMPK catalytic and regulatory subunits. . Diabetes. 2006 doi: 10.2337/db06-0175. In press. [DOI] [PubMed] [Google Scholar]
- 33.Zhou G., et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 2001;108:1167–1174. doi: 10.1172/JCI200113505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Musi N., et al. Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes. 2002;51:2074–2081. doi: 10.2337/diabetes.51.7.2074. [DOI] [PubMed] [Google Scholar]
- 35.Ruderman N.B., Saha A.K., Vavvas D., Witters L.A. Malonyl-CoA, fuel sensing, and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 1999;276:E1–E18. doi: 10.1152/ajpendo.1999.276.1.E1. [DOI] [PubMed] [Google Scholar]
- 36.Trumble G., Smith M., Winder W. Purification and characterization of rat skeletal muscle acetyl-CoA carboxylase. Eur. J. Biochem. 1995;231:192–198. doi: 10.1111/j.1432-1033.1995.tb20686.x. [DOI] [PubMed] [Google Scholar]
- 37.Winder W.W., Hardie D.G. Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am. J. Physiol. Endocrinol. Metab. 1996;270:E299–E304. doi: 10.1152/ajpendo.1996.270.2.E299. [DOI] [PubMed] [Google Scholar]
- 38.Winder W.W., et al. Phosphorylation of rat muscle acetyl-CoA carboxylase by AMP-activated protein kinase and protein kinase A. J. Appl. Physiol. 1997;82:219–225. doi: 10.1152/jappl.1997.82.1.219. [DOI] [PubMed] [Google Scholar]
- 39.Merrill G.F., Kurth E.J., Rasmussen B.B., Winder W.W. Influence of malonyl-CoA and palmitate concentration on rate of palmitate oxidation in rat muscle. J. Appl. Physiol. 1998;85:1909–1914. doi: 10.1152/jappl.1998.85.5.1909. [DOI] [PubMed] [Google Scholar]
- 40.Collier C.A., Bruce C.R., Smith A.C., Lopaschuk G., Dyck D.J. Metformin counters the insulin-induced suppression of fatty acid oxidation and stimulation of triacylglycerol storage in rodent skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2006 doi: 10.1152/ajpendo.00272.2005. [DOI] [PubMed] [Google Scholar]
- 41.Holloszy J.O. Exercise-induced increase in muscle insulin sensitivity. J. Appl. Physiol. 2005;99:338–343. doi: 10.1152/japplphysiol.00123.2005. [DOI] [PubMed] [Google Scholar]
- 42.Zierath J.R. Exercise effects of muscle insulin signaling and action. Invited review: exercise training-induced changes in insulin signaling in skeletal muscle. J. Appl. Physiol. 2002;93:773–781. doi: 10.1152/japplphysiol.00126.2002. [DOI] [PubMed] [Google Scholar]
- 43.Holloszy J. Adaptation of skeletal muscle to endurance exercise. Med. Sci. Sports. 1975;7:155–164. [PubMed] [Google Scholar]
- 44.Morino K., et al. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J. Clin. Invest. 2005;115:3587–3593. doi: 10.1172/JCI25151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Petersen K.F., Dufour S., Befroy D., Garcia R., Shulman G.I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Petersen K.F., et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. . Science. 2003;300:1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fisher J.S., Gao J., Han D.-H., Holloszy J.O., Nolte L.A. Activation of AMP kinase enhances sensitivity of muscle glucose transport to insulin. . Am. J. Physiol. Endocrinol. Metab. 2002;282:E18–E23. doi: 10.1152/ajpendo.2002.282.1.E18. [DOI] [PubMed] [Google Scholar]
- 48.Song X.M., et al. 5-Aminoimidazole-4-carboxamide ribonucleoside treatment improves glucose homeostasis in insulin-resistant diabetic (ob/ob) mice. Diabetologia. 2002;45:56–65. doi: 10.1007/s125-002-8245-8. [DOI] [PubMed] [Google Scholar]
- 49.Holmes B.F., Kurth-Kraczek E.J., Winder W.W. Chronic activation of 5′-AMP-activated protein kinase increases GLUT-4, hexokinase, and glycogen in muscle. J. Appl. Physiol. 1999;87:1990–1995. doi: 10.1152/jappl.1999.87.5.1990. [DOI] [PubMed] [Google Scholar]
- 50.Iglesias M.A., et al. AICAR administration causes an apparent enhancement of muscle and liver insulin action in insulin-resistant high-fat-fed rats. Diabetes. 2002;51:2886–2894. doi: 10.2337/diabetes.51.10.2886. [DOI] [PubMed] [Google Scholar]
- 51.Buhl E.S., et al. Chronic treatment with 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside increases insulin-stimulated glucose uptake and GLUT4 translocation in rat skeletal muscles in a fiber type-specific manner. Diabetes. 2001;50:12–17. doi: 10.2337/diabetes.50.1.12. [DOI] [PubMed] [Google Scholar]
- 52.Long Y.C., et al. Role of AMP-activated protein kinase in the coordinated expression of genes controlling glucose and lipid metabolism in mouse white skeletal muscle. Diabetologia. 2005;48:2354–2364. doi: 10.1007/s00125-005-1962-5. [DOI] [PubMed] [Google Scholar]
- 53.Nilsson E.C., et al. Opposite transcriptional regulation in skeletal muscle of AMPK gamma 3 R225Q transgenic versus knock-out mice. J. Biol. Chem. 2006;281:7244–7252. doi: 10.1074/jbc.M510461200. [DOI] [PubMed] [Google Scholar]
- 54.Barnes B.R., et al. Changes in exercise-induced gene expression in 5′-AMP-activated protein kinase γ3-null and γ3 R225Q transgenic mice. Diabetes. 2005;54:3484–3489. doi: 10.2337/diabetes.54.12.3484. [DOI] [PubMed] [Google Scholar]
- 55.Ojuka E.O., et al. Regulation of GLUT4 biogenesis in muscle: evidence for involvement of AMPK and Ca2+. Am. J. Physiol. Endocrinol. Metab. 2002;282:E1008–E1013. doi: 10.1152/ajpendo.00512.2001. [DOI] [PubMed] [Google Scholar]
- 56.Zong H., et al. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc. Natl. Acad. Sci. U. S. A. . 2002;99:15983–15987. doi: 10.1073/pnas.252625599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bergeron R., et al. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am. J. Physiol. Endocrinol. Metab. 2001;281:E1340–E1346. doi: 10.1152/ajpendo.2001.281.6.E1340. [DOI] [PubMed] [Google Scholar]
- 58.Puigserver P., Spiegelman B.M. Peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α): transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- 59.Winder W.W., et al. Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J. Appl. Physiol. 2000;88:2219–2226. doi: 10.1152/jappl.2000.88.6.2219. [DOI] [PubMed] [Google Scholar]
- 60.Jorgensen S.B., et al. Effects of alpha-AMPK knockout on exercise-induced gene activation in mouse skeletal muscle. FASEB J. . 2005;19:1146–1148. doi: 10.1096/fj.04-3144fje. [DOI] [PubMed] [Google Scholar]
- 61.Consoli A. Role of liver in pathophysiology of NIDDM. Diabetes Care. 1992;15:430–441. doi: 10.2337/diacare.15.3.430. [DOI] [PubMed] [Google Scholar]
- 62.Saltiel A.R. New perspectives into the molecular pathogenesis and treatment of type 2 diabetes. Cell. 2001;104:517–529. doi: 10.1016/s0092-8674(01)00239-2. [DOI] [PubMed] [Google Scholar]
- 63.Saltiel A.R., Kahn C.R. Insulin signalling and the regulation of glucose and lipid metabolism. . Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 64.Lochhead P.A., Salt I.P., Walker K.S., Hardie D.G., Sutherland C. 5-Aminoimidazole-4-carboxamide riboside mimics the effects of insulin on the expression of the 2 key gluconeogenic genes PEPCK and glucose-6-phosphatase. Diabetes. 2000;49:896–903. doi: 10.2337/diabetes.49.6.896. [DOI] [PubMed] [Google Scholar]
- 65.Andreelli F., et al. Liver adenosine monophosphate-activated kinase-alpha2 catalytic subunit is a key target for the control of hepatic glucose production by adiponectin and leptin but not by insulin. Endocrinology. 2006;147:2432–2441. doi: 10.1210/en.2005-0898. [DOI] [PubMed] [Google Scholar]
- 66.Koo S.-H., et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. . Nature. 2005;437:1109–1111. doi: 10.1038/nature03967. [DOI] [PubMed] [Google Scholar]
- 67.Shaw R.J., et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Foretz M., Carling D., Guichard C., Ferre P., Foufelle F. AMP-activated protein kinase inhibits the glucose-activated expression of fatty acid synthase gene in rat hepatocytes. J. Biol. Chem. 1998;273:14767–14771. doi: 10.1074/jbc.273.24.14767. [DOI] [PubMed] [Google Scholar]
- 69.Woods A., et al. Characterization of the role of AMP-activated protein kinase in the regulation of glucose-activated gene expression using constitutively active and dominant negative forms of the kinase. Mol. Cell. Biol. 2000;20:6704–6711. doi: 10.1128/mcb.20.18.6704-6711.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zang M., et al. AMP-activated protein kinase is required for the lipid-lowering effect of metformin in insulin-resistant human HepG2 cells. J. Biol. Chem. 2004;279:47898–47905. doi: 10.1074/jbc.M408149200. [DOI] [PubMed] [Google Scholar]
- 71.Groop L.C., et al. Glucose and free fatty acid metabolism in non-insulin-dependent diabetes mellitus. Evidence for multiple sites of insulin resistance. J. Clin. Invest. 1989;84:205–213. doi: 10.1172/JCI114142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bays H., Mandarino L., DeFronzo R.A. Role of the adipocyte, free fatty acids, and ectopic fat in pathogenesis of type 2 diabetes mellitus: peroxisomal proliferator-activated receptor agonists provide a rational therapeutic approach. J. Clin. Endocrinol. Metab. 2004;89:463–478. doi: 10.1210/jc.2003-030723. [DOI] [PubMed] [Google Scholar]
- 73.Garton A.J., et al. Phosphorylation of bovine hormone-sensitive lipase by the AMP-activated protein kinase. A possible antilipolytic mechanism. Eur. J. Biochem. 1989;179:249–254. doi: 10.1111/j.1432-1033.1989.tb14548.x. [DOI] [PubMed] [Google Scholar]
- 74.Sullivan J.E., et al. Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Lett. 1994;353:33–36. doi: 10.1016/0014-5793(94)01006-4. [DOI] [PubMed] [Google Scholar]
- 75.Daval M., et al. Anti-lipolytic action of AMP-activated protein kinase in rodent adipocytes. . J. Biol. Chem. 2005;280:25250–25257. doi: 10.1074/jbc.M414222200. [DOI] [PubMed] [Google Scholar]
- 76.Villena J.A., et al. Induced adiposity and adipocyte hypertrophy in mice lacking the AMP-activated protein kinase-alpha2 subunit. Diabetes. 2004;53:2242–2249. doi: 10.2337/diabetes.53.9.2242. [DOI] [PubMed] [Google Scholar]
- 77.Bergeron R., et al. Effect of 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside infusion on in vivo glucose and lipid metabolism in lean and obese Zucker rats. Diabetes. 2001;50:1076–1082. doi: 10.2337/diabetes.50.5.1076. [DOI] [PubMed] [Google Scholar]
- 78.Yin W., Mu J., Birnbaum M.J. Role of AMP-activated protein kinase in cyclic AMP-dependent lipolysis in 3T3-L1 adipocytes. J. Biol. Chem. 2003;278:43074–43080. doi: 10.1074/jbc.M308484200. [DOI] [PubMed] [Google Scholar]
- 79.Detimary P., Jonas J.C., Henquin J.C. Possible links between glucose-induced changes in the energy state of pancreatic B cells and insulin release. Unmasking by decreasing a stable pool of adenine nucleotides in mouse islets. J. Clin. Invest. 1995;96:1738–1745. doi: 10.1172/JCI118219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Detimary P., et al. The changes in adenine nucleotides measured in glucose-stimulated rodent islets occur in beta cells but not in alpha cells and are also observed in human islets. J. Biol. Chem. 1998;273:33905–33908. doi: 10.1074/jbc.273.51.33905. [DOI] [PubMed] [Google Scholar]
- 81.Aguilar-Bryan L., Bryan J. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr. Rev. 1999;20:101–135. doi: 10.1210/edrv.20.2.0361. [DOI] [PubMed] [Google Scholar]
- 82.Safayhi H., et al. L-type calcium channels in insulin-secreting cells: biochemical characterization and phosphorylation in RINm5F cells. Mol. Endocrinol. 1997;11:619–629. doi: 10.1210/mend.11.5.9922. [DOI] [PubMed] [Google Scholar]
- 83.Rutter G.A. Nutrient-secretion coupling in the pancreatic islet beta-cell: recent advances. Mol. Aspects Med. 2001;22:247–284. doi: 10.1016/s0098-2997(01)00013-9. [DOI] [PubMed] [Google Scholar]
- 84.Detimary P., Gilon P., Henquin J.C. Interplay between cytoplasmic Ca2+ and the ATP/ADP ratio: a feedback control mechanism in mouse pancreatic islets. Biochem. J. 1998;333:269–274. doi: 10.1042/bj3330269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Salt I.P., Johnson G., Ashcroft S.J., Hardie D.G. AMP-activated protein kinase is activated by low glucose in cell lines derived from pancreatic beta cells, and may regulate insulin release. Biochem. J. 1998;335:533–539. doi: 10.1042/bj3350533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.da Silva Xavier G., et al. Role of AMP-activated protein kinase in the regulation by glucose of islet beta cell gene expression. Proc. Natl. Acad. Sci. U. S. A. . 2000;97:4023–4028. doi: 10.1073/pnas.97.8.4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.da Silva Xavier G., et al. Role for AMP-activated protein kinase in glucose-stimulated insulin secretion and preproinsulin gene expression. . Biochem. J. 2003;371:761–774. doi: 10.1042/BJ20021812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Leclerc I., et al. Metformin, but not leptin, regulates AMP-activated protein kinase in pancreatic islets: impact on glucose-stimulated insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2004;286:E1023–E1031. doi: 10.1152/ajpendo.00532.2003. [DOI] [PubMed] [Google Scholar]
- 89.Zhang S., Kim K.H. Glucose activation of acetyl-CoA carboxylase in association with insulin secretion in a pancreatic beta-cell line. J. Endocrinol. 1995;147:33–41. doi: 10.1677/joe.0.1470033. [DOI] [PubMed] [Google Scholar]
- 90.Eto K., et al. Genetic manipulations of fatty acid metabolism in β-cells are associated with dysregulated insulin secretion. Diabetes. 2002;51(Suppl. 3):S414–S420. doi: 10.2337/diabetes.51.2007.s414. [DOI] [PubMed] [Google Scholar]
- 91.Rutter G.A., Da Silva Xavier G., Leclerc I. Roles of 5′-AMP-activated protein kinase (AMPK) in mammalian glucose homoeostasis. . Biochem. J. 2003;375:1–16. doi: 10.1042/BJ20030048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Halaas J.L., et al. Weight-reducing effects of the plasma protein encoded by the obese gene. . Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 93.Kahn B.B., Flier J.S. Obesity and insulin resistance. J. Clin. Invest. 2000;106:473–481. doi: 10.1172/JCI10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Unger R.H., Zhou Y.-T., Orci L. Regulation of fatty acid homeostasis in cells: novel role of leptin. Proc. Natl. Acad. Sci. U. S. A. . 1999;96:2327–2332. doi: 10.1073/pnas.96.5.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lee Y., et al. Liporegulation in diet-induced obesity. The antisteatotic role of hyperleptinemia. J. Biol. Chem. 2001;276:5629–5635. doi: 10.1074/jbc.M008553200. [DOI] [PubMed] [Google Scholar]
- 96.Muoio D.M., Dohm G.L., Fiedorek F.T., Tapscott E.B., Coleman R.A. Leptin directly alters lipid partitioning in skeletal muscle. Diabetes. 1997;46:1360–1363. doi: 10.2337/diab.46.8.1360. [DOI] [PubMed] [Google Scholar]
- 97.Kamohara S., Burcelin R., Halaas J.L., Friedman J.M., Charron M.J. Acute stimulation of glucose metabolism in mice by leptin treatment. Nature. 1997;389:374–377. doi: 10.1038/38717. [DOI] [PubMed] [Google Scholar]
- 98.Haque M., et al. Role of the sympathetic nervous system and insulin in enhancing glucose uptake in peripheral tissues after intrahypothalamic injection of leptin in rats. Diabetes. 1999;48:1706–1712. doi: 10.2337/diabetes.48.9.1706. [DOI] [PubMed] [Google Scholar]
- 99.Minokoshi Y., et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. 2002;415:339–343. doi: 10.1038/415339a. [DOI] [PubMed] [Google Scholar]
- 100.Fruebis J., et al. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc. Natl. Acad. Sci. U. S. A. . 2001;98:2005–2010. doi: 10.1073/pnas.041591798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tomas E., et al. Enhanced muscle fat oxidation and glucose transport by ACRP30 globular domain: acetyl-CoA carboxylase inhibition and AMP-activated protein kinase activation. Proc. Natl. Acad. Sci. U. S. A. . 2002;99:16309–16313. doi: 10.1073/pnas.222657499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yamauchi T., et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 103.Porte D., et al. , 1998Obesity, diabetes and the central nervous system. Diabetologia. 41863–881. [DOI] [PubMed] [Google Scholar]
- 104.Leibowitz S.F., Wortley K.E. Hypothalamic control of energy balance: different peptides, different functions. Peptides. 2004;25:473–504. doi: 10.1016/j.peptides.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 105.Schwartz M.W., Woods S.C., Porte D., Seeley R.J., Baskin D.G. Central nervous system control of food intake. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 106.Andersson U., et al. AMP-activated protein kinase plays a role in the control of food intake. . J. Biol. Chem. 2004;279:12005–12008. doi: 10.1074/jbc.C300557200. [DOI] [PubMed] [Google Scholar]
- 107.Minokoshi Y., et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 2004;428:569–574. doi: 10.1038/nature02440. [DOI] [PubMed] [Google Scholar]
- 108.Kim E.-K., et al. C75, a fatty acid synthase inhibitor, reduces food intake via hypothalamic AMP-activated protein kinase. J. Biol. Chem. 2004;279:19970–19976. doi: 10.1074/jbc.M402165200. [DOI] [PubMed] [Google Scholar]
- 109.Perrin C., Knauf C., Burcelin R. Intracerebroventricular infusion of glucose, insulin, and the adenosine monophosphate-activated kinase activator, 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside, controls muscle glycogen synthesis. . Endocrinology. 2004;145:4025–4033. doi: 10.1210/en.2004-0270. [DOI] [PubMed] [Google Scholar]
- 110.Viollet B., et al. The AMP-activated protein kinase α2 catalytic subunit controls whole-body insulin sensitivity. J. Clin. Invest. 2003;111:91–98. doi: 10.1172/JCI200316567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mu J., Barton E.R., Birnbaum M.J. Selective suppression of AMP-activated protein kinase in skeletal muscle: update on ‘lazy mice.’. Biochem. Soc. Trans. 2003;31:236–241. doi: 10.1042/bst0310236. [DOI] [PubMed] [Google Scholar]
- 112.Barnes B.R., et al. 5′-AMP-activated protein kinase regulates skeletal muscle glycogen content and ergogenics. Faseb J. 2005;19:773–779. doi: 10.1096/fj.04-3221com. [DOI] [PubMed] [Google Scholar]