

Abstract
The etiology of primary open angle glaucoma, a leading cause of age-related blindness, remains poorly defined, although elevated intraocular pressure (IOP) contributes to the disease progression. To better understand the mechanisms causing elevated IOP from aqueous humor circulation, we pursued proteomic analyses of trabecular meshwork (TM) from glaucoma and age-matched control donors. These analyses demonstrated that Cochlin, a protein associated with deafness disorder DFNA9, is present in glaucomatous but absent in normal TM. Cochlin was also detected in TM from the glaucomatous DBA/2J mouse preceding elevated IOP but found to be absent in three other mouse lines that do not develop elevated IOP. Histochemical analyses revealed co-deposits of Cochlin and mucopolysaccharide in human TM around Schlemm’s canal, similar to that observed in the cochlea in DFNA9 deafness. Purified Cochlin was found to aggregate after sheer stress and to induce the aggregation of TM cells in vitro. Age-dependent in vivo increases in Cochlin were observed in glaucomatous TM, concomitant with a decrease in type II collagen, suggesting that Cochlin may disrupt the TM architecture and render components like collagen more susceptible to degradation and collapse. Overall, these observations suggest that Cochlin contributes to elevated IOP in primary open angle glaucoma through altered interactions within the TM extracellular matrix, resulting in cell aggregation, mucopolysaccharide deposition, and significant obstruction of the aqueous humor circulation.
Glaucoma encompasses a group of blinding diseases classified generally as primary, for which there is no known etiology, or as secondary, in which a previous illness or injury is contributory. Primary open angle glaucoma (POAG)1 is the most common form of the disease, affecting 3 million Americans and over 70 million people worldwide (1). Vision loss in most but not all glaucoma cases is related to an increase in intraocular pressure (IOP) with subsequent damage to the optic nerve. The molecular basis of the pathology is understood poorly, but the risk for POAG clearly increases with age, and ethnicity plays a role (e.g. blacks exhibit a higher incidence of POAG than whites and at an earlier age of onset). Although specific genes have been implicated in glaucoma pathology, including for example, TIGR and its gene product of unknown function, myocilin, genetic studies to date remain inconclusive regarding glaucoma disease mechanisms (2).
Elevated IOP typically develops into glaucoma as a result of impeded aqueous humor outflow (3). Aqueous humor is actively produced by the ciliary epithelium in the posterior chamber of the eye and circulates through the pupil to the anterior chamber where it drains through the trabecular meshwork (TM) into Schlemm’s canal and the episcleral veins (4). Resistance to outflow occurs commonly in the TM, which has a complex extracellular matrix (ECM) composed of collagen beams lined with endothelium-like cells (5, 6). The mechanisms of resistance are not known; however, the pseudoendothelial cells in the TM produce a mucopolysaccharide (MPS) (7) that appears to function in attracting macrophages for phagocytic self-cleaning of the TM (8). A loss of control of MPS levels in the TM appears to disrupt the self-cleaning process and can result in large changes in IOP (9). In other sensory systems, MPS deposits in the cochlea have been associated with the late onset and progressive auditory and vestibular disorder DFNA9, which involves increased intracranial pressure (10, 11). Drugs for treating POAG slow the disease progression by reducing aqueous production or by increasing aqueous outflow but do not provide a cure. Trabeculectomy is the most common treatment of last resort for POAG and involves the surgical removal of a small amount of TM tissue and redirection of the aqueous flow through the conjunctiva to the episcleral vessels. To better understand the molecular mechanisms involved in glaucoma and specifically in the blockage of aqueous outflow, we initiated a classical proteomics study to compare the protein composition of glaucomatous TM obtained by trabeculectomy with that of normal TM. Here we present evidence that Cochlin, a protein associated with the auditory disorder DFNA9, is absent in normal TM but increases with age in glaucomatous TM in association with MPS deposits.
EXPERIMENTAL PROCEDURES
Tissue Procurement
Human eyes from 35 normal donors and 5 POAG donors, all between 40 and 85 years of age, were used in this study, and were obtained through the Cleveland Eye Bank. Eyes were enucleated within 12 h of death and stored at −80 °C until TM tissue was isolated by dissection. Normal control eyes were from donors with no visual field defects, no evidence of glaucoma, and without central nervous system abnormalities. Fixed human TM tissues used for immunohistochemistry were obtained from the Eye Donor Program of the Foundation Fighting Blindness (Owings Mills, MD). Glaucomatous eyes and tissues were from clinically documented POAG donors. Glaucomatous TM tissues (~1–2 mm3) were obtained by trabeculectomy from 30 POAG patients in the Cole Eye Institute, Cleveland Clinic Foundation, with institutional review board approval. Human tissue obtained by trabeculectomy consisted predominantly of TM, but possible contamination with small amounts of surrounding tissue (e.g. sclera) cannot be excluded. TM cells for cell culture were isolated from the rim tissue associated with corneas used for transplantation at the Cole Eye Institute and were obtained from healthy human eyes within 3 h of death and stored until use in Optisol-GS medium (Chiron Vision, Clairmont, CA).
Adult mice from inbred strains DBA/2J, BALBc/ByJ, CD1, and C57BL/6J were obtained from The Jackson Laboratory (Bar Harbor, ME) and bred in the Cole Eye Institute animal facility. TM samples were obtained surgically following sacrifice with carbon dioxide using animal procedures approved by the Institutional Animal Care and Use Committee of the Cleveland Clinic Foundation.
Protein Analyses
TM tissue from cadaver and trabeculectomy samples was extracted by homogenization in 100 mm Tris-Cl buffer, pH 7.8, containing 5 mm dithiothreitol, 1 mm SnCl2, 50 mm NaHPO4, 1 mm diethylenetriaminepentaacetic acid, 100 mm butylated hydroxy toluene, and 0.5% SDS. Insoluble material was removed by centrifugation (8000 × g for 5 min), and soluble protein was quantified by the Bradford assay (12), yielding ~15–20 μg of total soluble protein/trabeculectomy tissue sample (~1–2 mm3). Protein extracts were subjected to SDS-PAGE on 4–15% gradient gels (Bio-Rad), and the gels were used either for mass spectrometric proteomic analyses or for Western analyses (13). For protein identifications, gel slices were excised and digested in situ with trypsin, and peptides were analyzed by liquid chromatography electrospray tandem mass spectrometry using a CapLC system and a quadrupole time-of-flight mass spectrometer (QTOF2, Waters Corp., Milford, MA). Protein identifications from MS/MS data utilized the ProteinLynx™ Global Server (Waters Corp.) and Mascot (Matrix Science) search engines and the Swiss Protein and NCBI protein sequence data bases (13, 14). Western analysis on a polyvinylidene difluoride membrane utilized established protocols (13) and chicken polyclonal antibody against human Cochlin (at ~5 μg/ml) (15), mouse monoclonal anti-collagen II, and anti-glyceraldehyde-3-phosphate dehydrogenase (Chemicon International, Temecula, CA). For quantitative Western analyses, anti-mouse secondary antibody linked to 700 nm IR dye and anti-chicken secondary antibody coupled to 800 nm IR dye were used with analyses on an Odyssey infrared imaging system according to the manufacturer’s instructions (Li-Cor Biosciences, Lincoln, NE).
Histochemical Analyses
Immunohistochemical analyses to localize Cochlin in ocular tissue were performed with cadaver eyes enucleated within 8 h of death and fixed immediately with calcium acetate-buffered 4% para-formaldehyde. Paraffin-embedded tissue was blocked and sectioned (12 μm) in 5% BlokHen (Aves Labs, Inc., Tigard, OR) in phosphate-buffered saline, then incubated overnight with 10 ng of anti-Cochlin antibody (15) at 4 °C and subsequently with 10 ng of rhodamine-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) for 1 h at room temperature. Sections were sealed with Vectashield and analyzed with a Nikon EFD-3 fluorescence microscope attached to a charge-coupled device camera. Movat’s pentachrome staining for MPS was performed on multiple sections from each donor tissue according to a published protocol (16). Ten donor tissues each for control and glaucomatous TM were examined, with alternate sections used for Movat’s staining and immunohistochemistry. To ensure identical processing and uniform exposure, control and glaucomatous sections were examined side by side on the same slide. For confocal immunohistochemistry, 12-μm paraffin-embedded sections were labeled with antibodies specific to Cochlin and with a secondary antibody coupled to TRITC (red) and then analyzed using a Leica laser scanning confocal microscope (TCS-SP2, Leica, Exton, PA). A series of 1-μm thick x-y (en face) images were collected and added to obtain an image representing a three-dimensional projection of the entire 12-μm section. Confocal microscopic panels were composed using Adobe Photoshop 5.5.
Aggregation Assays
TM cells, from rim tissue associated with healthy corneas used for transplantation, were isolated for in vitro assays by dissection and extracellular matrix digestion (17). TM primary cell cultures were established in Dulbecco’s modified Eagle’s medium containing 10% bovine serum, 50 units/ml penicillin G sodium, and 50 μg/ml streptomycin sulfate (Invitrogen). For the aggregation assay, approximately 3000 cells were seeded in 4-well plates (35-mm wells, BD Biosciences) and grown for 5–7 days at 37 °C in humidified air containing 5% CO2. The cells were washed with PBS, the PBS was aspirated, and then the cells were treated with 5 μg of either purified recombinant Cochlin or purified recombinant Notch produced in COS-7 cells or with an equal volume of medium from empty vector-transfected COS-7 cells after HA-affinity fractionation. To evaluate whether antibody could neutralize the observed cell aggregation, Cochlin (5 μg) was preincubated for 30 min with either 20 μg of polyclonal chicken anti-Cochlin or nonspecific chicken IgG (Immun System, Uppsala, Sweden), and then the mixture was used to treat the primary TM cells in an identical manner. After 2 min, Dulbecco’s modified Eagle’s medium with 10% bovine serum was added, and cells were further incubated for 24–36 h as above. Cells were examined with an inverted microscope (model IM 35, Zeiss) and photographed using the Pix Cell II microscope system (Arcturus, Mountain View, CA) equipped with an Olympus camera, an Hitachi digital converter/Sony visual screen, and a Dell Oph Plex GX 110 computer. Recombinant Cochlin and recombinant Notch-1 were produced in COS-7 cells with 3-HA and 4-HA epitope tags, respectively, using the pcDNA 3.0 vector and SuperFect transfection reagent (Qiagen, Valencia, CA) (18, 19). The proteins were affinity-purified from the media of transiently transfected COS-7 cells using rabbit anti-HA antibody (Y-11, Santa Cruz Biotechnology, Inc., Santa Cruz, CA) coupled to protein A-agarose beads with dimethyl pimelimidate. Purified recombinant Cochlin was subjected to sheer stress by passing the protein (5 μg) through a Hamilton syringe (50 μl) five times in 50 mm Tris-Cl, pH 7.5, 125 mm NaCl, and 0.1% genapol and then was analyzed by non-reducing SDS-PAGE immunoblotting.
RESULTS
Cochlin Is Uniquely Associated with Glaucomatous TM
Protein extracts from six POAG and six normal TM tissues were subjected to SDS-PAGE, the gel slices were excised (Fig. 1A), and the proteins were identified using well established mass spectrometric and bioinformatic methods. Overall, 368 proteins were identified, of which 52 were detected only in glaucomatous TM but with variable frequency (see supplemental Table I). Cochlin, a protein of unknown function, was the component identified most frequently in glaucomatous TM. Indeed, Cochlin was detected by mass spectrometry in 5 of the 6 POAG samples (see supplemental Table I) and by immunoblotting analyses in 20 of 20 glaucomatous TM samples but not in the 20 normal control TM samples, irrespective of cadaver or trabeculectomy tissue source (Fig. 1B). Based on these findings from human TM, we probed Cochlin expression in an established animal model of glaucoma, the DBA/2J mouse. This mouse line exhibits increased IOP at 6–8 months of age, with progressive damage to the optic nerve and progressive hearing loss (20). Western analyses of TM extracts from 10-week-old DBA/2J mice and from age-matched C57BL/6J, CD1, or BALBc/ByJ control mice, which do not develop increased IOP, revealed Cochlin expression only in TM from the DBA/2J mice (Fig. 2A). Western analysis of DBA/2J mouse TM over the first 16 weeks of postnatal development detected Cochlin expression at 1–2 weeks of age and demonstrated progressively increased expression at 3, 10, and 16 weeks of age (Fig. 2B).
Fig. 1. SDS-PAGE and Cochlin Western analysis.
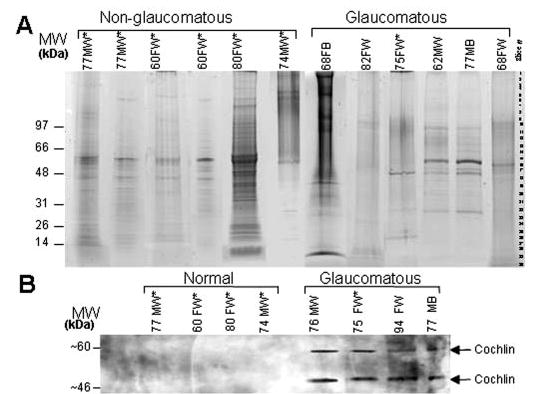
A, SDS-PAGE is shown of human TM protein (~10 μg/lane) from POAG (Glaucomatous) and normal (Non-glaucomatous) donors (Coomassie Blue staining). Gel slices were excised as indicated by slice number, and proteins were identified by LC MS/MS (see supplemental Table I). B, representative Western analyses are shown with polyclonal chicken anti-Cochlin of protein extracts from human TM demonstrating the presence of ~60-and ~46-kDa Cochlin isoforms (arrows) in glaucomatous tissues. Donor age, gender, and race are indicated in A and B (M, male; F, female; W, Caucasian; B, black). *, cadaver tissue; all other tissues are from trabeculectomy.
Fig. 2. Cochlin expression in DBA/2J mice.

Protein (~10 μg) extracted from mouse TM was subjected to Western analysis with anti-Cochlin antibody. A, Cochlin expression in TM from C57BL/6J, CD1, BALBc/ByJ, and DBA/2J mice (all 10 weeks old) is shown with chemiluminescence detection. B, Cochlin expression is shown in TM from DBA/2J mice (1–16 weeks old) and a 16-week-old C57BL/6J control mouse detected using 800 nm IR dye-coupled anti-chicken secondary IgG.
Cochlin Co-localizes with Mucopolysaccharide in Glaucomatous TM Deposits
Sheath-derived plaques and microfibrillar deposits have been reported previously (21, 22) in the TM of POAG donor eyes. To probe the possible association of Cochlin with TM deposits, we pursued histochemical localization of Cochlin in the anterior segment of the human eye by staining alternate tissue sections with anti-Cochlin antibody or hematoxylin-eosin (Fig. 3A). Immunohistochemical analyses of 10 glaucomatous and 10 normal donor eyes confirmed the presence of Cochlin in glaucomatous but not normal TM (Fig. 3, B–D). Additional lower magnification and confocal microscopic images supporting the unique presence of Cochlin in glaucomatous TM deposits are shown in supplemental Figs. 1 and 2, respectively. Cochlin was localized within glaucomatous TM to variably sized deposits extending 80–150 μm along the length of Schlemm’s canal (Fig. 3, C and D). Movat’s pentachrome staining revealed the presence of acidophilic MPS or glycosaminoglycan in the core of the Cochlin deposits (Fig. 3F) but not in control eyes (Fig. 3E). Cochlin and MPS co-localized throughout the inner portion of the glaucomatous deposits, around Schlemm’s canal, and in pseudoendothelial cells, but MPS staining was lacking around the periphery of the deposits. The dense, highly branched microfibrillar plaques in the cochlea that have been reported (18) in DFNA9 also contain both Cochlin and MPS or glycosaminoglycans.
Fig. 3. Histochemical localization of Cochlin in human TM.
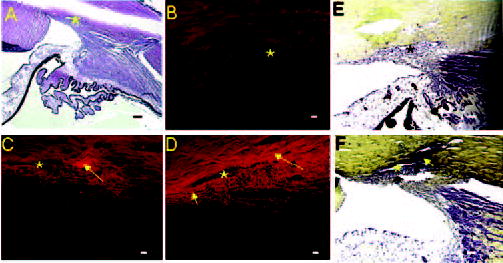
A, an anterior segment section stained with hematoxylin-eosin is shown. B–D, representative immunohistochemical analysis is shown with chicken polyclonal anti-Cochlin. B, normal TM from a 75-year-old Caucasian female donor; C and D, glaucomatous TM sections approximately 60 μm apart, obtained from a 77-year-old Caucasian female donor. Rhodamine-conjugated secondary antibody (anti-chicken) was used for immunofluorescence detection. Arrows, Cochlin deposits (C and D) that are ~80–150 μm; *, Schlemm’s canal. D, small and large arrows show spotty and large deposits, respectively. E and F, light microscopy of Movat’s pentachrome-stained tissue is shown. E, normal TM from a 75-year-old Caucasian female; F, glaucomatous TM from a 77-year-old Caucasian female. Arrow, MPS deposit around Schlemm’s canal. Bars = 100 μm (A, E, and F) and 40 μm (B–D).
Cochlin Causes in Vitro Aggregation of TM Cells
The Cochlin protein structure contains an amino-terminal factor C homology (FCH) domain, two von Willebrand factor A-like (VWFA) domains, and two potential N-linked glycosylation sites. The FCH domain is named after the horseshoe crab coagulation factor, which becomes activated on binding lipopolysaccharides, initiating a host defense coagulation cascade. The FCH domain exists with unknown function in several other proteins. VWFA domains exist in a number of ECM and immune system components, interact with collagen proteins such as GpIbα and integrin αIibβ3, and have been implicated in adherence to and aggregation of platelets and macrophages. To explore a possible role for Cochlin in ocular cell adhesion, we performed aggregation assays using human primary TM cells in culture and purified human recombinant Cochlin (Fig. 4). Aggregation of the TM cells was observed with the addition of exogenous Cochlin to the culture (Fig. 4A) but not with control protein Notch or with the empty recombinant vector-transfected medium (Fig. 4, B and C). Preincubating recombinant Cochlin with anti-Cochlin chicken antibody prior to adding it to the primary TM cell prevented cell aggregation, whereas preincubation with control chicken IgG did not (data not shown). Notably, the aggregation of purified recombinant Cochlin following sheer stress was also observed by non-reducing SDS-PAGE Western analysis, suggesting that disulfide bond formation may be involved in the aggregation mechanism (Fig. 4F). These in vitro observations are consistent with an extracellular role for Cochlin involving interactions with ECM components such as collagen.
Fig. 4. Aggregation of TM cells by Cochlin.
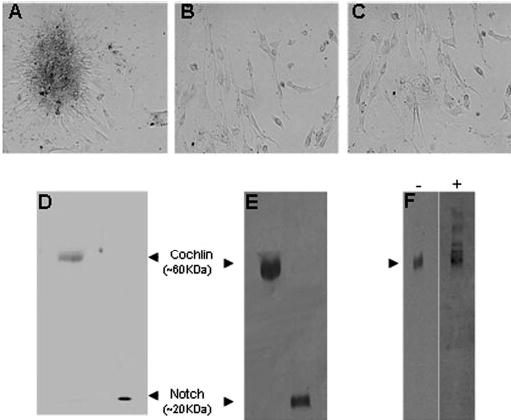
Primary TM cell cultures were established and treated as described under “Experimental Procedures” with: A, purified recombinant Cochlin (5 μg in 10 μl of PBS); B, purified recombinant Notch (5 μg in 10 μl of PBS); C, medium (10 μl) from mock empty vector-transfected COS-7 cells; D, Coomassie Blue-stained SDS-PAGE of purified recombinant Cochlin and Notch; E, Western analysis of purified recombinant Cochlin (~2 μg) and recombinant Notch (~1 μg) using polyclonal rabbit anti HA-antibody, and F, Western analysis after non-reducing 10% SDS-PAGE of purified recombinant Cochlin using anti-Cochlin antibody before (−) and after (+) sheer stress, revealing stress-induced higher mass aggregates (arrowhead).
Age-dependent Changes in the Cochlin and Collagen Content of Glaucomatous TM
To probe the molecular role of Cochlin in glaucomatous TM and a possible connection with ECM instability, expression levels of Cochlin and type II collagen were compared by quantitative Western analyses of glaucomatous and normal TM from donors of ages 40–85 years (Fig. 5). An age-dependent increase in Cochlin content was detected in glaucomatous TM, with a concomitant decrease in type II collagen. No significant change with age was apparent in the type II collagen content of normal TM. These results suggest that the increased presence of Cochlin may contribute to the alteration of ECM interactions in glaucomatous TM, perhaps resulting in the collapse or degradation of the collagen-containing microfibrils in TM beams.
Fig. 5. Cochlin increases and type II collagen decreases in glaucomatous TM.
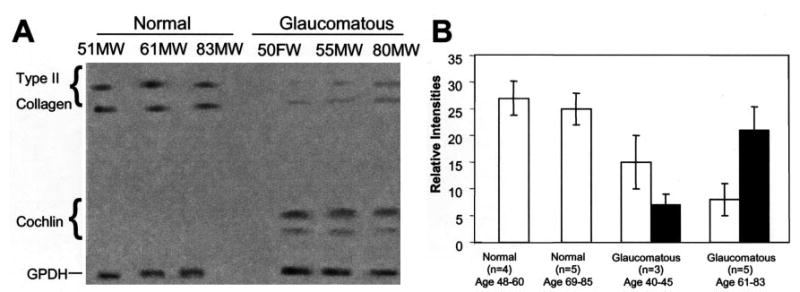
A, representative quantitative Western analyses of normal and glaucomatous TM protein extracts (5 μg each) are shown for Cochlin, type II collagen, and glyceraldehyde-3-phosphate dehydrogenase (GPDH) using IR dye-coupled secondary antibodies detected with an Odyssey infrared imaging system. Donor age, race, and gender are indicated (M, male; F, female; W, Caucasian). B, quantitative summary of multiple Western analyses as in A with normalization to glyceraldehyde-3-phosphate dehydrogenase staining intensity. Open bars, type II collagen; filled bars, Cochlin; error bars, ±S.D. for the indicated number of samples analyzed.
DISCUSSION
Classical proteomic methods initially detected Cochlin in the TM of glaucomatous but not normal human donors, irrespective of whether the tissue source was a cadaver or a fresh surgical specimen. Subsequently, Cochlin was found to be uniquely associated with glaucomatous human TM by Western and immunohistochemical analyses of additional POAG and normal TM donors. Western analyses also demonstrated the presence of Cochlin in TM from the DBA/2J glaucomatous mouse but not from three other mouse strains that do not develop elevated IOP. Proteomic analyses identified many other proteins in human TM (see supplemental Table I), including myocilin; however, the significance of proteins detected only in glaucomatous TM remains to be determined. A recent expressed sequence tag (EST) analysis (23) reported 1888 possibly expressed genes in normal human TM, including 198 of the 368 proteins (53%) identified in the present study. Other investigators (24) have demonstrated that protein and mRNA abundance levels in tissues are not directly proportional, and the lack of detection by LC MS/MS does not necessarily mean the absence of protein expression. Accordingly, comparisons of supplemental Table I data with the recent EST study results must be interpreted with caution. Our proteomic analyses were performed on individual, clinically documented TM samples, whereas the EST analysis was performed on mRNA pooled from 28 normal TM donors. The lack of rigorous quantitation further complicates the comparison of protein expression levels, which is exemplified by the EST detection of 28 clones of myocilin in normal donor TM in contrast to mass spectrometric detection of myocilin in 3 of 6 POAG TM analyzed but in none of 6 normal TM analyzed. Of relevance to the present study, two clones of Cochlin were observed in the EST analysis of pooled mRNA (23), implying a possible low level gene expression in normal TM. We have detected the COCH transcript in both normal and glaucomatous TM by reverse transcription PCR.2 Nevertheless, our Western analysis reproducibly failed to detect the protein in non-glaucomatous tissues regardless of the amount of TM analyzed, strongly suggesting that Cochlin was absent or in very low abundance in normal TM.
Cochlin is the product of the COCH gene, and its FCH domain is mutated in patients with the autosomal dominant nonsyndromic auditory and vestibular disorder DFNA9 (25, 26). COCH mutations have also been implicated in Ménière’s disease (with features of hearing loss and vertigo) (27) and in presbyacusis (age-related hearing loss) (28). These disorders are late onset and progressive in nature and parallel the clinical manifestations of POAG. Cochlin comprises the major non-collagen component of the ECM of the inner ear (29) but is also expressed in very low levels in the cerebellum and eye (25, 30). Localization of Cochlin within the eye has not been reported previously. The protein structure is highly conserved, with human Cochlin exhibiting 94 and 79% amino acid sequence identity, respectively, with the mouse and chicken proteins. Cochlin is a secreted protein with three glycosylated isoforms detectable in human cochlea, with apparent masses of ~40, ~46, and ~60 kDa (15, 18, 31). Two Cochlin isoforms (~46 and ~60 kDa) were detected in glaucomatous TM; however, the antibody we used recognizes only the higher molecular weight isoforms (18). Misfolded Cochlin has been implicated in the formation of cochlear deposits in DFNA9; interestingly, the ~40- and ~46-kDa isoforms contain VWFA-like domains but lack the amino-terminal FCH domain, which contains the disease-associated mutations (18). It has been suggested (25, 30) that Cochlin plays a structural role in the architecture of the cochlea by binding to components of the ECM. In patients with DFNA9, there is a marked decrease in cellularity and an accumulation of eosinophilic deposits that obstruct the cochlear and vestibular nerve channels (16, 25, 32). In patients with POAG, Cochlin expression in the TM increases with age, along with acidophilic Cochlin deposits around Schlemm’s canal, which are formed apparently by interactions with MPS and other ECM components.
Cochlin VWFA-like domains may contribute significantly to deposit formation in TM because hydrodynamic forces induce von Willebrand factor aggregation in suspension and may influence cell adhesion rates in the circulation (33). Although both wild type and mutant Cochlin expressed in COS-7 cells are processed and secreted normally, without apparent aggregation (18, 34), we found that sheer stress could induce the in vitro aggregation of purified recombinant Cochlin, possibly through disulfide bond formation. ECM interactions appear likely to underlie the formation of Cochlin deposits in glaucoma; however, hydrodynamic forces may exacerbate the process.
Cochlin may remain in the ECM of glaucomatous TM for a prolonged period and interact through its VWFA-like domains with fibrillar collagens. Altered interactions between fibrillar collagens and other ECM components have been suggested to trigger collagen degradation (35). Even simple dissociation between collagens and the surrounding ECM proteins may result in collagen degradation (36). Perturbations in collagenous fibrillar assembly caused by changes in collagen levels are known to result in a loss of tissue-specific morphology (36). Normally absent in healthy TM, Cochlin in combination with MPS may disrupt collagenous fibrillar assembly and contribute to collagen degradation in glaucomatous TM. The age-dependent increase in Cochlin and decrease in type II collagen that we observed in glaucomatous TM are consistent with an altered ECM. Although decreased collagen biosynthesis cannot be ruled out, the increased Cochlin may help to dissociate collagen from other TM proteins, rendering the ECM more susceptible to proteolytic degradation, collapse, and debris deposition. The large Cochlin deposits observed in glaucomatous TM could obstruct aqueous outflow across a wide region and thus have the potential to increase IOP.
Does Cochlin cause increased IOP or does increased IOP up-regulate Cochlin expression and deposition? Purified Cochlin exogenously added to TM primary cells resulted in the aggregation of the cells. Although the physiological relevance of this observation remains to be established, it is possible that Cochlin overexpression is associated with cell aggregation and MPS deposition, resulting in increased resistance and significant obstruction of the aqueous outflow. It is noteworthy that Cochlin is detectable in the DBA/2J mouse TM shortly after birth, and therefore, its expression precedes the earliest signs of elevated IOP at 6–8 months of age (20). Overall, the evidence presented here suggests that Cochlin may be involved in the intermediate events in glaucoma that lead to increased IOP. In any event, the presence of Cochlin-MPS deposits in TM from POAG patients opens new therapeutic avenues for treatment, for example, by targeted strategies for decreasing Cochlin expression or for increasing its degradation.
Supplementary Material
Acknowledgments
We thank Professors Ted Acott, M. Rosario Hernandez, Claude Burgoyne, and David Dueker for valuable discussions and comments on the manuscript, Marg Esser for technical assistance, Professor Joe Hollyfield for valuable discussions and for previously fixed histological samples, and Drs. Roger Langston, David Meisler, Bennie Jeng, and Victor Perez for control tissues.
Footnotes
This work was supported by the National Glaucoma Research Program of American Health Assistance Foundation (to S. K. B.), National Institutes of Health Grants DC03402 (to C. C. M.) and EY6603, EY14239, and EY015638 (to J. W. C.), a Merit Award from the Department of Veterans Affairs (to N. S. P.), a Research Center Grant from The Foundation Fighting Blindness (to J. W. C.), and funds from the Cleveland Clinic Foundation (to J. W. C.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table I and Figs. 1 and 2.
The abbreviations used are: POAG, primary open angle glaucoma; IOP, intraocular pressure; TM, trabecular meshwork; ECM, extracellular matrix; MPS, mucopolysaccharide; LC, liquid chromatography; MS/MS, tandem mass spectrometry; TRITC, tetramethylrhodamine isothiocyanate; HA, hemagglutinin; FCH, factor C homology; VWFA, von Willebrand factor A; EST, expressed sequence tag; PBS, phosphate-buffered saline.
S. K. Bhattacharya and J. W. Crabb, unpublished data.
References
- 1.Quigley HA. Br J Ophthalmol. 1996;80:389–393. doi: 10.1136/bjo.80.5.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen CS, Allingham RR. Curr Opin Ophthalmol. 2004;15:75–79. doi: 10.1097/00055735-200404000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Tomarev SI. Nat Med. 2001;7:294–295. doi: 10.1038/85432. [DOI] [PubMed] [Google Scholar]
- 4.Morrison, J. C., and Acott, T. S. (2003) in Glaucoma Science and Practice (Morrison, J. C., and Pollack, I. P., eds) pp. 34–41, Thieme Medical Publishers Inc., NY
- 5.Lutjen-Drecoll E. Prog Retin Eye Res. 1999;18:91–119. doi: 10.1016/s1350-9462(98)00011-1. [DOI] [PubMed] [Google Scholar]
- 6.Lutjen-Drecoll E. J Glaucoma. 2000;9:417–418. doi: 10.1097/00061198-200012000-00001. [DOI] [PubMed] [Google Scholar]
- 7.McMenamin PG, Steptoe RJ. J Anat. 1991;178:65–77. [PMC free article] [PubMed] [Google Scholar]
- 8.Sherwood ME, Richardson TM. Exp Eye Res. 1988;46:881–895. doi: 10.1016/s0014-4835(88)80040-x. [DOI] [PubMed] [Google Scholar]
- 9.Nickla DL, Wildsoet CF, Troilo D. Investig Ophthalmol Vis Sci. 2002;43:2519–2528. [PubMed] [Google Scholar]
- 10.Bitner-Glindzicz M. Br Med Bull. 2002;63:73–94. doi: 10.1093/bmb/63.1.73. [DOI] [PubMed] [Google Scholar]
- 11.Bom SJ, Kunst HP, Huygen PL, Cremers FP, Cremers CW. Br J Audiol. 1999;33:335–348. doi: 10.3109/03005369909090117. [DOI] [PubMed] [Google Scholar]
- 12.Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 13.Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H, Kamei M, Hasan A, Yan L, Rayborn ME, Salomon RG, Hollyfield JG. Proc Natl Acad Sci U S A. 2002;99:14682–14687. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.West KA, Yan L, Shadrach K, Sun J, Hasan A, Miyagi M, Crabb JS, Hollyfield JG, Marmorstein AD, Crabb JW. Mol Cell Proteomics. 2003;2:37–49. doi: 10.1074/mcp.d200001-mcp200. [DOI] [PubMed] [Google Scholar]
- 15.Robertson NG, Resendes BL, Lin JS, Lee C, Aster JC, Adams JC, Morton CC. Hum Mol Genet. 2001;10:2493–2500. doi: 10.1093/hmg/10.22.2493. [DOI] [PubMed] [Google Scholar]
- 16.Khetarpal U. Laryngoscope. 2000;110:1379–1384. doi: 10.1097/00005537-200008000-00030. [DOI] [PubMed] [Google Scholar]
- 17.Stamer WD, Roberts BC, Howell DN, Epstein DL. Investig Ophthalmol Vis Sci. 1998;39:1804–1812. [PubMed] [Google Scholar]
- 18.Robertson NG, Hamaker SA, Patriub V, Aster JC, Morton CC. J Med Genet. 2003;40:479–486. doi: 10.1136/jmg.40.7.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nam Y, Aster JC, Blacklow SC. Curr Opin Chem Biol. 2002;6:501–509. doi: 10.1016/s1367-5931(02)00346-0. [DOI] [PubMed] [Google Scholar]
- 20.Chang B, Smith RS, Hawes NL, Anderson MG, Zabaleta A, Savinova O, Roderick TH, Heckenlively JR, Davisson MT, John SW. Nat Genet. 1999;21:405–409. doi: 10.1038/7741. [DOI] [PubMed] [Google Scholar]
- 21.Ueda J, Wentz-Hunter K, Yue BY. Investig Ophthalmol Vis Sci. 2002;43:1068–1076. [PubMed] [Google Scholar]
- 22.Lutjen-Drecoll E, Shimizu T, Rohrbach M, Rohen JW. Exp Eye Res. 1986;42:457–465. doi: 10.1016/0014-4835(86)90005-9. [DOI] [PubMed] [Google Scholar]
- 23.Tomarev SI, Wistow G, Raymond V, Dubois S, Malyukova I. Investig Ophthalmol Vis Sci. 2003;44:2588–2596. doi: 10.1167/iovs.02-1099. [DOI] [PubMed] [Google Scholar]
- 24.Gygi SP, Rochon Y, Franza BR, Aebersold R. Mol Cell Biol. 1999;19:1720–1730. doi: 10.1128/mcb.19.3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robertson NG, Lu L, Heller S, Merchant SN, Eavey RD, McKenna M, Nadol JB, Jr, Miyamoto RT, Linthicum FH, Jr, Lubianca Neto JF, Hudspeth AJ, Seidman CE, Morton CC, Seidman JG. Nat Genet. 1998;20:299–303. doi: 10.1038/3118. [DOI] [PubMed] [Google Scholar]
- 26.Kamarinos M, McGill J, Lynch M, Dahl H. Hum Mutat. 2001;17:351. doi: 10.1002/humu.37. [DOI] [PubMed] [Google Scholar]
- 27.Fransen E, Verstreken M, Verhagen WI, Wuyts FL, Huygen PL, D’Haese P, Robertson NG, Morton CC, McGuirt WT, Smith RJ, Declau F, Van de Heyning PH, Van Camp G. Hum Mol Genet. 1999;8:1425–1429. doi: 10.1093/hmg/8.8.1425. [DOI] [PubMed] [Google Scholar]
- 28.de Kok YJ, Bom SJ, Brunt TM, Kemperman MH, van Beusekom E, van der Velde-Visser SD, Robertson NG, Morton CC, Huygen PL, Verhagen WI, Brunner HG, Cremers CW, Cremers FP. Hum Mol Genet. 1999;8:361–366. doi: 10.1093/hmg/8.2.361. [DOI] [PubMed] [Google Scholar]
- 29.Ikezono T, Omori A, Ichinose S, Pawankar R, Watanabe A, Yagi T. Biochim Biophys Acta. 2001;1535:258–265. doi: 10.1016/s0925-4439(00)00101-0. [DOI] [PubMed] [Google Scholar]
- 30.Robertson NG, Skvorak AB, Yin Y, Weremowicz S, Johnson KR, Kovatch KA, Battey JF, Bieber FR, Morton CC. Genomics. 1997;46:345–354. doi: 10.1006/geno.1997.5067. [DOI] [PubMed] [Google Scholar]
- 31.Ikezono T, Shindo S, Li L, Omori A, Ichinose S, Watanabe A, Kobayashi T, Pawankar R, Yagi T. Biochem Biophys Res Commun. 2004;314:440–446. doi: 10.1016/j.bbrc.2003.12.106. [DOI] [PubMed] [Google Scholar]
- 32.Merchant SN, Linthicum FH, Nadol JB., Jr Adv Otorhinolaryngol. 2000;56:212–217. doi: 10.1159/000059105. [DOI] [PubMed] [Google Scholar]
- 33.Shankaran H, Alexandridis P, Neelamegham S. Blood. 2003;101:2637–2645. doi: 10.1182/blood-2002-05-1550. [DOI] [PubMed] [Google Scholar]
- 34.Grabski R, Szul T, Sasaki T, Timpl R, Mayne R, Hicks B, Sztul E. Hum Genet. 2003;113:406–416. doi: 10.1007/s00439-003-0992-7. [DOI] [PubMed] [Google Scholar]
- 35.Pareti FI, Niiya K, McPherson JM, Ruggeri ZM. J Biol Chem. 1987;262:13835–13841. [PubMed] [Google Scholar]
- 36.Marchant JK, Hahn RA, Linsenmayer TF, Birk DE. J Cell Biol. 1996;135:1415–1426. doi: 10.1083/jcb.135.5.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.