

Abstract
A potential anticancer drug delivery polymeric micelle system with an in vitro degradation half-life of about 48 hours that releases its drug upon application of ultrasound was synthesized. This vehicle was composed of an amphiphilic copolymer poly(ethylene oxide)-b-poly(N-isopropylacrylamide-co-2-hydroxyethyl methacrylate-lactaten). The degree of polymerization of lactate side group, n, was 0, 3 or 5. The molar ratio of NIPAAm to HEMA-lactaten to PEO in polymerization was optimized to produce an in vitro polymeric micelle half-life of about 48 hour at 40°C. 1,6-diphenyl-1,3,5-hexatriene (DPH) was used as a fluorescent probe to study the hydrophobicity of the cores of the polymeric micelles. The results showed that the cores of the polymeric micelles were hydrophobic enough to sequester DPH and the anti-cancer drug Doxorubicin (Dox). Dox was encapsulated into the polymeric micelles having molar feed ratio of NIPAAm to HEMA-lactate3 to PEO equal to 20 : 5 : 1; this drug was released upon the application of low-frequency ultrasound. The Dox release was about 2 % at room temperature and 4 % at body temperature, and the drug returned to the polymeric micelles when insonation ceased.
Keywords: poly(ethylene oxide)-b-poly(N-isopropylacrylamide-co-2-hydroxyethyl methacrylate-lactaten), micelle, drug delivery, drug release, doxorubicin, ultrasound
Introduction
To use polymeric micelles for controlled drug delivery, they must be stable (must not dissociate) in the blood for a sufficient amount of time to carry the drug to the desired target site. However, polymeric micelles normally dissolve when their concentration drops below the critical micelle concentration (CMC). The CMC values are fairly high for many amphiphilic copolymers, such as the copolymer Pluronic® P105 (Mw 6,500), which has CMC values in water around 1 wt% at 25°C and 0.1 wt% at 37°C [1]. Thus this and most polymeric micelles would dissolve quickly when injected into the blood because of dilution below the CMC. Unfortunately the dissolution of the drug carrier would quickly release the drug into the circulatory system, thus precluding controlled drug delivery.
Another requirement for an ideal drug delivery vehicle is that it is biodegradable so that it will not build up in the body, but will eventually be cleared.
A potential candidate from which to construct a temporarily stable drug carrier is the block copolymer poly(ethylene oxide)-b-poly(N-isopropylacrylamide-co-2-hydroxyethyl methacrylate-lactaten) (abbreviated as PEO-b-poly(NIPAAm-co-HEMA-lactaten)), which at fairly low concentrations can self–assemble to a core–shell structure in aqueous solutions, and thus form polymeric micelles. Such polymeric micelles would be expected to sequester hydrophobic drugs like Doxorubicin (Dox) into their hydrophobic core, and their PEO outer corona would reduce clearance of the polymeric micelles by the mononuclear phagocytic system (MPS) in vivo [2]. The formation and stability of the PEO-b-poly(NIPAAm-co-HEMA-lactaten) micelles also would depend on their concentration and surrounding temperature, with the polymeric micelles forming at temperatures above the critical micelle temperature (CMT). Such thermal behavior arises because the PEO block of the copolymer remains hydrophilic over a wide temperature range, but the NIPAAm-co-HEMA-lactaten block becomes sufficiently hydrophobic only above the CMT. Fortunately, even at dilute concentration, the CMT of this copolymer is lower than 37°C, and thus it forms polymeric micelles at body temperature [3,4]. We postulate that after the polymeric micelles are injected into body, the lactate ester side groups of the copolymer will be hydrolyzed gradually, and the PEO-b-poly(NIPAAm-co-HEMA-lactaten) copolymer will transform into PEO-b-poly(NIPAAm-co-HEMA) copolymer, which has a CMT greater than 37 °C; and thus the polymeric micelles will spontaneously dissolve as the hydrolysis progresses. If the individual copolymer chains have a molecular weight of less than 20, 000 Daltons, they will be cleared by the renal system.
In addition to such an amphiphilic copolymer, ultrasound (US) is another interesting tool for drug delivery [5]. US-mediated drug delivery has the advantage that US is non-invasive, it is easily focused within the body, and it enhances drug action [6–8].
This research reveals how the in vitro degradation half-life of this polymeric micelle system is controlled by varying the length of the lactate side group (indicated by the value of n) and the molar ratio of NIPAAm to HEMA-lactaten to PEO in the copolymer, which controls the length of time before the CMT shifts above body temperature. The release of anticancer drug Doxorubicin (Dox) from these polymeric micelles using low-frequency ultrasound was investigated.
Materials and Methods
Materials
2-hydroxyethyl methacrylate, 3, 6-dimethyl-1, 4-dioxane-2, 5-dione, tin (II) 2-ethylhexanoate, methoxypoly(ethylene glycol) (Mw 2,000), N-isopropylacrylamide, 4,4′-azobis(4-cyanopentanoic acid), 1,4-dioxane and thionyl chloride were obtained from Sigma-Aldrich and used without further purification. Doxorubicin hydrochloride was obtained from Pharmacia & Upjohn Company, Kalamazoo MI in dosage form. 1,6-diphenyl-1,3,5-hexatriene was obtained from Molecular Probes (Eugene, Oregon).
Synthesis
Oligolactate esters of 2-hydroxyethyl methacrylate (HEMA-lactaten), (n=number of lactate units in oligolactate) were synthesized by ring-opening oligomerization of lactide using HEMA as the initiator and stannous octoate as a catalyst as described by Cadee et al [9]. In brief, a mixture of lactide and HEMA was stirred at 110°C under nitrogen purging until the lactide was molten. Subsequently, catalytic amounts of stannous octoate dissolved in toluene were added dropwise to the mixture. The mixture reacted for 1 hour. The target stoichiometry was 3 or 5 lactate units per HEMA.
Incorporation of PEO into the block copolymer requires the synthesis of a PEO macroinitiator that was produced in two steps. In the first step, 4,4′-azobis(4-cyanopentanoic acid) (ABCPA) was treated with SOCl2 at 100 °C for 20 minutes and converted to the corresponding acid chloride: 4,4′azobis(4-cyanopentanoyl chloride) (ABCPC). In the second step, the PEO macroinitiator was prepared by a condensation reaction of ABCPC and PEO in dry dichloromethane in the presence of an excess amount of triethylamine for 24 hr as described by Virtanen et al [10].
The PEO-b-poly(NIPAAm-co-HEMA-lactaten) was obtained by radical polymerization of NIPAAm with HEMA-lactaten obtained above using the PEO macroinitiator. The copolymerization was conducted in 1, 4-dioxane at 80 °C for 24 hr in a nitrogen atmosphere. The solution was then cooled to room temperature and concentrated under reduced pressure. The formed solid was dissolved in distilled water to form 100 mg/ml solution and centrifuged 0.5 hour (10,000 rpm, Eppendorf 5415C) at 40°C to separate the unreacted PEO. Colorimetric analysis was carried out with Ce(IV) as a colorimetric reagent to verify that no unreacted PEO remained [11]. The precipitated copolymer products were dried under reduced pressure.
1H NMR spectra of the copolymers were recorded using a Varian Unity 300 MHz instrument in DMSO-d6. The molar composition of the block copolymer was determined from the relative proton integral intensities at 3.4 ppm (HEMA-lactate terminal hydroxyl), 3.5 ppm (-CH2CH2O of PEO block), 3.8 ppm (-NHCH(CH3)2 of pNIPAAm block), 4.0–4.5 ppm (-OCH2CH2O- and COCH(OH)CH3 of p(HEMA-lactaten) block), and 5.0–5.2 ppm (-COCH(CH3)O- of p(HEMA-lactaten) block). The data showed that the molar ratios of the recovered polymer calculated from NMR were similar to the feed molar ratios, but with about 25% less HEMA-lactate than predicted from the feed ratios. The number average molecular weight was determined from the NMR data, based on the assumption that the PEO block has a molecular weight of 2,000 as stated by the supplier.
Analysis
The sizes of the copolymer micelles at different aqueous solution concentrations were measured using dynamic light scattering at 90º (Brookhaven 90Plus submicron particle size analyzer). The CONTIN algorithm that gives the intensity distribution was used to analyze the data. For each sample, 3 measurements were taken and each measurement took 2 minutes. The average count rate of the background was 15 kcps and that of each measurement was between 200~500 kcps. The temperature was controlled at 40°C.
The CMTs of the copolymer solutions, before and after hydrolysis, were quantified by measuring their optical density at 600 nm at various temperatures with a Beckman Coulter DU 640 spectrophotometer. To hydrolyze the copolymer, samples were dissolved in phosphate buffered saline (PBS, pH=7), in a concentration of 1 mg/ml and incubated at 40 °C. The CMTs of the copolymer solutions were measured at 0, 2 and 7 days of hydrolysis.
The temporal stabilities of the various polymeric micelle systems were studied using a luminescence spectrometer (Perkin-Elmer LS50B) with 1, 6-diphenylhexatriene (DPH) as a fluorescent probe. A stock solution of DPH in tetrahydrofuran (THF) was added to an empty glass vial, and the THF was evaporated. The copolymer solution with a specific micelle concentration was then added to the vial. The vial was kept in a water bath at 40°C for 30 min to thoroughly dissolve the fluorescent probe. In all measurements, the concentration of DPH was 0.1 μg/ml. Fluorescence intensity was measured with λexcitation = 360 nm and λemission= 430 nm.
The anticancer drug Doxorubicin (Dox) was introduced into PEO-b-poly(NIPAAm-co-HEMA-lactaten) micelles by simple mixing as described previously [12]. The Dox concentration in the solution was 6.67 μg/ml. The release of the drug upon application of ultrasound was measured in a 70 kHz ultrasonic bath using the apparatus shown in Figure 1. The 488 nm line of an argon ion laser (Ion Laser Technology, Model 5500 A) was directed through a dielectric-interface beam splitter. The intensity of the split portion of the beam was measured by a photo detector (Newport Model 818-SL with 835 display) and was used to monitor the laser power throughout the measurements. The other portion of the beam was guided into one branch of a bifurcated fiber optic bundle (part DF13036M, Edmund Optics, Barrington, NJ) that guided the light into an acoustically transparent plastic (cellulose butyrate) tube of 2.54 cm diameter filled with the Dox solution. The laser light exited the fiber optic bundle in a cone of light. Dox within the cone of light absorbed at 488 nm and isotropically emitted fluorescence centered at 580 nm. In the same fiber optic bundle were fibers that collected and directed the fluorescence to a detector. The geometry of the fiber optic is such that 99% of the collected fluorescence originated from within 3 mm of fiber optic tip. The fluorescence signal was directed through the second branch of the fiber optic bundle through a multinode dielectric band pass filter (Omega Optical Model 535DF35) to a silicon detector (EGSG Model 450-1). The filter was used to cut off any emissions with a wavelength below 517 nm, including any Rayleigh-scattered laser light. The photo detector signal was recorded with an oscilloscope and then stored on a computer for further processing.
Figure 1.
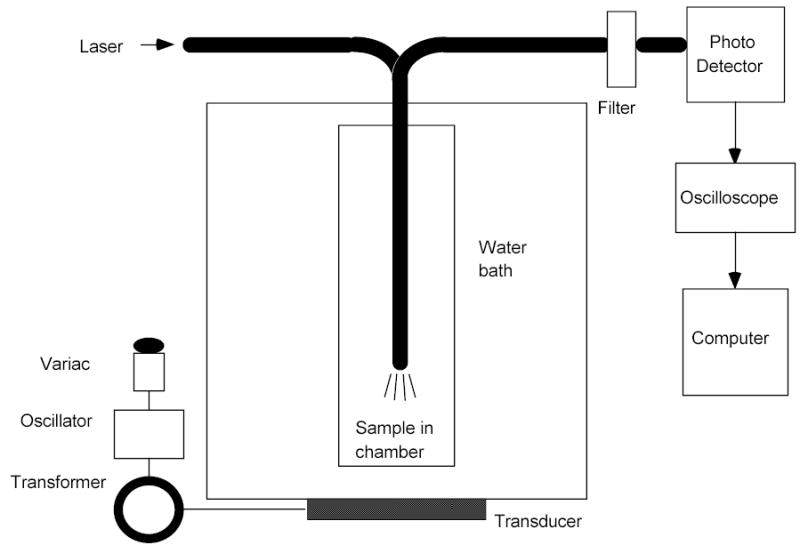
Schematic of the experimental apparatus used to measure the release of Dox from the drug carrier.
As ultrasound from ultrasonic bath released Dox from the polymeric micelles, the fluorescence of the Dox was quenched. The collection fiber optic recorded this change in fluorescence intensity, and the amount of drug released was computed from the change in fluorescence [13].
The acoustically transparent tube and fiber optic were positioned in the ultrasonic bath such that the end of the fiber optic bundle was in an ultrasonically intense region of the bath. The ultrasonic intensity was controlled by adjusting the 60 Hz voltage that powered the ultrasonic bath using a variable AC transformer (Variac). The ultrasonic intensity was measured by placing a hydrophone (8103, Bruel and Kjaer, Naerum, DK) at the same location as the end of the fiber optic.
To execute these measurements, solutions of either PBS, Dox in PBS or Dox-copolymer in PBS were placed in the plastic tube, which was placed in the ultrasonic bath. The ultrasound was applied at various intensities, and the changes in fluorescence were measured and converted to a % drug released, as described previously [12]. In brief, the % drug released equals the change in fluorescence intensity obtained during the ultrasound pulse compared to that observed when the drug is in PBS without copolymer.
Results
Polymer and Particle Size
The number average molecular weights (Mn) were determined from the NMR analysis. The Mn values for the PEO-b-poly(NIPAAm-co-HEMA-lactate5) copolymers are given in Table 1 and are between 5,300 and 7,400 Daltons. Thus these polymers are small enough to be cleared by the renal system. Table 1 also shows that the observed molar ratios have less of the HEMA-lactate segment compared to the feed ratio. A similar observation was made in the copolymerization of HEMA and NIPAAm (without PEO) [14]. For simplicity we will use the feed ratio to identify the polymers in the following sections.
Table 1.
Molecular weights and molar ratios of NIPAAM : HEMA-lactate5 : PEO.
| Molar feed ratio | Molar ratio in polymer1 | Molecular weight1 |
|---|---|---|
| 17.5 : 7.5 : 1 | 20.7 : 6.1 : 1 | 7,400 |
| 20 : 5 : 1 | 16.6 : 4.8 : 1 | 6,300 |
| 22.5 : 2.5 : 1 | 22.8 : 1.2 : 1 | 5,300 |
Based on proton NMR spectra
Diameters of the polymeric micelles were measured by DLS at three concentrations : 1 mg/ml, 0.1 mg/ml and 0.01 mg/ml. Figure 2 shows the changes of diameters as the molar ratios of NIPAAm to HEMA-lactaten to PEO was varied. In Figure 2A and 2B, n equals 3 and 5 respectively. The molar ratio of NIPAAm to PEO increases along the horizontal axis in each figure. The data in these figures shows that the diameters of the polymeric micelles increase when the molar ratios of NIPAAm to PEO increase. The polymeric micelles with the NIPAAm : PEO ratio of 45 : 1 had diameters over 150 nm at all three dilutions (Figure 2A), indicating that large micelles had formed. These larger sizes may be due to fairly long poly(NIPAAm) hydrophobic blocks formed during polymerization. The diameters of the polymeric micelles with all other NIPAAm to PEO ratios were less than 130 nm. Compared with other polymeric micelles at 0.01 mg/ml dilutions, there are larger error bars of the polymeric micelles with NIPAAm : HEMA-lactate3 : PEO ratio of 17.5 : 7.5 : 1 or 20.0 : 5.0 : 1, indicating that they were not very stable at this low concentration, and thus the DLS data was noisy. Comparison of figures 2A and 2b shows that with the same NIPAAm to HEMA-lactaten to PEO molar ratio, the polymeric micelles with the longer lactate block in the oligomer (n=5) were larger than the ones with the shorter lactate block (n=3). This is because the larger hydrophobic lactate blocks are incorporated into hydrophobic cores, and thus increased the overall sizes of the polymeric micelles.
Figure 2.
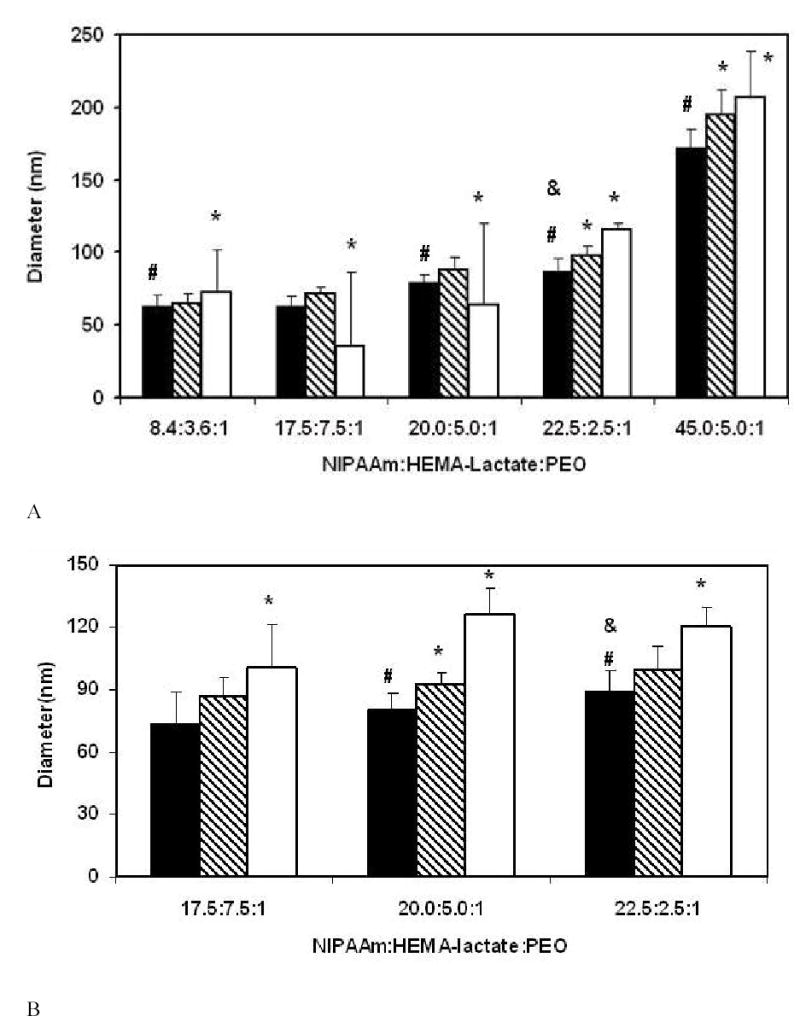
Diameters of PEO-b-poly(NIPAAm-co-HEMA-lactaten) micelles at 40°C. (A) n=3, (B) n=5. The black bars represent data at 1 mg/ml solution, the hatched bars represent data at 0.1 mg/ml dilution, and the white bars represent data at 0.01 mg/ml dilution. Error bars represent the standard deviations (n=3). The * indicates that data of 0.1mg/ml and 0.01mg/ml are statistically different (p<0.05) from that of 1 mg/ml within each NIPAAm : HEMA-lactaten : PEO ratio. The # indicates that the data are statistically different (p<0.05) from data of polymers having a NIPAAm : HEMA-lactaten : PEO ratio equal to 17.5 : 7.5 : 1 in 1mg/ml solution. The & indicates that the data are statistically different (p<0.05) from data of polymers having a NIPAAm : HEMA-lactaten : PEO ratio equal to 20.0 : 5.0 : 1 in 1 mg/ml solution. Statistical comparisons were made using the double-sided t-distribution test (n=3).
Critical Micelle Temperature characterization
Figure 3(A) shows all six copolymers had CMTs far below 37 °C immediately upon dissolving in PBS, indicating that they all would form polymeric micelles at body temperature at this concentration. When the ratio of NIPAAm : HEMA-lactaten : PEO was the same, the copolymers with shorter lactate blocks (open symbols at day 0) had higher CMTs than those with longer lactate blocks (closed symbols at day 0). This is because the lactate block is hydrophobic and contributes toward lowering the CMT. Within each lactate block length set (n=3 or n=5), the CMTs increased as the molar ratio of NIPAAm to HEMA-lactaten increased. We presume that this occurred because when the molar ratio of NIPAAm to HEMA-lactaten increased, there was a smaller proportion of hydrophobic lactate blocks in the copolymer.
Figure 3.
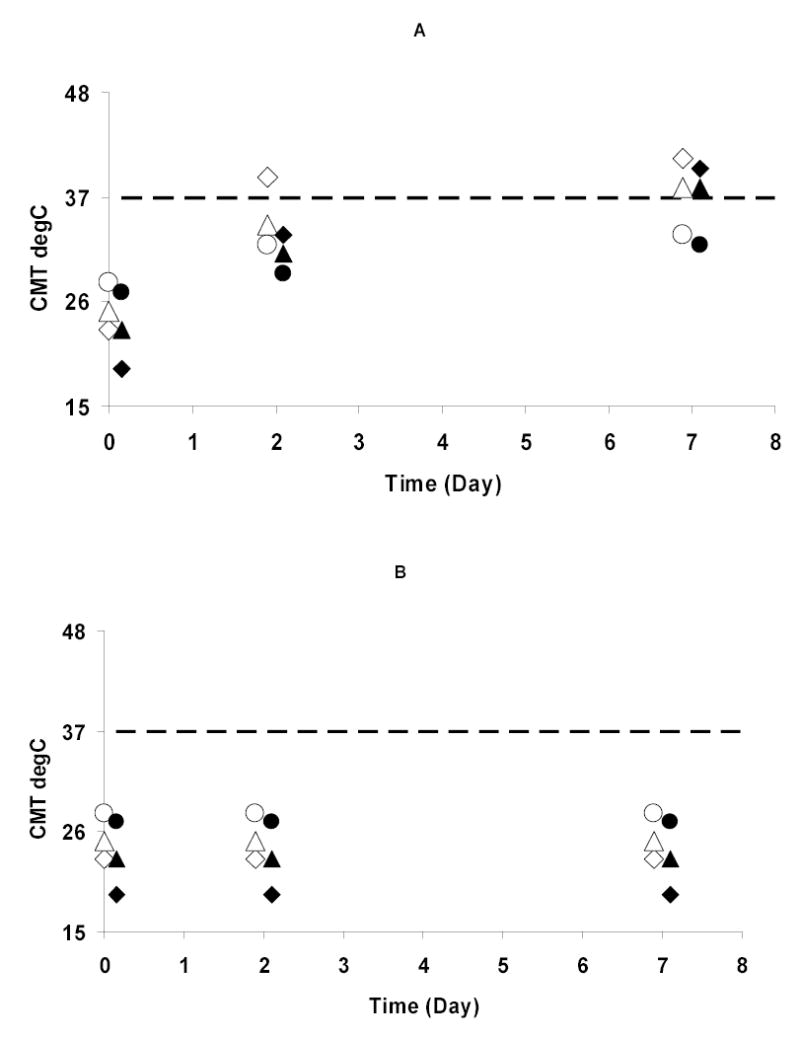
CMTs of copolymers at 1 mg/ml as a function of time: (A) Hydrolyzed copolymers; and (B) Unhydrolyzed copolymers stored as solids. Closed symbols indicate n=5; open symbols indicate n=3. Diamonds indicate the NIPAAm : HEMA-lactaten : PEO ratio is 17.5 : 7.5 : 1. Triangles indicate the NIPAAm : HEMA-lactaten : PEO ratio is 20.0 : 5.0 : 1; Circles indicate the NIPAAm : HEMA-lactaten : PEO ratio is 22.5 : 2.5 : 1. The data are slightly offset in time for clarity.
After hydrolysis for 2 days, the CMTs of the copolymers with n=5 increased but were still below 37 °C (closed symbols at day 2). The copolymers with n=3 and having molar ratios of NIPAAm : HEMA-lactate3 : PEO of 20.0 : 5.0 : 1 or 22.5 : 2.5 : 1 had a similar change in their CMTs (open triangle and open circle at day 2). When NIPAAm : HEMA-lactate3 : PEO was 17.5 : 7.5 : 1, the copolymers had CMTs above 37 °C after hydrolysis for 2 days, and thus would no longer form polymeric micelles at body temperature (open diamond at day 2). This indicates that the stabilities of the polymeric micelles are controlled by the hydrolysis of the lactate groups. In hydrolyzed copolymers, those with the greater NIPAAm : HEMA-lactaten ratios have the lower CMT, while in unhydrolyzed polymers, those with the lesser NIPAAm : HEMA-lactaten ratios have the lower CMT. This seems to indicate that when lactate is present, it controls the CMT, but when it has been hydrolyzed away, the ratio of NIPAAm to PEO controls the CMT.
After hydrolysis for 7 days, the copolymers with NIPAAm : HEMA-lactaten : PEO ratio of 22.5 : 2.5 : 1 still had their CMTs below 37 °C, whether n=3 or n=5 (open and closed circles at day 7). The CMTs of copolymers with NIPAAm : HEMA-lactaten : PEO ratios of 20.0 : 5.0 : 1 shifted above 37 °C (open and closed triangles at day 7). This indicates that polymeric micelles formed from the latter copolymers would dissolve within 7 days at body temperature. The copolymer with a NIPAAm : HEMA-lactate5 : PEO ratio of 17.5.0 : 7.5 : 1 displayed a similar change in its CMT (closed diamond at day 7).
Figure 3(B) shows the CMTs of all six copolymers that were unhydrolyzed (stored as solids) were same at 0, 2 and 7 days. This proves that the changes of the CMTs of the copolymers in Figure 3(A) were caused by hydrolysis upon exposure to water.
Figure 4 shows the CMTs of all three sets of copolymers (n=0, n=3 or n=5) hydrolyzed for 7 days at different dilutions. The CMTs of the copolymers with n=3 or n=5 reached the values of the copolymers with n=0 at corresponding concentrations after hydrolysis for 7 days. These data indicate that the lactate blocks of the copolymers with n=3 or n=5 were completely hydrolyzed in seven days. At each concentration and within each lactate block length set, the CMTs decreased with the increase of NIPAAm to HEMA-lactaten ratio. We postulate that when there are no lactate blocks remaining attached to the hydrolyzed copolymers, the CMTs of those copolymers are lower as the amount of hydrophobic NIPAAm blocks is greater.
Figure 4.
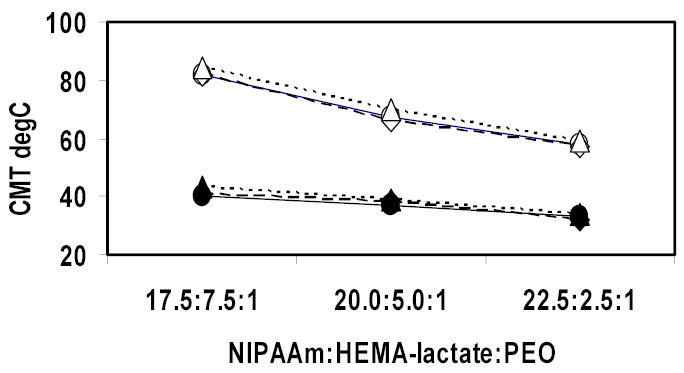
CMTs of copolymers after hydrolysis for 7 days as a function of the ratio of NIPAAm : HEMA-lactaten : PEO. Closed symbols represent 1 mg/ml copolymer solutions and open symbols represent 0.1 mg/ml copolymer solutions. Circles represent copolymer with n=5. Diamonds represent copolymers with n=3. Triangles represent copolymers without any lactate block.
Fluorescence Measurements
DPH has a fluorescent emission at 430 nm when it is excited at 360 nm. The emission intensity of DPH is strongly dependent on the hydrophobicity of the local environment. It has almost no fluorescence in an aqueous solution while it is highly fluorescent in a hydrophobic environment. DPH also has very low solubility in aqueous solutions. When DPH is mixed with polymeric micelle solutions, it is trapped in the hydrophobic cores of the polymeric micelles, and the emission intensities of DPH indicate the relative volume of hydrophobic material in a sample. Once the polymeric micelle loses its stability and dissolves, DPH is released to the aqueous environment and the emission intensity goes to near zero. Thus we can monitor the formation of polymeric micelles and how fast they are dissolving.
The DPH emission intensities of PEO-b-poly(NIPAAm-co-HEMA-lactaten) at 40°C right after dissolving in double distilled water are shown in Figure 5. The data show that the DPH emission intensities of all six kinds of polymeric micelles in all three dilutions were higher than that in pure water. This proved the formation of the polymeric micelles and is consistent with the DLS data.
Figure 5.
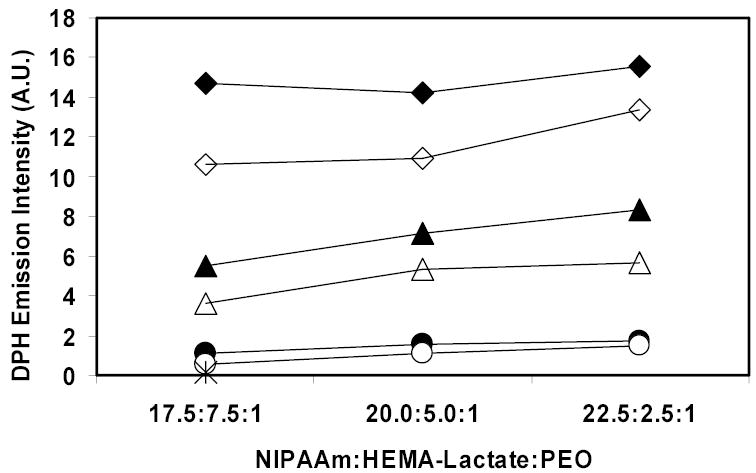
DPH emission intensities of PEO-b-poly(NIPAAm-co-HEMA-lactaten) micelles at 40°C immediately after dissolving in double distilled water. Closed symbols represent data with n=5, and open symbols represent data with n=3. Diamonds represent data at 1 mg/ml solution, triangles represent data at 0.1 mg/ml dilution, and circles represent data at 0.01 mg/ml dilution. The star indicates the emission intensity of DPH in double distilled water.
Pruitt et al. reported that the DPH emission intensities of another stabilized polymeric micelle, Plurogel®, in 1 mg/ml and 0.1 mg/ml solutions with the same DPH concentration (0.1μg/mL) were 20 and 3 respectively [15]. Compared with Pruitt’s results, the PEO-b-poly(NIPAAm-co-HEMA-lactaten) micelles provide similar hydrophobic core environments as Plurogel®. As shown in Figure 5, at each concentration the fluorescent intensity increased as the molar ratio of NIPAAm to PEO increased. This is because as the molar ratio of NIPAAm to PEO increases, the hydrophobic part of the copolymer increases and the cores of the polymeric micelles became more compact and perhaps larger. We postulate that less water was able to penetrate into the core, and thus there was less quenching of the DPH fluorescence. The copolymers with longer lactate blocks showed higher DPH emission intensities than those with shorter lactate blocks, which was expected because the lactate block is hydrophobic, thus allowing more DPH fluorescence inside the polymeric micelles.
Doxorubicin encapsulation and release upon exposure to ultrasound
The anticancer drug Dox has a fluorescent emission at 590 nm when it is excited at 488 nm. It has less fluorescence in an aqueous solution than in a hydrophobic environment. When Dox is mixed with polymeric micelle solutions, it is encapsulated into the hydrophobic cores of the polymeric micelles and can be released from polymeric micelles upon exposure to low-frequency ultrasound.
Polymeric micelles formed from the copolymer with NIPAAm : HEMA-lactate3 : PEO ratio of 20.0 : 5.0 : 1 were used at a concentration of 10 wt% in this analysis. The diameters of these polymeric micelles were smaller than 100 nm; they were stable up to 48 hours and degraded within 7 days; a hydrophobic environment was present at a very low concentration of drug carrier. These data suggest that this kind of polymeric micelle should be capable of sequestering hydrophobic drugs like Dox and might be used as an anti-caner drug carrier.
Low-frequency (70 kHz) continuous ultrasound was used to release Dox from freshly suspended PEO-b-poly(NIPAAm-co-HEMA-lactate3) micelles in PBS as described elsewhere [16]. Release occurred within 1 second of initiation of the insonation, an the amount of Dox released was 2% at room temperature and was 4% at 37 ºC, which values are lower than the Dox released from Plurogel®. Upon cessation of ultrasound, the Dox promptly (within 1 second) returned to the micelles. Plurogel® exhibits about 9.1% release of the Dox at 37 ºC under similar ultrasonic exposure conditions [17].
Discussion
PEO-b-poly(NIPAAm-co-HEMA-lactaten) micelles have been synthesized as potential anticancer drug delivery carriers. Synthesis was achieved by copolymerization of NIPAAm with HEMA-lactaten initiated by a PEO-ABCPC macroinitiator. The molar ratio of NIPAAm to HEMA-lactaten to PEO in the polymerizations was varied over a wide range.
The diameters of PEO-b-poly(NIPAAm-co-HEMA-lactaten) micelle as measured by DLS increased with the molar ratio of NIPAAm to PEO (Figure 2). The diameters of the polymeric micelles with NIPAAm to PEO molar ratios less than 45.0 : 1 were less than 130 nm. Such a size is slightly larger than Plurogel® micelles [15] but still small enough to be used in vivo. It is purported that polymeric micelles with diameters less than 100 nm would be suitable for IV administration of drug carriers [18]. In our experiment, polymeric micelles with NIPAAm to PEO ratio less than 22.5 : 1 at concentrations greater than or equal to 0.1 mg/ml fall in this category.
Within each lactate length set, the diameters of the polymeric micelles increased when the molar ratio of NIPAAm to PEO increased. We presume that the lower amounts of PEO provide less PEO to cover the surface of the polymeric micelle, so the aggregates are larger in order to accommodate a smaller surface to volume ratio.
PNIPAAm homopolymer has a CMT of about 32°C. As hydrophilic monomers are copolymerized with NIPAAm, the CMT increases, while copolymerization with hydrophobic monomers decreases the CMT. In this research, we were adding both a hydrophilic PEO segment and a hydrophobic HEMA- lactaten comonomer. Figure 3 shows the CMTs of the copolymers immediately upon dissolution in PBS decreased as the molar ratio of HEMA-lactaten to PEO increased. The CMTs decreased to about 19°C when the molar ratio of HEMA-lactate5 to PEO was 7.5 : 1. Thus polymer with greater amounts of hydrophobic HEMA-lactaten block displayed a greater decrease in the CMTs.
As the lactate ester side groups of the copolymers hydrolyzed gradually, the copolymer became more hydrophilic and thus the CMTs of the copolymers increased [19]. Comparing the CMTs of the copolymers after hydrolysis for 2 days to those of the copolymers immediately after dissolution in water, the copolymers with greater HEMA-lactaten to PEO molar ratio produced a larger increase in CMTs. The CMTs of the copolymers with molar ratios of HEMA-lactaten : PEO of 5.0 : 1 or 2.5 : 1 increased but were still below 37 °C at 2 days. Such data suggest that polymeric micelles formed from these copolymers would be stable at body temperature up to 48 hours; this is a longer time than the stability of Plurogel®, which has an in vitro degradation half-life in dilute aqueous solution of only about 18 hours [15].
Figure 4 shows that after hydrolysis for 7 days, the CMTs of all three sets of copolymers (n=0, n=3 or n=5) with same NIPAAm : HEMA-lactaten : PEO ratios were similar at the same solution concentration, which indicates that the lactate blocks were completely hydrolyzed in seven days. The CMTs of these hydrolyzed copolymers varied between 55~85 °C, depending on the NIPAAm : HEMA-lactaten : PEO ratios. Neradovic et al. reported that poly(NIPAAm-co-HEMA) had a lower critical solution temperature (LCST) of about 30°C in 0.1 mg/ml solution for a series of copolymers in which the molar ratio of HEMA to NIPAAm varied from 0 to 1 : 1 [19]. The difference between the CMTs of Neradovic and those of our copolymers is attributed to the large hydrophilic PEO block. Though PEO is a LCST polymer too, it is reported that a linear PEO (Mw = 2,270, which was similar to the PEO used in this research) has an LCST of over 160 °C at 1 mg/ml solution [20]. Experiment carried out in our lab also showed that in a 0.1 mg/ml solution, the LCST of pure PEO (Mw = 2,000) was not detectable up to 85 °C. These data suggest that PEO is very hydrophilic under the condition of this research and apparently increase the CMTs of the PEO-b-poly(NIPAAm-co-HEMA) copolymers.
Low-frequency ultrasound caused the anticancer drug Dox to be released from PEO-b-poly(NIPAAm-co-HEMA-lactate3) micelles with NIPAAm : HEMA-lactate3 : PEO equal to 20.0 : 5.0 : 1. But the amount released was less than previously observed from Plurogel®. Currently, we can only speculate on the cause of this difference. Because the glass transition temperature (Tg) of PPO is −185°C, the lightly crosslinked PPO core of Plurogel® may be a rubbery gel at the experiment temperature (room temperature or 37°C) [21]. The Tg of polylactide is around 57~60°C, and the poly(NIPAAm-HEMA-lactaten) core may be more of a glassy solid at the experiment temperature [22]. Shearing forces from cavitating bubbles may open micelles with gel cores more easily than those with solid cores. More work remains to be done to test this hypothesis and to investigate other unique properties of this novel potential micellar drug carrier
Conclusion
Particle size analysis, CMT characterization and fluorescence measurement showed that with a suitable molar ratio of NIPAAm to HEMA-lactaten to PEO in polymerization, temporally stable PEO-b-poly(NIPAAm-co-HEMA-lactaten) micelles could be obtained. Some formulations were stable beyond 48 hours, and all lactate apparently was hydrolyzed within 7 days in vitro. Control of the in vitro degradation half-life of these polymeric micelles will make PEO-b-poly(NIPAAm-co-HEMA-lactaten) a potential drug carrier not only for ultrasonic activated delivery, but also in other systems in which the in vitro degradation half-life of a biodegradable carrier must be carefully controlled.
Acknowledgments
Financial support provided by Cancer Research Center of Brigham Young University, and an NIH grant (CA 98138). We thank Dr. G. A. Husseini and Mario Diaz for assistance with the ultrasonic drug-releasing experiments.
References
- 1.Rapoport N, Caldwell K. Colloids and Surfaces B: Biointerfaces. 1994;3:217–228. [Google Scholar]
- 2.De Jaeghere F, Allemann E, Leroux JC, Stevels W, Feijen J, Doelker E, Gurny R. Pharmaceutical Research. 1999;16:859–866. doi: 10.1023/a:1018826103261. [DOI] [PubMed] [Google Scholar]
- 3.Neradovic D, Hinriches WLJ, Kettenes-van den Bosch JJ, Hennink WE. Macromol Rapid Commun. 1999;20:577–581. [Google Scholar]
- 4.Neradovic D, van Nostrum CF, Hennink WE. Macromolecules. 2001;34:7589–7591. [Google Scholar]
- 5.Koch S, Pohl P, Cobet U, Rainov NG. Ultrasound Med Biol. 2000;26:897–903. doi: 10.1016/s0301-5629(00)00200-3. [DOI] [PubMed] [Google Scholar]
- 6.Pitt WG. Am J Drug Deliv. 2003;1:27–42. [Google Scholar]
- 7.Pitt WG, Husseini GA, Staples BJ. Expert Opin Drug Delivery. 2004;1:37–56. doi: 10.1517/17425247.1.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mitragotri S. Nature Reviews Drug Discovery. 2005;4:255–260. doi: 10.1038/nrd1662. [DOI] [PubMed] [Google Scholar]
- 9.Cadee JA, De Kerf M, De Groot CJ, Den Otter W, Hennink WE. Polymer. 1999;40:6877–6881. [Google Scholar]
- 10.Virtanen J, Holappa S, Lemmetyinen H, Tenhu H. Macromolecules. 2002;35:4763–4769. [Google Scholar]
- 11.Lin HH, Cheng YL. Macromolecules. 2001;34:3710–3715. [Google Scholar]
- 12.Husseini GA, Rapoport NY, Christensen DA, Pruitt JD, Pitt WG. Coll Surf B: Biointerfaces. 2002;24:253–264. [Google Scholar]
- 13.Husseini GA, Myrup GD, Pitt WG, Christensen DA, Rapoport NY. J Contr Release. 2000;69:43–52. doi: 10.1016/s0168-3659(00)00278-9. [DOI] [PubMed] [Google Scholar]
- 14.Lee BH, Vernon B. Macromolecular Bioscience. 2005;5:629–635. doi: 10.1002/mabi.200500029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pruitt JD, Husseini G, Rapoport N, Pitt WG. Macromolecules. 2000;33:9306–9309. [Google Scholar]
- 16.G.A. Husseini; M.A. Diaz; Y. Zeng; D.A. Christensen; W.G. Pitt. submitted to Ultrasound Med. Biol., (2005).
- 17.Husseini GA, Christensen DA, Rapoport NY, Pitt JWG. Controlled Rel. 2002;83:302–304. doi: 10.1016/s0168-3659(02)00203-1. [DOI] [PubMed] [Google Scholar]
- 18.Ha TS, Kim D. Journal of Macromolecular Science-Pure and Applied Chemistry. 1999;A36:1031–1044. [Google Scholar]
- 19.Neradovic D, Hinrichs WLJ, Kettenes-van den Bosch JJ, Hennink WE. Macromolecular Rapid Communications. 1999;20:577–581. [Google Scholar]
- 20.Kjellander R, Florin E. J Chem Soc Faraday trans 1. 1981;77:2053–2077. [Google Scholar]
- 21.Volegova IA, Konyukhova EV, Godovsky YK. Journal of Thermal Analysis and Calorimetry. 2000;59:123–130. [Google Scholar]
- 22.Perepelkin KE. Fibre Chemistry. 2002;34:85–100. [Google Scholar]