

Abstract
Purpose
Eyes with age-related macular degeneration (AMD) demonstrate accumulation of specific deposits and extracellular matrix (ECM) molecules under the retinal pigment epithelium (RPE). Metalloproteinases (MMP) are crucial regulators of basement membrane and ECM turnover. Accordingly, loss of RPE MMP activity likely leads to excessive accumulation of collagen and other ECM, a potential mechanism for deposit formation. We have previously shown that MMP-2 activity, but not pro-MMP-2 protein, decreases following RPE oxidative injury, indicating that oxidant injury disrupts the enzymatic cleavage of pro-MMP-2. Activation of MMP-2 requires the formation of a trimolecular complex of pro-MMP-2, MMP-14 and Tissue Inhibitor of Metalloproteinases (TIMP) -2. Therefore we investigated the impact of oxidant injury on the interaction between these three molecules.
Methods
Human GFP-RPE cells were oxidant injured by transient exposure to H2O2 and myeloperoxidase and time course of recovery performed. Supernatants and cell lysates were collected for analysis of MMP-2, MMP-14 and TIMP-2 activity, mRNA and protein expression. In some studies over-expression with either MMP-14 or TIMP-2 was performed to revert the cells to a pre-injury phenotype.
Results
Transient injury resulted in a decrease of both MMP-14 and TIMP-2 activity and protein. Over-expression of each single molecule failed to prevent the injury-induced decrease of MMP-2 activity. On the other hand, over-expression of MMP-14 together with the addition of exogenous TIMP-2 did prevent the loss of MMP-2 activation.
Conclusions
Loss of MMP-2 activity after oxidant injury is caused by down regulation of MMP-14 and TIMP-2. Over-expression of either MMP-14 or TIMP-2 alone prior to oxidant injury is not enough to prevent loss of MMP-2 activity. All three components of the trimolecular complex must be present to preserve normal MMP-2 activity after oxidant injury.
Age-related macular degeneration (AMD) is the most important cause of lost central vision in the elderly1. Histopathology of early AMD demonstrates accumulation of specific lipid-rich deposits under the retinal pigmented epithelium (RPE). Many studies suggests a role for oxidant injury to the RPE as a putative mechanism for AMD pathogenesis, and several different potential oxidants have been suggested, such as those induced by light exposure, endogenous metabolism and inflammatory cells. We hypothesize that two specific “nonlethal” injury responses are especially relevant to early deposit formation in AMD. In previous studies, we have demonstrated that oxidant injury to the RPE causes nonlethal blebbing, characterized by extrusion of cell membrane and cytosol. However, dysregulated production and breakdown of extracellular matrix (ECM) components in Bruch’s membrane is another specific injury response relevant to AMD.
The normal anatomy and physiology of ECM in most tissues requires continuous turnover of collagen and other matrix components by a tightly regulated balance in production of matrix molecules like collagen IV, matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) 2. Relatively small dysregulation in the ratio of these factors can produce profound changes in the ECM, including thickening and deposit formation. We and others have previously shown that RPE express high levels of MMP-2, a gelatinase important for degradation of collagen IV, fibronectin and other matrix components relevant to Bruch’s membrane and subRPE deposit formation 3–6. In recent work, we have shown that transient nonlethal RPE injury with the macrophage-derived pro-oxidant enzyme, myeloperoxidase, plus its substrate, hydrogen peroxide, resulted in cell membrane blebbing associated with increased release of pro-MMP-2 from the cell surface, but decreased activation of pro-MMP-2 into the active form 7. Since most MMPs, including MMP-2, are synthesized as pro-molecules that require post-translational cleavage to produce maximum enzymatic activity, oxidant injury may alter production, expression or activity of enzyme regulators of MMP activation to explain the decrease in MMP-2 activity.
Two important regulators of pro-MMP activation include membrane type 1 matrix metalloproteinase (MT1-MMP or MMP-14), and tissue inhibitor of metalloproteinases (TIMP-2) 8,9. Active cell surface bound MMP-14 binds the amino-terminal domain of TIMP-2 while its carboxy terminal domain interacts with pro-MMP2 10. Activation of the tethered pro-MMP-2 is executed by a second active MMP-14. Conversely, binding of a second TIMP-2 results in inhibition of pro-MMP activation by MMP-14. Since we and others have previously shown that RPE express high levels of pro-MMP-2 and active MMP-2, and TIMP-2 3–6,11, we investigated whether MMP-14 was expressed in RPE cells and if MMP-14 and TIMP-2 mRNA and protein expression were altered after oxidant injury as a mechanism for changes in MMP-2 activation.
We found that both MMP-14 and TIMP-2 mRNA and protein were expressed and active on non-injured cultured RPE cells. 6–8 hours following removal of transient oxidative injury, both MMP-14 and TIMP-2 protein expression and activity decreased, resulting in decreased MMP-2 activation. The changes returned to normal by 96 hours after removal of oxidant injury. Importantly, transfection studies revealed that both MMP-14 and TIMP-2 were necessary to prevent oxidant-mediated loss of MMP-2 activation. Loss of regulation of MMP activation may contribute to AMD pathophysiology and these molecules might be targets for therapeutic intervention.
Materials and Methods
Cell culture conditions
ARPE-19 cells stably expressing green fluorescent protein (GFP, GFP-RPE), were grown in 10% FBS DMEM: F12 and exposed to 10% CO2 12. For experiments, cells were plated in 24 or 6 well plates (NUNCLON, Nalge Nunc, Rochester, NY) and allowed to grow to confluence. At the time of confluence, cells were exposed to 10% FBS DMEM: F12 phenol-red free for 48 hours, followed by 10% charcoal dextran stripped serum (Hyclone, Logan Utah) phenol-red free for 24 hours, and 1.0% charcoal dextran stripped serum and phenol-red free medium for an additional 24 hours. All cell culture reagents were purchased from Gibco-Invitrogen (Grand Island, NY).
Oxidant injury
GFP-RPE cells were treated with 10 microunits of MPO followed by 100μM H202 as previously described 5. 6 hours later, cells were washed and exposed to 0.1% charcoal stripped serum. 24 hours after treatment GFP-RPE cells were collected and processed. For time course experiments, cells were collected 6, 20, 48, 72, 96, 144 hrs following injury.
MMP-2 activation assay and Zymography
Ten μg of GFP-RPE cell lysate, 10mU of pro-MMP-2 and reaction buffer (50mM Tris-HCl, 5mM CaCl2, 1mM ZnCl2, 0.05% NaN3) were incubated for 1h at 37°C. Gelatin zymography was performed as previously described using 10ul aliquots of this reaction mixture13. Samples were applied to an SDS polyacrylamide gel copolymerized with 10% gelatin. Gels were rinsed twice after electrophoresis in 2.5% Triton X-100 and then incubated for 18h at 37°C in incubation buffer (50mM Tris-HCl, 5mM CaCl2, 1uM ZnCl2, 0.05% NaN3). The gelatin gels were stained with Coomassie brilliant blue and destained with10% acetic acid and 10% isopropanol and dried.
MMP-14 activity
GFP-RPE cells were plated in 24 well plates and grown to confluence at which time cells were treated as above. After treatment the media was removed and replaced with 250il of extraction buffer provided with the MMP-14 Biotrak Activity Assay System (Amersham Biosciences, Piscataway, NJ) and incubated at 4°C for 15 minutes. The supernatant was assayed for MMP-14 according to manufacturer’s directions for lower endogenous MMP-14 levels (assay range 0.125–4 ng/ml).
Reverse Zymography
TIMP expression was determined as previously described 13. Briefly, conditioned medium was prepared as for zymography and loaded onto a polyacrylamide gel containing gelatin and recombinant gelatinase A. The gels were processed as for zymography and after staining with Coomassie brilliant blue, areas of inhibition of gelatinase A activity were visualized as blue staining regions on a clear background.
Western blot analysis
GFP-RPE cell lysates were extracted and protein assessed using the Pierce BCA protein assay kit (Rockford, IL). Equal amounts of protein were applied to precast SDS polyacrylamide gels (Invitrogen) as previously described5. Electrotransfer of proteins from the gel to the nitrocellulose was performed by electroelution and immunoblotting. Blots were exposed to antibodies against MMP-14 and TIMP-2 (Chemicon, Temecula, CA) followed by chemiluminescence solution (Santa Cruz Biotechnology, Santa Cruz, California) and exposure to X-Omat AR film (Eastman Kodak Co., Rochester, NY). The film was scanned for densitometric analysis using Image J software from NIH. For immunoprecipitation experiments, 100 μg of protein extract was incubated with MMP-14 antibody (Chemicon) or normal goat IgG for 1 hour at 4°C, followed by the addition of protein G-Agarose overnight. The resulting protein G-antibody conjugate was centrifuged at 4°C and washed 4 times with lysis buffer, pH 7.4. The final pellet was resuspended and analyzed as above. Human MMP-14 or of TIMP-2 were used as positive controls for the antibodies.
Real time PCR
Amplification and measurement of target RNA was performed on the ABI PRISM 7700 Sequence Detection System as previously described 14. Quantitative RT-PCR was performed in a single one-step buffer system according to the manufacturer’s instructions. The sequence of the primers and probes for TIMP-2 and MMP-14 were respectively published15,16. All primers and probes were synthesized by Applied Biosystems (Foster City, CA). PCR assays were conducted in duplicate for each sample.
Transfection studies
A PCR3.1 expression vector containing a full-length, membrane-anchored MMP-14 17 was transfected into GFP-RPE cells using transfast (Promega Corp, Madison, WI) as previously described 5. To determine if MMP-14 was over-expressed, protein was collected and Western analysis performed as described above. Transfected cells were exposed to increasing concentrations of TIMP-2 followed by the standard injury protocol as described above. Supernatants were collected for zymography and reverse zymography and cell number counted for loading normalization. Control cells were transfected with an empty control plasmid. Alternatively cells were infected with a replication-deficient adenoviral (Ad) vector encoding the cDNA for hTIMP-2 under the transcriptional control of cytomegalovirus. Ad vector infection in vitro was carried out with 5 pfu/cell (multiplicity of infection).
Statistical analysis
The mean and standard deviation values for the measures were calculated and p values (students t-test) performed using Prism. Values of p<0.05 were considered statistically significant for all forms of statistical analysis used.
Results
GFP-RPE express pro-MMP-2, TIMP-2 and MMP-14
MMP-2 activation requires a trimolecular complex between pro-MMP-2, MMP-14 and TIMP-2. We have previously shown the presence of MMP-2 in GFP-RPE cells 5, and in this study western analysis of GFP-RPE cell lysates revealed a band at approximately 72kDa representing MMP-14 (Fig. 1A, lane 2) and 22kDa for TIMP-2 (Fig. 1B, lane 2). To determine if MMP-14 and TIMP-2 were bioactive, we performed an activity assay to measure the activation of exogenously added pro-MMP2 into active MMP-2 (Fig. 2). The data demonstrate a doubling of specific activity in the presence of GFP-RPE cell lysate compared to medium alone which was not exposed to cells. We conclude that bioactive MMP-14 and TIMP-2 are produced by cultured GFP-RPE.
Figure 1. MMP-14 and TIMP-2 are expressed by GFP-RPE cells.
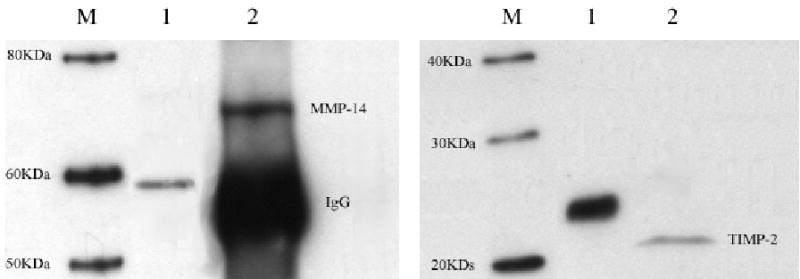
Human GFP-RPE cell lysates were collected and analyzed by Western blot for the presence of MMP-14 (A) and TIMP-2 (B) as described in methods. A. MMP-14 protein expression M: marker; lane 1: positive control expressing only the catalytic domain; lane 2: Endogenous expression of MMP-14 in GFP-RPE cell lines. B. TIMP-2 protein expression. M: marker, lane 1: positive control lane human TIMP-2 protein 2: Endogenous expression of TIMP-2 in GFP-RPE cells. Western blots are representative of three individual experiments.
Figure 2. ProMMP-2 is activated by RPE cells.
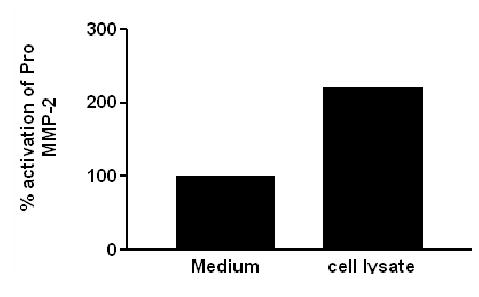
Graphic depiction of a two fold increase in pro MMP-2 activation in the presence of GFP-RPE cell lysates compared to medium alone. Ten μg of GFP-RPE cell lysates and 10mU of pro MMP-2 were incubated and analyzed by zymography. Data are expressed as percent activation of pro-MMP-2. Results are representative of two individual experiments performed in duplicate on cultured cells.
Oxidative injury decreases activity of MMP-14 and TIMP-2 in GFP-RPE cells
To test our hypothesis that oxidative injury changed surface activation of MMP-2 and thereby led to ECM dysregulation, we injured GFP-RPE cells by exposure to MPO/H202. As previously described by our group, MMP-2 activity decreased by over 60 % after oxidant injury (33 ± 12% of control, p<0.005 n =3). Time course of recovery (Fig 3A and Table 1) indicated that the maximal loss occurred within 20 hrs after removal of the oxidant, followed by a slow return to normal within 96 hours. TIMP-2 activity demonstrated a partial decrease that remained constant throughout the experiment. (Fig 3B and Table 1). Loss of MMP-14 activity decreased in a pattern that paralleled MMP-2 (Table 1). Importantly, MMP-14 activity was preserved at the 6 hour time point, suggesting that direct oxidation of the molecule was not the specific mechanism for loss of activity.
Figure 3. A. Time Course of MMP-2 and TIMP-2 activity following injury.
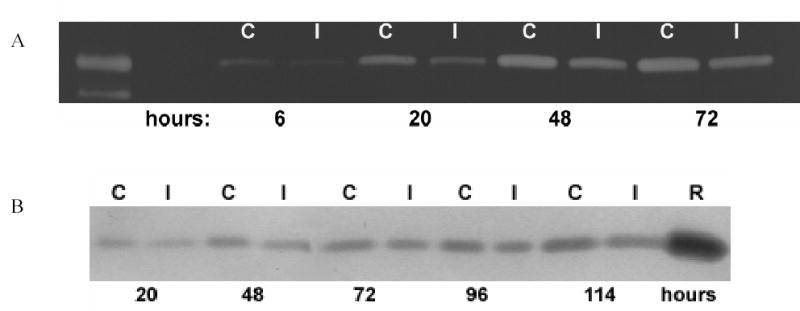
A. Representative zymography of MMP-2 activity time course before (C) and after injury (I) from 6 to 72 hours. Data is representative of three individual experiments performed in duplicate B. Representative reverse zymography of TIMP-2 activity time course before (C) and after injury (I) from 20 –114 hours, R; recombinant TIMP-2 protein. Data represents three individual experiments performed in duplicate.
Table I.
Time course of recovery of MMP-2, MMP-14 and TIMP-2 activity
| Time after removal of oxidant injury (hours) | % of control activity MMP-2 | % of control activity MMP-14 | % of control activity TIMP-2 |
|---|---|---|---|
| 6 | 38 ± 12 | 77 ± 18 | ND |
| 20 | 73 ± 16 | 49± 26 | 70 ± 4.5 |
| 48 | 81 ± 16 | 68± 0.2 | 72 ± 10 |
| 72 | 78 ± 13 | 81± 13 | 78 ± 2.3 |
| 96 | NC | 84 ± 17 | 66 ± 7.9 |
Data are mean ± of three experiments performed in duplicate. NC=No change from control, ND=not determined.
Oxidative injury decreases MMP-14 protein and mRNA expression
MMPs demonstrate complex transcriptional and translational regulation. We investigated the relative protein and mRNA expression of MMP-2, MMP-14 and TIMP-2 after oxidant injury. As shown by us previously, oxidant injury actually increased pro-MMP-2 protein expression, consistent with a loss of bioactivation 5. However, TIMP-2 protein demonstrated a partial reduction in expression after injury (Table 2). More striking, we found oxidant injury caused a decline in MMP-14 protein expression (Table 2). The time course of recovery indicated that decreased MMP-14 protein expression persisted for 48 hours after removal of oxidant injury, and then returned to baseline (Table 2).
Table II.
Time course of recovery of MMP-2, MMP-14 and TIMP-2 protein expression
| Time after removal of oxidant injury (hours) | % of control protein content MMP-2 | % of control in protein content MMP-14 | % of control protein content TIMP-2 |
|---|---|---|---|
| 6 | 64 ± 0.8 | 51 ± 3.7 | 75 ± 15 |
| 20 | 87 ± 1.2 | ND | 63 ± 2 |
| 48 | NC | 62 ± 24 | 70 ± 15 |
| 72 | ND | 85 ±1.4 | 66 ± 14 |
| 96 | NC | 92 ± 7.7 | 91 ± 8 |
Data are mean ± of three experiments performed in duplicate. NC=No change from control, ND=not determined.
Real time RT-PCR (table 3) demonstrated that after oxidant injury there was a decrease in MMP-14 mRNA content at 20 hrs, followed by recovery. MMP-2 mRNA and TIMP-2 mRNA acted in a similar manner. Taken together, the data suggest that oxidant-induced loss of MMP-2 activity occurred concomitantly with loss of MMP-14 activity which, in turn, was related to significant downregulation of MMP-14 protein synthesis.
Table III.
Time course of recovery of MMP-2, MMP-14 and TIMP-2 mRNA expression
| Time after removal of oxidant injury (hours) | % of control mRNA content MMP-2 | % of control mRNA content MMP-14 | % of control in mRNA content TIMP-2 |
|---|---|---|---|
| 6 | ND | ND | ND |
| 20 | 55 ± 13 | 76 ± 14 | 61 ± 18 |
| 48 | 77 ± 13 | 70 ± 24 | 72 ± 15 |
| 72 | 70± 19 | NC | 75 ± 27 |
Data are mean ± of three experiments performed in triplicate. NC= No change from control, ND= not determined
Overexpression of MMP-14 or TIMP-2
MMP-14 was over-expressed in GFP-RPE cells and oxidant injury performed as described. Cell lysate and supernatant were collected approximately 24 hours after injury. MMP-14 over-expression after transfection was detected by western blot analysis. Although over-expression of MMP-14 prevented the injury-induced decrease in MMP-14 protein and activity (Fig 4A), it did not prevent the injury-induced decrease of MMP-2 activity (Fig 5, lane 5). Therefore increasing concentrations of human recombinant TIMP-2 were added to GFP-RPE cells prior to injury (Fig 5, lanes 6–10). TIMP-2 enhanced the activation of MMP-2 when MMP-14 was over-expressed following injury suggesting that both TIMP-2 and MMP-14 were necessary to restore MMP-2 activation following oxidant injury.
Figure 4. Overexpression of MMP-14 and TIMP-2.
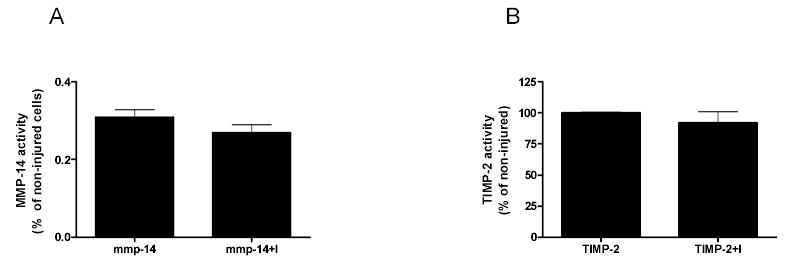
Activity of MMP-14 (A) as measured with the Biotrak activity kit and TIMP-2 (B) as measured by reverse zymography is not significantly decreased after overexpression and injury. (I=injury) Data represents two individual experiments performed in duplicate. Data are graphed as % activity of non-injured cells.
Figure 5. Restoration of MMP-2 activity requires both MMP-14 and TIMP-2.
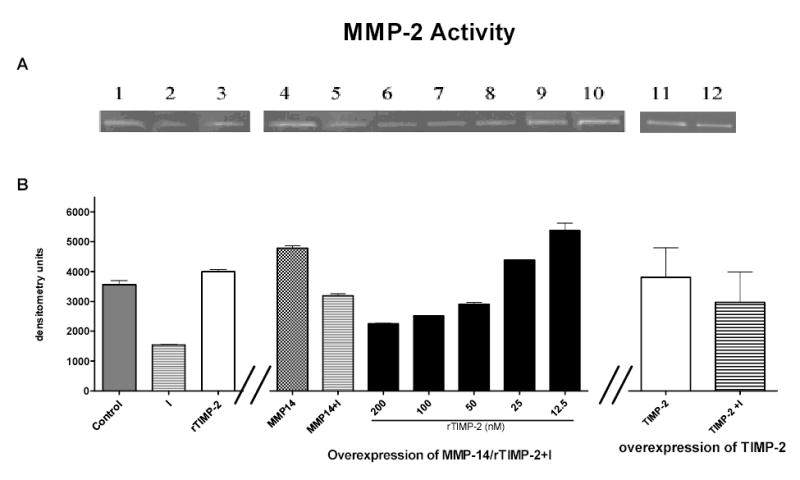
Representative zymogram (A) and graph depicting the mean ± SEM of duplicate experiments (B) of RPE cells transiently transfected with either a full length transcript of MMP-14 (lanes 4 –10) or TIMP-2 (lanes 11 and 12). Cells were treated with decreasing concentrations of recombinant TIMP-2 (rTIMP-2, nM) and injured according to the standard protocol described in methods (lanes 6–10). Only those cells with MMP-14 and the appropriate concentration of TIMP-2 had restored MMP-2 activity (lanes 9 and 10). Lane 3: recombinant TIMP-2 treatment to cells without injury (C=control, I= injury). Data are expressed as arbitrary densitometry units and represents three individual experiments performed in duplicate.
In complementary experiments, over-expression of TIMP-2 prevented the injury induced decrease of TIMP-2 (Fig 4B) as measured by reverse zymography, However recombinant TIMP-2 alone (Fig 5, lane 3) or overexpression of TIMP-2 in the absence (Fig 5, lane 11 and 12) or presence of recombinant MMP-14 did not prevent the injury mediated MMP-2 activity decrease (30% decrease of MMP-2 activity compared to control cells).
Discussion
AMD, the most important cause of central vision loss in the elderly, is characterized by progressive thickening and accumulation of ECM deposits under the RPE1,18. We have previously shown that loss of RPE MMP-2 activity correlated with deposit severity in vivo when mice were exposed to blue light, a surrogate for sunlight and acute oxidant injury 19. We therefore hypothesized that dysregulated production of MMP-2 following oxidant injury could contribute to matrix accumulation. The regulation of MMP-2 activation is well studied in other tissues, however little information is available regarding MMP-2 activation in human GFP-RPE cells 5,7.
Activation of MMP-2 is a membrane associated event requiring the presence of TIMP-2 and surface MMP-14 binding. This complex acts as a receptor for pro-MMP-2 and subsequent conversion to its active form. In this study, we demonstrated that GFP-RPE cells have active MMP-14 on the cell surface and that it is required for MMP-2 activation. Although the presence of MMP-14 has been reported previously in human RPE cells 4,20, to our knowledge there have been no reports on its ability to activate MMP-2 in vitro in retinal epithelial cells. Since TIMP-2 performs dual roles of either promoting or inhibiting activation of MMP-2, we also investigated its presence in human RPE cells. We were able to detect TIMP-2 protein by western analysis and reverse zymography as previously reported in rat and human RPE cells 6,21.
To mimic an oxidant mediated injury relevant to AMD, we exposed the cells to MPO/H2O2. MMP-14 protein expression and activity were significantly decreased to 50% of control 6 hours following injury and correlated with a decrease of MMP-2 activity. Not unexpectedly, TIMP-2 activity and protein expression were also significantly decreased by 6 hours.
Regulation of MMPs is controlled by gene transcription, activation of pro-MMPs and interaction of secreted MMPs with inhibitors 22. Therefore time course experiments were performed to assess transcriptional and translational responses to injury. We found the maximum decrease in MMP-14 activity and protein expression at 48 hours at which time recovery began and returned to near baseline levels by 96 hours. Importantly, mRNA expression began to increase at 20 hrs prior to recovery of activity. These data partially confirm that the injury, although limited to the cell surface, was not merely damaging cell surface associated proteins.
MMP-14 activity is crucial for both physiological and disease processes. It was initially identified as the activator of MMP-2 8,23, but recently has been shown to be important for wound healing, angiogenesis and inflammation 24 . It is over-expressed in cancers leading to migration, invasion and metastasis 25. MMP-14 activation occurs via a proprotein convertase 26,27 and recent data suggests that its function is modified by glycosylation, internalization and recycling 28,29. Finally MMP-14 directly degrades ECM molecules including collagen type I, III, laminin and fibronectin 30,31. MMP-14 in our system clearly plays a crucial role in regulating MMP-2 and subsequent ECM changes, although its direct influence on accumulation of deposits in AMD remained to be explored in future experiments.
TIMP-2 protein and activity recovered in a coordinate manner although in our experiments, activity levels never fully reached control levels. Despite this, TIMP-2 activity levels were sufficient to form a ternary complex with pro-MMP-2 and MMP-14 and promote recovery of MMP-2 activity.
TIMP-2 has a dual role as mediator of activation and inhibitor of MMP-2 32. Translational regulation of TIMP-2 is not well described as its expression is constitutive. Sp1 and NF-Y have been shown to be involved in a cAMP-dependent TIMP-2 gene transcription activation 33. Yamamoto et al showed that oxidant injury to an RPE cell line increased cAMP production 36 hours later which could lead to increased TIMP-2 gene expression 34. This appears consistent with our mRNA expression data. Alternatively, TNFα activation of the ERK-MAPK pathway following oxidant injury may play a role in TIMP-2 protein regulation. Alexander et al showed that treatment of human and porcine trabecular cells with TNFα increased MMP expression but decreased TIMP-2 activity 35. This may be relevant as oxidative stress has been shown to cause ERK activation in RPE cells 36.
It is likely that the amount of TIMP-2 protein produced after injury is precisely regulated for the RPE to maintain ECM balance. In fact it has been proposed for TIMP-1, that there is an exquisite equilibrium between the rate of mRNA degradation and the rate of translational initiation and elongation.
To further test our hypothesis that MMP-2 changes were occurring due to changes in its regulator molecules, we transiently transfected human RPE cells to overexpress MMP-14 prior to oxidant injury. Over-expression of MMP-14 prevented the injury-induced decrease of MMP-14 activity and protein, but did not prevent injury-induced decrease of MMP-2 activity. Therefore we pre-treated transfected cells with increasing concentrations of recombinant TIMP-2 and found that both MMP-14 and TIMP-2 were necessary to restore MMP-2 activation to pre injury levels. This effect of TIMP-2 was concentration dependent. Interestingly, when we over-expressed TIMP-2 we were unable to restore MMP-2 activity after injury even with the addition of recombinant MMP-14. We presume that in our system over-expression of TIMP-2 produced a ratio of MMP-14/TIMP-2 favoring inhibition rather than activation of MMP-2. These paradoxical effects of TIMP-2 have been shown in other cell types and it appears that in RPE cells as well prevention of dysregulation of ECM requires a precise ratio of MMP-14/TIMP-2.
A recent report eloquently showed that there is an alternative pathway for MMP-2 activation through another surface MMP, MMP-15 which is TIMP-2 independent 37. The expression of MMP-15 has been reported in human RPE cells, but in our studies this molecule clearly did not compensate for lack of MMP-14 and TIMP-2 following injury 4. Although there have been reports of other MMP/TIMP molecules produced by RPE cells, we focused on the components of the trimolecular complex. In fact, MMP-14 was found to be the most abundant molecule in microarray analysis of human RPE samples 21. This does not exclude the relevance of RPE synthesized MMP-9/TIMP-1, which are regulated when exposed to cytokines 4. In fact we detected a decrease in TIMP-1 activity by reverse zymography (data not shown) after injury. Additionally, we were able to visualize TIMP-3 although there were no changes in activity after injury. Mutations in the gene for TIMP-3 cause Sorsby’s fundus dystrophy, a degenerative disease of the macula 38 and possible implications for its role in wet AMD have been suggested 39.
We propose that in dry AMD RPE injury or stimulation with macrophage-derived factors, especially MPO and TNF-α lead to dysregulated RPE production of matrix molecules that might influence turnover of BrM, accumulation of deposits and development of CNV. Prevention of dysregulation of the components of the trimolecular complex may lead to possible therapeutic interventions.
Footnotes
Grants: This work was supported in part by National Institutes of Health National Eye Institiute Grant RO1 EY14477-02 (SE and SWC)
References
- 1.Berger JW, Fine SL, Maguire MG. Age-related macular degeneration. St. Louis: Mosby; 1999.
- 2.Corcoran ML, Hewitt RE, Kleiner DE, Jr, Stetler-Stevenson WG. MMP-2: expression, activation and inhibition. Enzyme Protein. 1996;49:7–19. doi: 10.1159/000468613. [DOI] [PubMed] [Google Scholar]
- 3.Ahir A, Guo L, Hussain AA, Marshall J. Expression of metalloproteinases from human retinal pigment epithelial cells and their effects on the hydraulic conductivity of Bruch’s membrane. Invest Ophthalmol Vis Sci. 2002;43:458–465. [PubMed] [Google Scholar]
- 4.Eichler W, Friedrichs U, Thies A, Tratz C, Wiedemann P. Modulation of matrix metalloproteinase and TIMP-1 expression by cytokines in human RPE cells. Invest Ophthalmol Vis Sci. 2002;43:2767–2773. [PubMed] [Google Scholar]
- 5.Marin-Castano ME, Elliot SJ, Potier M, Karl M, Striker LJ, Striker GE, Csaky KG, Cousins SW. Regulation of estrogen receptors and MMP-2 expression by estrogens in human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2003;44:50–59. doi: 10.1167/iovs.01-1276. [DOI] [PubMed] [Google Scholar]
- 6.Padgett LC, Lui GM, Werb Z, LaVail MM. Matrix metalloproteinase-2 and tissue inhibitor of metalloproteinase-1 in the retinal pigment epithelium and interphotoreceptor matrix: vectorial secretion and regulation. Exp Eye Res. 1997;64:927–938. doi: 10.1006/exer.1997.0287. [DOI] [PubMed] [Google Scholar]
- 7.Marin-Castano ME, Csaky K, Cousins SW. Nonlethal oxidant injury to Human Retinal Pigment Epithelium cells causes cell membrane blebbing but decreased MMP-2 activity. Invest Ophthalmol Vis Sci. 2005;46:3331–3340. doi: 10.1167/iovs.04-1224. [DOI] [PubMed] [Google Scholar]
- 8.Strongin AY, Collier I, Bannikov G, Marmer BL, Grant GA, Goldberg GI. Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J Biol Chem. 1995;270:5331–5338. doi: 10.1074/jbc.270.10.5331. [DOI] [PubMed] [Google Scholar]
- 9.Kinoshita T, Sato H, Okada A, Ohuchi E, Imai K, Okada Y, Seiki M. TIMP-2 promotes activation of progelatinase A by membrane-type 1 matrix metalloproteinase immobilized on agarose beads. J Biol Chem. 1998;273:16098–16103. doi: 10.1074/jbc.273.26.16098. [DOI] [PubMed] [Google Scholar]
- 10.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamei M, Hollyfield JG. TIMP-3 in Bruch’s membrane: changes during aging and in age-related macular degeneration. Invest Ophthalmol Vis Sci. 1999;40:2367–2375. [PubMed] [Google Scholar]
- 12.Strunnikova N, Baffi J, Gonzalez A, Silk W, Cousins SW, Csaky KG. Regulated heat shock protein 27 expression in human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2001;42:2130–2138. [PubMed] [Google Scholar]
- 13.Elliot SJ, Striker LJ, Stetler-Stevenson WG, Jacot TA, Striker GE. Pentosan polysufate decreases proliferation and net extracellular matrix production in mouse mesangial cells. J Am Soc Nephrol. 1999;10:62–68. doi: 10.1681/ASN.V10162. [DOI] [PubMed] [Google Scholar]
- 14.Potier M, Elliot SJ, Tack I, Lenz O, Striker GE, Striker LJ, Karl M. Expression and regulation of estrogen receptors in mesangial cells: influence on matrix metalloproteinase-9. J Am Soc Nephrol. 2001;12:241–251. doi: 10.1681/ASN.V122241. [DOI] [PubMed] [Google Scholar]
- 15.Dahan M, Nawrocki B, Elkaim R, Soell M, Bolcato-Bellemin AL, Birembaut P, Tenenbaum H. Expression of matrix metalloproteinases in healthy and diseased human gingiva. J Clin Periodontol. 2001;28:128–136. doi: 10.1034/j.1600-051x.2001.028002128.x. [DOI] [PubMed] [Google Scholar]
- 16.Sun HB, Yokota H. Reduction of cytokine-induced expression and activity of MMP-1 and MMP-13 by mechanical strain in MH7A rheumatoid synovial cells. Matrix Biol. 2002;21:263–270. doi: 10.1016/s0945-053x(02)00003-3. [DOI] [PubMed] [Google Scholar]
- 17.Hiraoka N, Allen E, Apel IJ, Gyetko MR, Weiss SJ. Matrix metalloproteinases regulate neovascularization by acting as pericellular fibrinolysins. Cell. 1998;95:365–377. doi: 10.1016/s0092-8674(00)81768-7. [DOI] [PubMed] [Google Scholar]
- 18.Green WR. Histopathology of age-related macular degeneration. Mol Vis. 1999;5:27. [PubMed] [Google Scholar]
- 19.Cousins SW, Espinosa-Heidmann DG, Alexandridou A, Sall J, Dubovy S, Csaky K. The role of aging, high fat diet and blue light exposure in an experimental mouse model for basal laminar deposit formation. Exp Eye Res. 2002;75:543–553. doi: 10.1006/exer.2002.2047. [DOI] [PubMed] [Google Scholar]
- 20.Smine A, Plantner JJ. Membrane type-1 matrix metalloproteinase in human ocular tissues. Curr Eye Res. 1997;16:925–929. doi: 10.1076/ceyr.16.9.925.5044. [DOI] [PubMed] [Google Scholar]
- 21.Leu ST, Batni S, Radeke MJ, Johnson LV, Anderson DH, Clegg DO. Drusen are Cold Spots for Proteolysis: Expression of Matrix Metalloproteinases and Their Tissue Inhibitor Proteins in Age-related Macular Degeneration. Exp Eye Res. 2002;74:141–154. doi: 10.1006/exer.2001.1112. [DOI] [PubMed] [Google Scholar]
- 22.Jones CB, Sane DC, Herrington DM. Matrix metalloproteinases: a review of their structure and role in acute coronary syndrome. Cardiovasc Res. 2003;59:812–823. doi: 10.1016/s0008-6363(03)00516-9. [DOI] [PubMed] [Google Scholar]
- 23.Sato H, Takino T, Okada Y, Cao J, Shinagawa A, Yamamoto E, Seiki M. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature. 1994;370:61–65. doi: 10.1038/370061a0. [DOI] [PubMed] [Google Scholar]
- 24.Zucker S, Pei D, Cao J, Lopez-Otin C. Membrane type-matrix metalloproteinases (MT-MMP) Curr Top Dev Biol. 2003;54:1–74. doi: 10.1016/s0070-2153(03)54004-2. [DOI] [PubMed] [Google Scholar]
- 25.Seiki M, Yana I. Roles of pericellular proteolysis by membrane type-1 matrix metalloproteinase in cancer invasion and angiogenesis. Cancer Sci. 2003;94:569–574. doi: 10.1111/j.1349-7006.2003.tb01484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pei D, Weiss SJ. Furin-dependent intracellular activation of the human stromelysin-3 zymogen. Nature. 1995;375:244–247. doi: 10.1038/375244a0. [DOI] [PubMed] [Google Scholar]
- 27.Pei D, Weiss SJ. Transmembrane-deletion mutants of the membrane-type matrix metalloproteinase-1 process progelatinase A and express intrinsic matrix-degrading activity. J Biol Chem. 1996;271:9135–9140. doi: 10.1074/jbc.271.15.9135. [DOI] [PubMed] [Google Scholar]
- 28.Remacle A, Murphy G, Roghi C. Membrane type I-matrix metalloproteinase (MT1-MMP) is internalised by two different pathways and is recycled to the cell surface. J Cell Sci. 2003;116:3905–3916. doi: 10.1242/jcs.00710. [DOI] [PubMed] [Google Scholar]
- 29.Wu YI, Munshi HG, Sen R, Snipas SJ, Salvesen GS, Fridman R, Stack MS. Glycosylation broadens the substrate profile of membrane type 1 matrix metalloproteinase. J Biol Chem. 2004;279:8278–8289. doi: 10.1074/jbc.M311870200. [DOI] [PubMed] [Google Scholar]
- 30.Itoh Y, Seiki M. MT1-MMP: an enzyme with multidimensional regulation. Trends Biochem Sci. 2004;29:285–289. doi: 10.1016/j.tibs.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 31.Zucker S, Hymowitz M, Conner CE, DiYanni EA, Cao J. Rapid trafficking of membrane type 1-matrix metalloproteinase to the cell surface regulates progelatinase a activation. Lab Invest. 2002;82:1673–1684. doi: 10.1097/01.lab.0000041713.74852.2a. [DOI] [PubMed] [Google Scholar]
- 32.Holmbeck K, Bianco P, Yamada S, Birkedal-Hansen H. MT1-MMP: a tethered collagenase. J Cell Physiol. 2004;200:11–19. doi: 10.1002/jcp.20065. [DOI] [PubMed] [Google Scholar]
- 33.Zhong ZD, Hammani K, Bae WS, DeClerck YA. NF-Y and Sp1 cooperate for the transcriptional activation and cAMP response of human tissue inhibitor of metalloproteinases-2. J Biol Chem. 2000;275:18602–18610. doi: 10.1074/jbc.M001389200. [DOI] [PubMed] [Google Scholar]
- 34.Yamamoto M, Sato N, Tajima H, Furuke K, Ohira A, Honda Y, Yodoi J. Induction of human thioredoxin in cultured human retinal pigment epithelial cells through cyclic AMP-dependent pathway; involvement in the cytoprotective activity of prostaglandin E1. Exp Eye Res. 1997;65:645–652. doi: 10.1006/exer.1997.0370. [DOI] [PubMed] [Google Scholar]
- 35.Alexander JP, Acott TS. Involvement of the Erk-MAP kinase pathway in TNFalpha regulation of trabecular matrix metalloproteinases and TIMPs. Invest Ophthalmol Vis Sci. 2003;44:164–169. doi: 10.1167/iovs.01-1201. [DOI] [PubMed] [Google Scholar]
- 36.Garg TK, Chang JY. Oxidative stress causes ERK phosphorylation and cell death in cultured retinal pigment epithelium: Prevention of cell death by AG126 and 15-deoxy-delta 12, 14-PGJ2. BMC Ophthalmol. 2003;3:5. doi: 10.1186/1471-2415-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morrison CJ, Butler GS, Bigg HF, Roberts CR, Soloway PD, Overall CM. Cellular activation of MMP-2 (gelatinase A) by MT2-MMP occurs via a TIMP-2-independent pathway. J Biol Chem. 2001;276:47402–47410. doi: 10.1074/jbc.M108643200. [DOI] [PubMed] [Google Scholar]
- 38.Jacobson SG, Cideciyan AV, Bennett J, Kingsley RM, Sheffield VC, Stone EM. Novel mutation in the TIMP3 gene causes Sorsby fundus dystrophy. Arch Ophthalmol. 2002;120:376–379. doi: 10.1001/archopht.120.3.376. [DOI] [PubMed] [Google Scholar]
- 39.Langton KP, McKie N, Curtis A, Goodship JA, Bond PM, Barker MD, Clarke M. A novel tissue inhibitor of metalloproteinases-3 mutation reveals a common molecular phenotype in Sorsby’s fundus dystrophy. J Biol Chem. 2000;275:27027–27031. doi: 10.1074/jbc.M909677199. [DOI] [PubMed] [Google Scholar]