

Abstract
Digestive fluid of the araneid spider Argiope aurantia is known to contain zinc metallopeptidases. Using anion-exchange chromatography, size-exclusion chromatography, sucrose density gradient centrifugation, and gel electrophoresis, we isolated two lower-molecular-mass peptidases, designated p16 and p18. The N-terminal amino acid sequences of p16 (37 residues) and p18 (20 residues) are 85% identical over the first 20 residues and are most similar to the N-terminal sequences of the fully active form of meprin (β subunits) from several vertebrates (47–52% and 50–60% identical, respectively). Meprin is a peptidase in the astacin (M12A) subfamily of the astacin (M12) family. Additionally, a 66-residue internal sequence obtained from p16 aligns with the conserved astacin subfamily domain. Thus, at least some spider digestive peptidases appear related to astacin of decapod crustaceans. However, important differences between spider and crustacean metallopeptidases with regard to isoelectric point and their susceptibility to hemolymph-borne inhibitors are demonstrated. Anomalous behavior of the lower-molecular-mass Argiope peptidases during certain fractionation procedures indicates that these peptidases may take part in reversible associations with each other or with other proteins. A. aurantia digestive fluid also contains inhibitory activity effective against insect digestive peptidases. Here we present evidence for at least thirteen, heat-stable serine peptidase inhibitors ranging in molecular mass from about 15 to 32 kDa.
Keywords: Argiope, Astacin, Crayfish, Meprin, Metalloproteinase, Metzincin, Procambarus, Serine protease inhibitor, Spider, Zinc metalloprotease
1. Introduction
The digestive peptidases of orb-weaving spiders are necessary not only for consumption of prey, but also for the spider’s recycling of its old orb webs into new (Tillinghast and Kavanagh 1977; Kavanagh and Tillinghast 1983; Townley and Tillinghast 1988). For some orb-weavers, including the araneid Argiope aurantia Lucas 1833, peptidases also play a role in the emergence of young spiders from the egg sac (Foradori et al. 2002). The study of spider digestive enzymes spans more than a century (early studies reviewed in Pickford 1942), but has been sporadic and remains a largely unexplored area. The most comprehensive studies to date are those of Mommsen (1978a,b,c,d, 1980).
In earlier studies (Tillinghast and Kavanagh 1977; Kavanagh 1979; Kavanagh and Tillinghast 1983), two peptidases were isolated from the digestive fluid of A. aurantia (though additional peptidases were demonstrated and partially characterized). Designated Argiope protease B and Argiope protease D, they had estimated molecular masses of 17.6 and 20.2 kDa, respectively, slightly alkaline pH optima, were cationic at physiological pH (about 7.8), and were shown to be metallopeptidases. Argiope protease B, but not protease D, was observed to cleave spider major ampullate silk, the principal component of draglines and of non-sticky lines in orb webs.
Relatively low-molecular-mass peptidases such as these, found in spider digestive fluid, bear a resemblance to small peptidases described from spider venom (Mebs 1970, 1972). Mebs found three peptidases in the venom of the theraphosid Vitalius roseus (Mello-Leitão 1923) (formerly Pamphobeteus roseus). By size-exclusion chromatography, all three had molecular masses of about 11 kDa (but see Discussion regarding peptidase molecular masses estimated by this method), and they, like the A. aurantia digestive peptidases (Tillinghast and Kavanagh 1977; Tugmon and Tillinghast 1995), had alkaline pH optima, did not cleave trypsin substrates, and were unaffected by serine peptidase inhibitors.
The venom gland origin of the peptidases described by Mebs (1970, 1972) is questionable as there are reasons to suspect that his venom sample was contaminated by digestive fluid regurgitated at the time of venom collection. First, Mebs acknowledged that the venom was a gift (i.e., he did not collect it). Second, though there appear to be exceptions (Young and Pincus 2001; da Silva et al. 2004), very little peptidase activity has been observed in spider venoms that have been collected so as to avoid contamination by regurgitated digestive fluid (see Perret 1977, Geren and Odell 1984, and Kuhn-Nentwig et al. 1994 for reviews of this problem). And third, as indicated above, the digestive fluid peptidases share several characteristics with the peptidases Mebs described. Mebs (1970, 1972) drew attention to the similarity of the V. roseus ‘venom’ peptidases to a peptidase reported in crayfish gastric fluid (Pfleiderer et al. 1967), now known as astacin and the eponymous prototype of a family of zinc metallopeptidases (Stöcker and Zwilling 1995). In this study, we concentrated our efforts on the lower-molecular-mass digestive peptidases of A. aurantia and present amino acid sequence data that indicate a link between spider and crayfish digestive peptidases. However, our observations also point to important differences between them.
In addition to digestive hydrolases, the digestive fluid of A. aurantia contains inhibitors of insect digestive peptidases (Tugmon and Tillinghast 1995). In this report we present evidence for at least thirteen, heat-stable serine peptidase inhibitors in the digestive fluid of A. aurantia.
2. Materials & Methods
2.1. Collection of Biological Fluids
Adult female A. aurantia were collected in New Hampshire, Maine, Pennsylvania, Virginia, North Carolina, and Georgia, USA. Spider digestive fluid (SDF) was obtained by holding the spider in one hand and putting a micropipet to its mouthparts. Under these circumstances spiders regurgitated SDF that was drawn up into the micropipets, pooled, and stored at –20°C. The SDF was then centrifuged for 2 min at 15,996 x g to remove particulates and dialyzed overnight against the buffer in which it would be fractionated. All dialyses in this study were performed using 3500MWCO Slide-A-Lyzer dialysis cassettes (Pierce Biotechnology, Rockford IL, USA).
Crayfish gastric fluid (CGF) was obtained by inserting the needle of a 5 mL syringe into the stomachs of anesthetized crayfish (Procambarus clarkii (Girard), Carolina Biological Supply, Burlington NC, USA) and withdrawing fluid.
Hemolymph was obtained from both species by clipping a leg and drawing fluid into micropipets.
2.2. Fractionation of SDF: Peptidases
The fractionation of SDF was evaluated by size-exclusion chromatography, ion-exchange chromatography followed by size-exclusion chromatography, and sucrose density gradient centrifugation. We used the SDF endogenous α-amylase and N-acetyl-β-d-glucosaminidase activities as internal indicators of the effectiveness of our methods. The molecular weights of both have been determined in digestive fluid from spiders (Mommsen 1978b,d, 1980; see Fig. 1), though not A. aurantia. Presumably, the molecular weights of these enzymes are at least similar in A. aurantia. Size-exclusion columns were calibrated with various combinations of blue dextran (Sigma-Aldrich D4772, St. Louis MO, USA; MW 2,000 kDa), bovine serum albumin (Sigma A8531; 66 kDa), carbonic anhydrase (Sigma C7025; 29 kDa), cytochrome C (Sigma C7150; 12.4 kDa), and vitamin B12 (Sigma V2876; 1.355 kDa), run separately from SDF components.
Fig. 1.
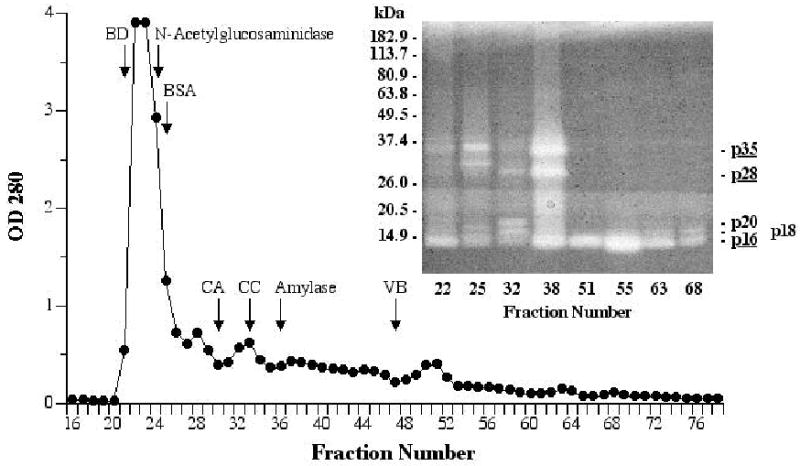
Fractionation of 0.7 ml SDF by size-exclusion chromatography on Sephacryl S-100 HR (1.4 x 100 cm column, 50 mM Tris, pH 7.7, 12.8 mL/hr, 3.2 mL/fraction). Fractions were assayed for endogenous N-acetyl-β-glucosaminidase (108–110 kDa; Mommsen 1978b, 1980) and α-amylase (58–63 kDa; Mommsen 1978b, 1978d) activity, with the fraction containing the highest concentration of each indicated on the chromatogram. Selected fractions were examined for peptidase activity by 4–16% SDS-PAGE zymography (inset). Distances migrated by molecular mass standards are indicated on the left; peptidase bands are labeled on the right (those underlined appear to be most abundant). In separate runs, blue dextran (BD; 2000 kDa) was chromatographed with one of four other standards: bovine serum albumin (BSA; 66 kDa), carbonic anhydrase (CA; 29 kDa), cytochrome C (CC; 12.4 kDa), or vitamin B12 (VB; 1.355 kDa).
Size-exclusion chromatography
SDF (0.7 ml) was dialyzed against 50 mM Tris buffer, pH 7.7, then fractionated in a column (1.4 x 100 cm) of Sephacryl S-100 HR (fractionation range 1–100 kDa; Amersham Biosciences, Piscataway NJ, USA) eluted with the same buffer at a flow rate of 12.8 mL/hr with 3.2 mL fractions collected. The fractions were assayed for α-amylase and N-acetyl-β-glucosaminidase by methods described below. Peptidases were visualized by zymography using small-format Novex 4–16% SDS-PAGE Blue Casein gels (Invitrogen EC6415, Carlsbad CA, USA) run under non-reducing conditions in a Novex Xcell II Mini-Cell electrophoresis unit (Invitrogen). Pre-stained protein standards (BenchMark, Invitrogen 10748-010 or SeeBlue Plus2, Invitrogen LC5925) were run concurrently.
In addition, selected fractions were pooled, reduced in volume in a Savant Speed-Vac concentrator, and evaluated by isoelectric focusing (IEF). Small-format Novex pH 3-10 IEF gels (Invitrogen EC6655A) were used in a Novex XCell SureLock Mini-Cell (Invitrogen) to separate sample proteins and Serva IEF protein markers 3–10 (Invitrogen 39212-01).
Anion-exchange/size-exclusion chromatography
SDF (0.75 ml) was made to 3.0 mL with 25 mM Tris, pH 8.1, containing 5 mM CaCl2 (Buffer A), then dialyzed against the same buffer. A 1.5 mL aliquot was injected into a 5 mL Econo-Pac High Q cartridge (Bio-Rad, Hercules CA, USA) and eluted with: 20 mL Buffer A, 50 mL 0 to 0.25 M NaCl gradient in Buffer A, 10 mL 0.25 to 0 M NaCl gradient in Buffer A, and 20 mL Buffer A. Two mL fractions were collected and the OD280 of each was measured. Fractions were then assayed in pooled groups of five for protein, peptidases, α-amylase, N-acetyl-β-glucosaminidase, and serine peptidase inhibitors by methods described below.
The first 10 fractions were pooled, lyophilized to reduce volume, dialyzed against 50 mM Tris buffer, pH 7.6, then fractionated on a 1.4 x 100 cm column of Sephacryl S-100 HR as described above (12.8 mL/hr; 3.2 mL fractions). The OD280 was recorded and selected fractions were assayed for peptidase activity by radial diffusion (section 2.6). Peptidases were visualized by 4–16% SDS-PAGE zymography as described above.
Sucrose density gradient centrifugation
A 200 μL sample of SDF was centrifuged at 735 x g for 3 min, then 15 μL aliquots of the supernatant were mixed with 180 μL 50 mM Tris-HCl, pH 7.5, and carefully laid upon a 5 mL linear gradient of 5–30% (w/v) sucrose in the same buffer. The samples were centrifuged at 4°C for 21 h at 160,000 x g in a Beckman L8-70 ultracentrifuge using a SW65 rotor. Spun tubes were placed in a holder, punctured at the bottom with a needle, and 0.25 mL fractions were collected. All fractions were assayed for peptidases, serine peptidase inhibitors, α-amylase, and N-acetyl-β-glucosaminidase, while selected fractions were examined for peptidases by zymography as described above. Fractions were then pooled into groups of four or five and these were examined by non-reducing SDS-PAGE using small-format 4–20% Novex Tris-glycine gels (Invitrogen EC6028) with pre-stained protein standards run concurrently (Invitrogen 10748-010).
2.3. Fractionation of SDF: Serine peptidase inhibitors
SDF (1 ml) was mixed with 2 mL 20 mM Bis-Tris (Sigma B7535) buffer, pH 6.8, then applied to a 1.4 x 20.3 cm anion-exchange column of Q Sepharose Fast Flow (Amersham Biosciences 17-0510-01) and eluted with 100 mL of the same buffer. This was followed by a 400 mL 0 to 500 mM NaCl gradient in the same buffer and then 100 mL of 500 mM NaCl in the same buffer, all at a flow rate of 1.3 mL/min with 3.4 mL fractions collected and the OD280 monitored continuously. Peak fractions in the OD280 trace and fractions between peaks were pooled separately, lyophilized, and desalted on PD-10 columns (Amersham 17-0851-01). The 3.5 mL high-molecular-weight fraction off the PD-10 columns was reduced to about 500 μL in a Savant Speed-Vac concentrator, boiled for 4 min, and centrifuged at 15,996 x g for 5 min. The supernatants were further concentrated to about 250 μL and 20 μL aliquots were used to assay serine peptidase inhibitor activity by radial diffusion (section 2.6). Fractions containing the highest activity (56–67 and 68–91) were examined by native and denaturing PAGE using samples of 7.5 to 15 μL/lane.
We used the high pH discontinuous system of Ornstein-Davis as described in Hames (1981) to separate serine peptidase inhibitors by native PAGE on large-format 8% resolving gels cross-linked with AcrylAide (FMC BioProducts, Rockland ME, USA, discontinued). Serine peptidase inhibitors were also separated by non-reducing SDS-PAGE on large-format 13% resolving gels cross-linked with AcrylAide using Laemmli’s discontinuous system as given in Hames (1981). Samples were boiled for 1 min after addition of sample buffer. No inhibitors were seen if the sample buffer contained 2-mercaptoethanol. Lanes loaded with molecular mass standards (Gibco-BRL, discontinued) were cut off after SDS-PAGE and stained with Coomassie blue R-250.
Serine peptidase inhibitors were visualized in gels by the method of Uriel and Berges (1968). Following non-denaturing PAGE, gels were placed in a solution of trypsin (TPCK-treated, Sigma T8642, replaced by T1426; 0.04 mg/mL in PBS, pH 7.4) for 10 min at 37°C with gentle shaking. The solution was then decanted and the residual trypsin was allowed to penetrate the gels for 30 min at 37°C. Gels were then covered by the following two solutions that were mixed together just prior to staining. The first consisted of 50 mg of the tryptic substrate N-acetyl-dl-phenylalanine-β-naphthyl ester (Sigma A7512) dissolved in 20 mL N, N -dimethylformamide; the second consisted of 100 mg tetrazotized o-dianisidine (Sigma D3502, replaced by D9805) dissolved in 180 mL PBS, pH 7.4. The stain was removed after 1 h of gentle shaking at room temperature and the gels were stored in 2% acetic acid. The same procedure was followed with SDS-PAGE gels except that SDS was removed from the gels prior to the incubation in trypsin solution. This was done using a modified version of the wash sequence described by Coughlin and Green (1983): 15 min in 40 mM Tris HCl, pH 9.0, 5 min in distilled water, 25 min in the same Tris buffer, and 10 min in distilled water, all at 37°C with continuous agitation.
Some inhibitor bands seen on native gels were cut out from adjacent, unstained sample lanes and eluted using a Bio-Rad 422 Electro-Eluter. They were then re-electrophoresed under denaturing or non-denaturing conditions.
2.4. Amino acid sequencing
For N-terminal sequencing, selected peptidase-containing fractions off size-exclusion or anion-exchange columns or sucrose density gradients were subjected to non-reducing SDS-PAGE in Novex 14% Tris-glycine gels (Invitrogen EC6484). The proteins were then electroblotted to polyvinylidene difluoride (PVDF) membranes for 45 min to 1 h using a Novex XCell SureLock Mini-Cell with an Xcell II blot module (Invitrogen). The PVDF membranes were stained with a 50% methanol, 0.1% Coomassie blue R-250 solution for 5 min and destained with 50% methanol, 10% acetic acid (LeGendre and Matsudaira 1989). Selected protein bands were cut out with a razor blade and sent to the University of Texas Medical Branch Protein Chemistry Laboratory (UTMB-PCL) for Edman sequencing on Applied Biosystems Procise 494/HT and 494 cLC Protein Sequencing Systems (Foster City CA, USA).
Internal sequencing was carried out using a single group of pooled fractions off a size-exclusion column (fractions 49–64, see section 3.1) in which the dominant protein component was SDF peptidase p16 (Fig. 2C). A cyanogen bromide digest was prepared at UTMB-PCL directly from an aliquot of the pooled fractions. Modified trypsin (Promega V5113, Madison WI, USA) and asparaginylendopeptidase (Takara Bio Inc. 7319, Otsu, Japan) digests were also prepared (Williams and Stone 1995) after aliquots of the pooled fractions were electrophoresed on 14% SDS-PAGE gels (as above) and gel slices containing p16 were sent to UTMB-PCL. Peptides in the three digests were separated and blotted onto PVDF using an Applied Biosystems 173A Microblotter and then sequenced as above.
Fig. 2.
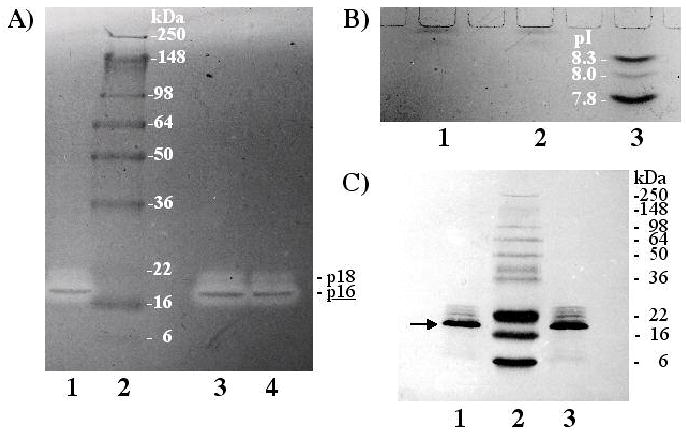
Examination of fractions 49–64 from the Sephacryl S-100 HR run shown in Fig. 1. (A) By 4–16% SDS-PAGE zymography, two peptidases were evident and are labeled on the right (major, underlined, p16; minor, p18). Lanes 1, 3, and 4 were each loaded with 2.5 μl of the same sample, but the aliquots in lanes 3 and 4 were heated for 2 min at 85°C prior to loading. Molecular mass standards are shown in lane 2. (B) By IEF, two bands, stained with Coomassie blue, were observed close to the well. Lanes 1 and 2 were both loaded with 5 μl of the same sample electrophoresed in (A). pI standards are shown in lane 3. (C) By 14% SDS-PAGE followed by blotting onto PVDF membrane, one major band (arrow, identified as p16) and four minor bands were observed with Coomassie blue staining. Lanes 1 and 3 were both loaded with 10 μl of the same sample electrophoresed in (A). Molecular mass standards are shown in lane 2 and labeled on the right.
The protein sequence data reported in this paper appear in the UniProt knowledgebase under accession numbers P84748 and P84749.
2.5. Comparison of spider and crayfish digestive peptidases
Spider and crayfish digestive peptidases were compared by native electrophoresis and hemolymph peptidase inhibitor studies. Hemolymph of A. aurantia is known to contain inhibitors that effectively block spider digestive peptidases (Tugmon and Tillinghast 1995).
Horizontal 1% agarose gels were prepared with barbital buffer, pH 8.6. Twenty μL samples of SDF and CGF were electrophoresed at 20 mA (constant) for 1 h. To visualize peptidase activity, lanes were laid upon 1% agarose gels containing 1% Carnation nonfat dry milk in Buffer A.
Radial diffusion was used to compare the effect of spider or crayfish hemolymph on SDF and CGF. Gels containing 1% agarose and 1% casein were prepared with 110 mM Tris, pH 7.3. Three equidistant wells in a linear arrangement were cut into the gels and 2 μL aliquots of SDF or CGF, made to 20 μL with the same buffer, were added to the central well. To the wells on either side of this central well were added 20 μL of spider or crayfish hemolymph. After a two-day incubation at room temperature, the ability of hemolymph components to inhibit casein digestion by SDF or CGF peptidases was evaluated. Acetic acid (3%) was used to stop the digestion and enhance the non-digested areas.
2.6. Assays
Peptidase activity was assayed by radial diffusion as described by Schumacher and Schill (1972) or by the method of Kunitz (1947) using 1% Hammerstein casein (Schwarz/Mann, Orangeburg NY, USA, discontinued) in Buffer A as the substrate. Removal of precipitated undigested casein in the latter procedure was accomplished by filtration through Whatman No. 1 filter paper rather than by centrifugation. SDF serine peptidase inhibitors were assayed using radial diffusion in casein/agarose gels as described by Tugmon and Tillinghast (1995). α-Amylase activity was assayed by Noelting’s 3,5-dinitrosalicylic acid method (Meyer et al. 1947; Bernfeld 1955) using starch as substrate. N-acetyl-β-d-glucosaminidase activity was assayed as described in Peters et al. (1998) using 4-nitrophenyl-N-acetyl-β-d-glucosaminide (Sigma N9376) as substrate. Protein was measured by the method of Lowry et al. (1951) as modified by Peterson (1977) (Sigma kit P5656, replaced by TP0300).
3. Results
In what follows we refer to separate digestive peptidase bands on electrophoretic gels or blots by making reference to their molecular masses. In an earlier study (Foradori et al. 2001), four major SDF peptidases were estimated to have apparent molecular masses of 12, 30, 38, and 120 kDa. For the three smallest peptidases, our best estimates of their molecular masses have shifted to 16, 28, and 35 kDa, respectively, and consequently we refer to them here as p16, p28, and p35. Given that the molecular mass standards electrophoresed with the peptidases were pre-stained, we should still consider these values only approximate (as recommended by the manufacturer; Shi and Jackowski 1998:26–27). Indeed, we have obtained different molecular masses using aliquots of the same peptidase solution electrophoresed on different dates with different sets of pre-stained standards. An example of this is described in section 3.1 and is the reason a peptidase’s designation may appear at odds with its position on an individual gel presented in this paper. The designation is an average calculated from several gels employing two different sets of standards. Also, we have not ruled out the possibility that one or more of the bands of proteolytic activity is due to more than one peptidase, but for the present we will refer to all bands that have migrated the same relative distance in a gel by the same designation.
3.1. Fractionation of SDF by size-exclusion chromatography
The fractionation of SDF on Sephacryl S-100 HR is presented in Fig. 1. A peak in peptidase activity was seen by radial diffusion assay at fraction 38 where peptidases p35 and p28 were observed by zymography at highest concentration (Fig. 1 inset). Peptidase p16 appeared in every fraction after the void volume, out at least to fraction 68 (Fig. 1 inset), though in highest concentration at about fraction 54. p120 was not observed in any fractions off Sephacryl columns. In one experiment, fractions 49–64 were pooled, reduced in volume, and analyzed by zymography, IEF, and SDS-PAGE. Zymography revealed one major peptidase (p16) and one more minor and slightly larger peptidase (p18) (Fig. 2A). By IEF, only two protein bands were observed in sample lanes, neither of which migrated as far as the standard with an isoelectric point (pI) at pH 8.3. Thus, both p16 and p18 appear to have a pI above 8.3 (Fig. 2B). By SDS-PAGE, five closely spaced bands were visible, with p16 predominating. This was also evident after electroblotting the proteins onto PVDF membrane (Fig. 2C). Aliquots of the same pooled fractions 49–64 were used to obtain N-terminal and internal amino acid sequences of p16 (see sections 2.5 and 3.5). We should note that the pooled fractions sampled to produce Fig. 2 were part of the same chromatographic run presented in Fig. 1. But while p16 and p18 had apparent molecular masses of 14.2 and 15.5 kDa, respectively, in the gel shown in Fig. 1 (inset), in Fig. 2 we obtained molecular masses of 18.2 and 20.6 kDa for these two peptidases. These discrepancies might be due to the use of two different sets of pre-stained standards.
3.2. Fractionation of SDF by anion-exchange/size-exclusion chromatography
About half the peptidase activity and the α-amylase eluted in the first 10 fractions during anion-exchange chromatography at pH 8.1, whereas serine peptidase inhibitors and N-acetyl-β-glucosaminidase appeared only after application of the salt gradient (Fig. 3). In other fractionations by anion-exchange chromatography at pH 9, p120, but not p16, p28, or p35, was retained on the column and eluted by the salt gradient, indicating that the pI of p120 is lower than those of the other three major peptidases.
Fig. 3.
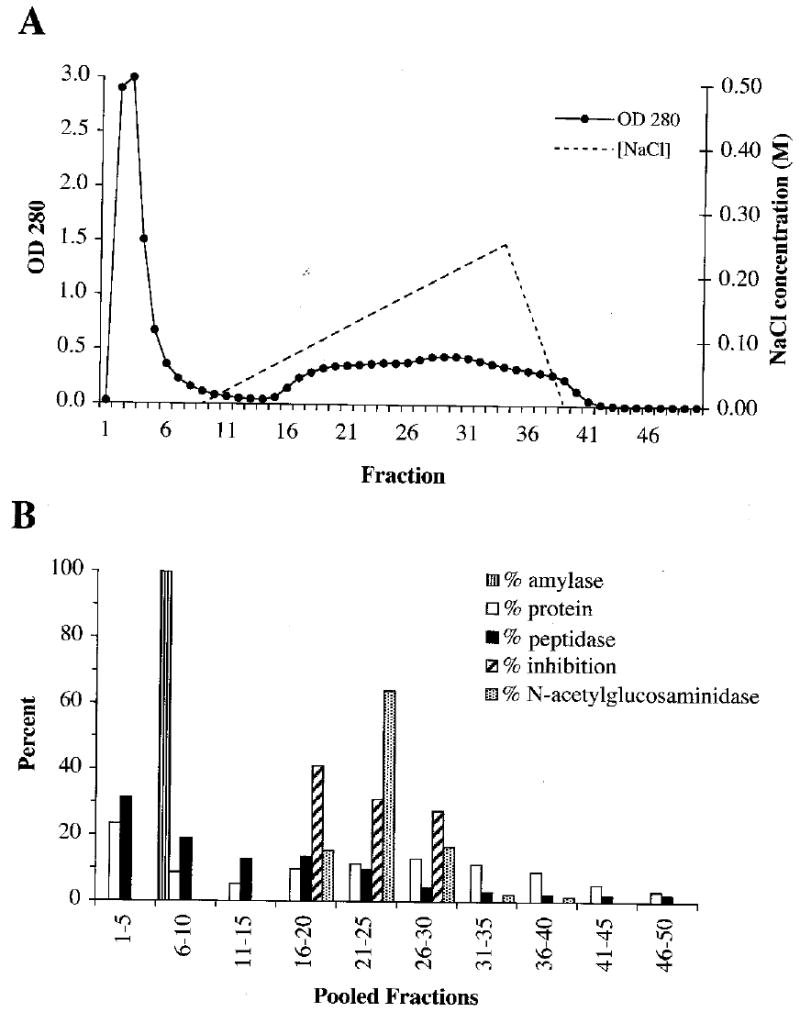
Fractionation of 0.75 ml SDF by anion-exchange chromatography on an Econo-Pac High Q cartridge (25 mM Tris, pH 8.1, 5 mM CaCl2, 50 ml 0-0.25 M NaCl gradient, 2.0 mL/fraction). (A) The OD280 of each fraction was measured and (B) pooled groups of five fractions were assayed for α-amylase activity, protein, peptidase activity, serine peptidase inhibitors, and N-acetyl-β-glucosaminidase activity. Percentages given are the percentages of the total seen over all fractions.
When the first 10 fractions at pH 8.1 were pooled and passed through Sephacryl S-100 HR, α-amylase peaked in fraction 36 whereas p16, p18, and p20 began eluting prior to it and p35 and p28 peaked just after it. Peptidase p16 was present in all fractions inspected after the void volume (fraction 21) but was most abundant at about fraction 45 (Fig. 4).
Fig. 4.
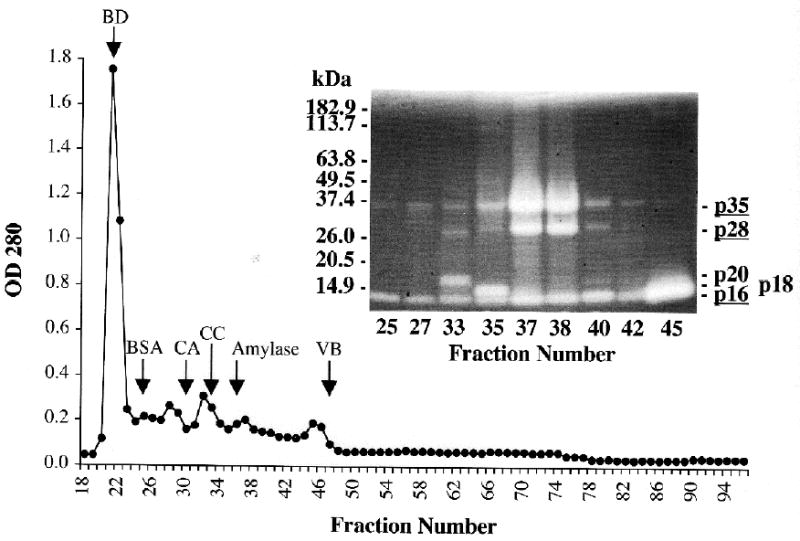
Fractionation by size-exclusion chromatography of early-eluting peptidases from the anion-exchange run shown in Fig. 3. The first ten fractions off the anion-exchange cartridge were pooled, concentrated, and fractionated on a Sephacryl S-100 HR column (1.4 x 100 cm) as in Fig. 1. Abbreviations on chromatogram also as in Fig. 1. Selected fractions were examined for peptidase activity by 4–16% SDS-PAGE zymography (inset). Note that p16 occurs in all these fractions. Positions of molecular mass standards are indicated on the left; peptidase bands are labeled on the right (the most abundant are underlined).
3.3. Fractionation of SDF by sucrose density gradient centrifugation
By sucrose gradient centrifugation, the spider’s N-acetyl-β-glucosaminidase, α-amylase, and serine peptidase inhibitors migrated as relatively discrete bands in accordance with their molecular masses, while peptidase activity appeared in every fraction (Fig. 5). In part, this broad distribution can be explained by the widely different molecular masses of the individual peptidases. However, peptidases p120, p35, p28, and p16 were found together in most of the fractions above fraction 3 (Fig. 6).
Fig. 5.
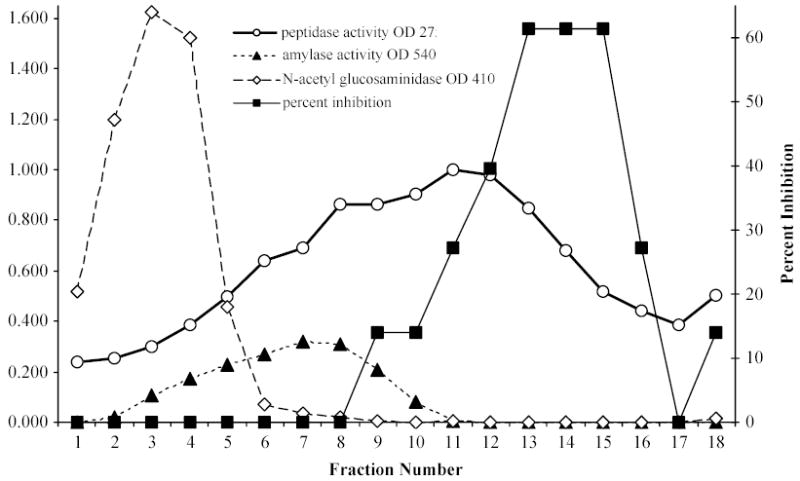
Fractionation of SDF by sucrose density gradient centrifugation. Fifteen μL SDF, mixed with 180 μL 50 mM Tris-HCl, pH 7.5, was applied to 5 mL of a 5–30% sucrose density gradient in the same buffer and centrifuged for 21 hr at 160,000 x g at 4°C. Fractions (0.25 ml) were assayed for peptidase, α-amylase, and N-acetyl-β-glucosaminidase activities and serine peptidase inhibitory activity.
Fig. 6.
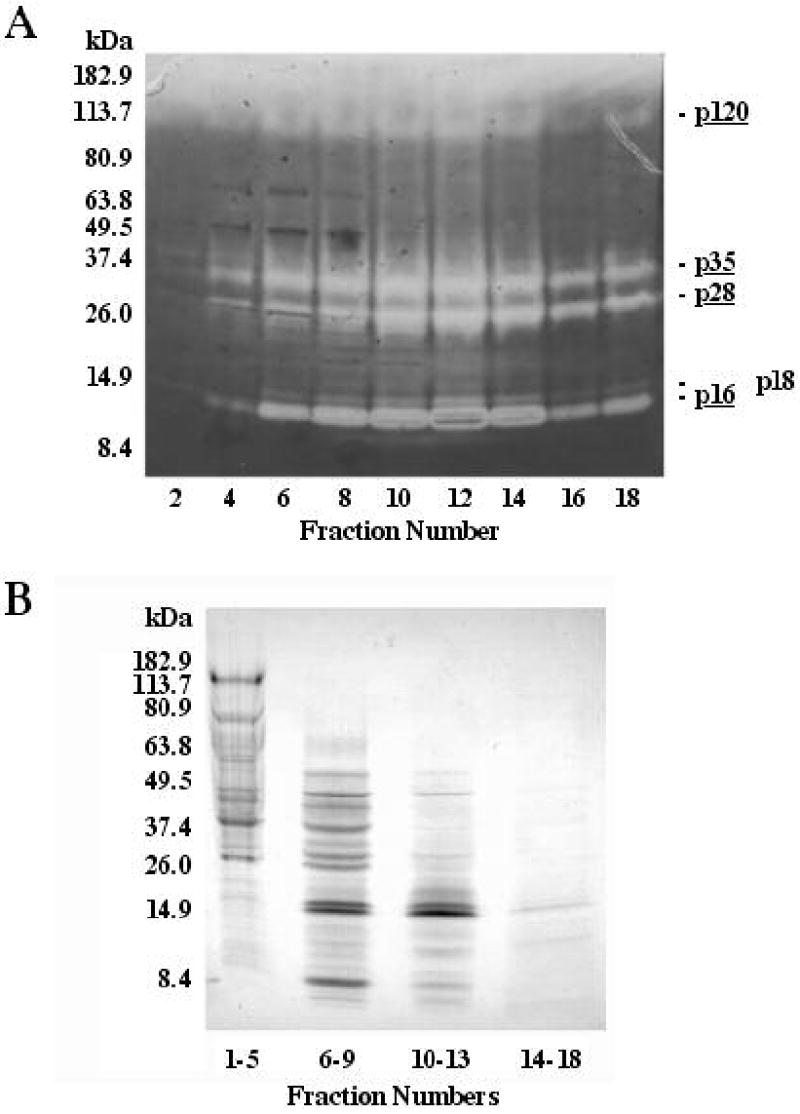
SDF peptidases and total protein in fractions produced by sucrose density gradient centrifugation of SDF. The peptidases in 5 μL of each even-numbered fraction are shown in the 4–16% SDS-PAGE zymogram in (A). Note the persistence of peptidases p16, p28, p35, and p120 throughout most of the fractions. Coomassie blue-stained proteins in 20 μL aliquots of pooled fractions are shown in the 4–20% SDS-PAGE gel in (B).
3.4. SDF serine peptidase inhibitors
The majority of the serine peptidase inhibitory activity eluted in the earlier part of the salt gradient during anion-exchange chromatography on both the Econo-Pac High Q cartridge (Fig. 3) and the Q Sepharose Fast Flow column. With the latter, the highest activity was observed in pooled fractions 56–67 and 68–91. When aliquots of these fractions were examined by non-denaturing PAGE, 10 prominent serine peptidase inhibitor bands were seen. These were designated A, B, C, D, E, E’, F, G, G’, and H, with E’ and G’ evident in fractions 68–91 but not in 56–67. Conversely, A and B appeared in fractions 56–67, but not in 68–91 (Fig. 7).
Fig. 7.
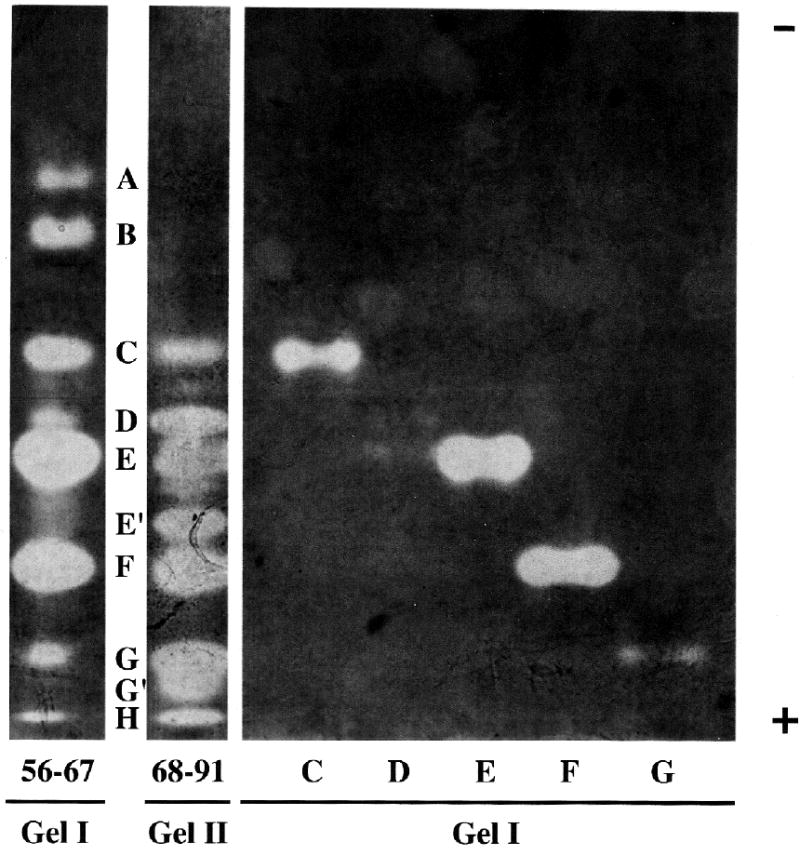
Separation of SDF serine peptidase inhibitors by native 8% PAGE. SDF (1 ml) was fractionated by anion-exchange chromatography on Q Sepharose Fast Flow (1.4 x 20.3 cm column, 100 mL 20 mM Bis-Tris, pH 6.8, followed by 400 mL 0–500 mM NaCl gradient in the same buffer, 1.3 mL/min, 3.4 mL/fraction). The highest inhibitor activity was in pooled fractions 56–67 and 68–91, which together contain ten prominent inhibitors. Inhibitors C, D, E, F, and G were electroeluted individually from unstained lanes loaded with aliquots of fractions 56–67 and re-electrophoresed. In this instance, inhibitor D was only faintly visible upon re-electrophoresis. Lanes loaded with fractions 56–67 and individual inhibitor bands (C, D, E, F, G) are from the same gel (Gel I) while the lane loaded with fractions 68–91 is from a different gel (Gel II). That the alignment of Gels I and II is correct was confirmed in two other native gels in which fractions 56–67 and 68–91 were run concurrently in adjacent lanes. Aliquots of the same solutions containing individual inhibitors C, E, F, and G, run in Gel I, were also electrophoresed by SDS-PAGE in Gel III shown in Fig. 8.
We isolated some of these inhibitors by loading aliquots of pooled fractions 56–67 in multiple lanes of 8% native PAGE gels. After electrophoresis, one lane was stained for inhibitors and used as a guide for cutting bands from unstained lanes. After electroeluting these bands the inhibitors were re-electrophoresed on native 8% PAGE gels (Fig. 7) and/or on 13% SDS-PAGE gels (Fig. 8). Under non-reducing, denaturing conditions, inhibitors C, E, and F each produced two bands of inhibition while inhibitors B and D produced single bands (though a second very faint band was seen in B). Our observations thus far indicate that inhibitors A and G also produce single bands of inhibition by SDS-PAGE, but as recovery of these inhibitors from native gel pieces was low, this will need to be confirmed. Apparent molecular masses of these inhibitors (in kDa), obtained by non-reducing SDS-PAGE, were: A, 14.7; B, 16.7 and 21.3 (very faint); C, 16.7 and 21.2; D, 20.5; E, 23.2 and 32.1; F, 27.7 and 31.0; G, 24.1.
Fig. 8.

Separation of SDF serine peptidase inhibitors by 13% SDS-PAGE. Aliquots of fractions 56–67 and individual inhibitor bands (A through G, eluted from native polyacrylamide gels) were separated under denaturing, but non-reducing conditions. See Fig. 7 and text for the origins of these samples. Lanes containing individual inhibitor bands C, E, F, and G in Gel III are aliquots of the same solutions electrophoresed in native Gel I in Fig. 7. The individual inhibitor band lanes (A-E) in Gel IV contain inhibitors eluted from a different native gel. Note that inhibitors C, E, and F have each now separated into two bands of inhibition. Recovery of inhibitors A (white arrow) and G (black arrow) from native gel pieces was low. Positions of molecular mass standards are indicated to the left of each gel.
3.5. Amino acid sequencing of p16 and p18
A peptidase identified as p16 was isolated by each of the three SDF fractionation protocols described above, followed by SDS-PAGE and blotting onto PVDF. All three had identical N-terminal amino acid sequences over the first 20 residues. One of these (from fractions 49–64, see section 3.1) was in sufficient quantity to sequence the first 34 residues. A fourth sample identified as p16 was isolated by anion-exchange chromatography and SDS-PAGE without an intervening gel filtration step. Its N-terminal sequence differed from the other three at 1 of 20 residue positions (Arg at position 10 instead of Trp) and with residue 18 unassignable. A peptidase isolated by sucrose density gradient centrifugation and SDS-PAGE, identified as p18, was 85% identical and 90% similar to the consensus p16 sequence (Fig. 9).
Fig. 9.

N-terminal amino acid sequences of A. aurantia peptidases p16 and p18, and an internal sequence from p16. For comparison, aligned portions of zebrafish meprin A β-subunit (Mep A-β□□□□ Bank accession no. BX571842), nematode astacin 15 (NAS-15; GenBank accession no. AJ561208), and the astacin conserved domain consensus sequence (Ast CDCS; CD database no. 24541, www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=cdd) are also shown. Residues identical to those in p16 are shown in bold. Dashes represent gaps inserted to optimize alignment. Mep A-β and NAS-15 show especially high similarity to the N-terminal and internal sequences of p16, respectively. The sequence in p16 between the two arrows is a tryptic peptide, the position of which relative to other peptides was presumed on the basis of homology to other astacin subfamily members rather than by benefit of overlapping peptides from other digests.
Sequencing of peptides from CNBr, tryptic, and asparaginylendopeptidyl digests of p16 both confirmed the 34-residue N-terminal sequence obtained from the intact protein and extended this sequence by 3 residues. We were also able to assemble a 66-residue internal sequence from p16, relying primarily on overlaps among peptides from the different digests, but also, in two instances, on similarity to homologous peptidases (Fig. 9).
BLAST searches (Altschul et al. 1997; Tatusova and Madden 1999; Marchler-Bauer and Bryant 2004) of nucleotide (translated) and protein sequence databases (www.ncbi.nlm.nih.gov/blast/) revealed that the N-terminal sequence of p16 aligns most significantly with the β subunit of vertebrate meprin A, a zinc metallopeptidase in the astacin (M12A) subfamily within the astacin (M12) family (Rawlings and Barrett 1995; Wolz and Bond 1995; Rawlings et al. 2004, merops.sanger.ac.uk). More specifically, the p16 N-terminal sequence aligns with the N-terminal sequence of fully active meprin β subunit, i.e., the β subunit after cleavage of signal peptide and propeptide sequences (Gorbea et al. 1993). This N-terminus forms the N-terminal portion of the astacin or peptidase domain of the meprin β subunit (as opposed to one of the non-proteolytic domains of meprin) (reviewed in Wolz and Bond 1995). With single 1-residue gaps introduced in both the p16 and meprin sequences to optimize alignment, the p16 sequence is 47–52% identical and 63–65% similar to chicken, zebrafish, dog, human, rat, and mouse meprin A β-subunits (GenBank accession nos. XM_419181, BX571842, XM_547619, X81333, NM_013183 and NM_008586, respectively).
The 66-residue internal sequence from p16 provided further evidence that we are dealing with astacin subfamily metallopeptidases as a BLAST search showed that this sequence aligns with the astacin (M12A) conserved domain consensus sequence. With a single 1-residue gap introduced in the p16 sequence, the two sequences are 51% identical and 66% similar. Alignment of the two sequenced portions of p16 with the astacin conserved domain indicates that there are 58 residues between these two sequences yet to be determined (Fig. 9).
3.6. Comparison of spider and crayfish digestive peptidases
When SDF and CGF were co-electrophoresed at pH 8.6 in a native agarose gel, the crayfish digestive peptidases moved rapidly toward the anode, while the spider digestive peptidases migrated toward the cathode (Fig. 10A). In radial diffusion studies, the spider’s hemolymph readily inhibited SDF peptidases, but was much less effective inhibiting CGF peptidases (Fig. 10B). Likewise, in the converse experiment, crayfish hemolymph readily inhibited CGF peptidases, but its potency against SDF peptidases was considerably less (Fig. 10B).
Fig. 10.
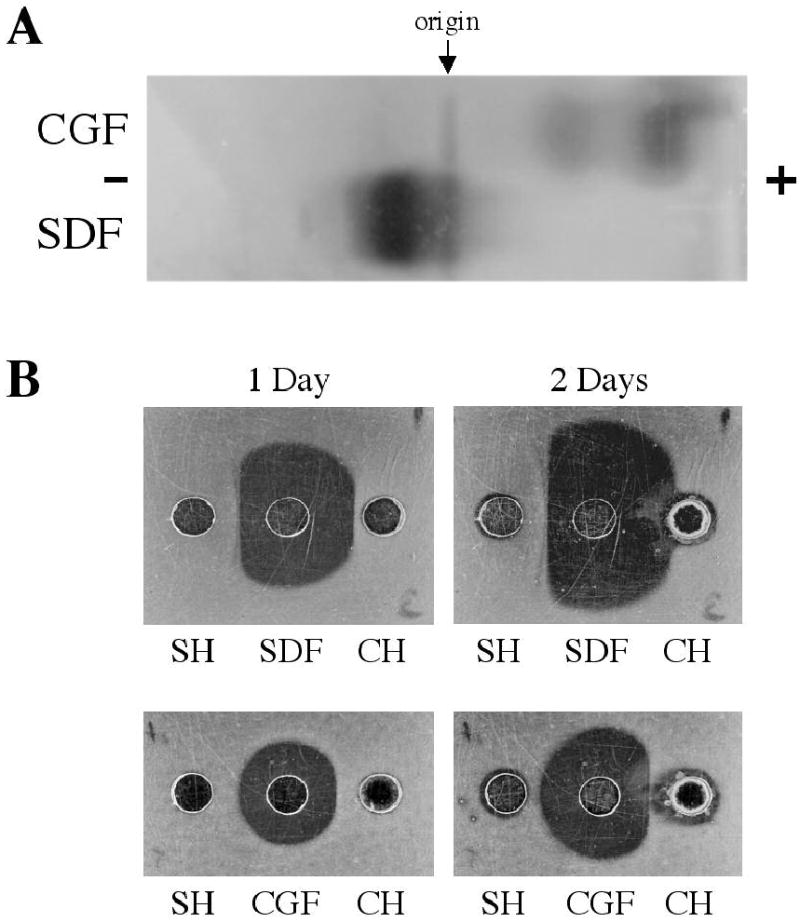
Comparison of spider and crayfish digestive peptidases by (A) agarose electrophoresis and (B) radial diffusion against hemolymph-borne inhibitors. (A) Twenty μL samples of spider digestive fluid (SDF) and crayfish gastric fluid (CGF) were electrophoresed in a 1% agarose gel, pH 8.6, for 1 hr at 20 mA. This gel was then placed on a 1% nonfat dry milk/1% agarose gel, pH 8.1, to visualize caseinolytic peptidases. Note that the bulk of the spider peptidases moved slowly toward the cathode whereas the crayfish peptidases moved rapidly toward the anode. (B) The ability of 20 μL samples of spider hemolymph (SH) or crayfish hemolymph (CH) to inhibit peptidases in 2 QL samples of SDF or CGF was photographed after 1 and 2 days incubation at room temperature. The casein/agarose gels shown after 1 day on the left are the same gels shown after 2 days on the right.
4. Discussion
4.1. SDF peptidases
The peptidases in the digestive fluid of A. aurantia exhibited anomalous behavior on size-exclusion columns. Peptidase p16 especially eluted over a wide range of fractions (Figs. 1, 4), first appearing much earlier than anticipated given its relatively small mass, as though it had a tendency to form associations, with itself and/or other proteins. We explored additional chromatographic and preparative electrophoretic steps beyond those recorded here, but they only dispersed p16 further. p16 showed a similar anomalous distribution following sucrose density gradient centrifugation (Fig. 6).
Our experience is reminiscent of that of Mebs (1970, 1972). He also observed anomalous elution patterns of peptidases after passing spider venom (possibly contaminated with digestive fluid, see Introduction) through a series of size-exclusion media. Thus, while he isolated three peptidases from different chromatographic fractions, all had molecular masses of about 11 kDa, none cleaved tryptic or chymotryptic substrates, and none were affected by serine peptidase inhibitors. He suspected that he had isolated just a single peptidase and that reversible association accounted for its unexpected elution pattern. We believe that Mebs’ hypothesis provides the best explanation for our data and also explains why previous efforts to isolate SDF peptidases have resulted in low yields (Kavanagh and Tillinghast 1983). Future efforts may benefit from finding conditions of pH, ionic environment, and temperature that favor the dissociation of the peptidases prior to and during fractionation.
It is possible that reversible association of spider digestive peptidases has a functional advantage during the consumption of prey. The spider’s digestive enzymes must function initially within the body of its prey and having two or more peptidases with different cleavage specificities in close proximity may help to rapidly inactivate prey peptidases, thereby sparing amylases, lipases, and other spider digestive enzymes. Under these conditions the SDF would not be as dilute as during the chromatographic methods employed here, and the higher concentration should favor greater association of peptidases.
Mebs (1970, 1972) also drew attention to similarities in molecular mass and catalytic properties between the crayfish (Astacus) digestive peptidase, astacin (Pfleiderer et al. 1967), and the spider peptidases he isolated. [It has since been determined that the molecular mass of astacin is not 11 kDa, but 23 kDa, the discrepancy being attributed to anomalous retention of the peptidase on the size-exclusion columns used to estimate molecular mass (Stöcker and Zwilling 1995; Zwilling 1997). The 11 kDa molecular mass of the spider ‘venom’ peptidases was also estimated by gel filtration (Mebs 1970, 1972) and may have been subject to the same error.] If, as suggested above, the spider venom peptidases were actually from SDF, then subsequent observations have confirmed the relevance of this comparison. Like astacin, the principal spider digestive peptidases are zinc metallopeptidases (Kavanagh 1979; Kavanagh and Tillinghast 1983; Foradori et al. 2001). Moreover, in this study, database searches of two extended sequences from p16 and the N-terminus of p18 demonstrated that these peptidases belong to the astacin (M12A) subfamily of the astacin (M12) family (Rawlings et al. 2004, merops.sanger.ac.uk). We note that a peptidase of the astacin family has apparently been detected in venom uncontaminated by digestive fluid in the spider Kukulcania hibernalis, but this peptidase is a member of the adamalysin (= reprolysin, M12B) subfamily (Jackson et al. 1995, GenBank accession no. I15012).
The p16 and p18 N-terminal sequences align with the N-terminal end of the astacin or peptidase domain of M12A subfamily members, including meprin β chain, to which they show the greatest similarity. However, given the relatively low masses of p16 and p18, they must lack the multidomain structures of meprins and some other astacin subfamily members. Virtually unequivocal proof of an astacin subfamily relationship for p16 was obtained with the sequencing of a 66-residue internal segment, showing significant alignment with the astacin (M12A) conserved domain consensus sequence.
While these data demonstrate that the spider peptidases are related to astacin of decapod crustaceans, substantial differences must exist. Thus, while spider hemolymph efficiently inhibited spider digestive peptidases, as observed previously (Tugmon and Tillinghast 1995), it had much less of an effect on crayfish digestive peptidases. Similarly, crayfish hemolymph was a much more potent inhibitor of crayfish digestive peptidases than of spider digestive peptidases (Fig. 10B). This low-level cross-inhibition raises suspicion that non-specific interactions (e.g., involving an α2-macroglobulin, Stöcker et al. 1991a and references therein) may account for much or all of the inhibition that was seen between these arthropods. And whereas spider digestive peptidases migrated toward the cathode when electrophoresed at pH 8.6, the crayfish digestive peptidases moved toward the anode (Fig. 10A). This observation is consistent with (1) the electrophoretic migration of astacin and crayfish trypsin-like peptidase toward the anode at pH 8.6 and 8.0 (Pfleiderer et al. 1967; Pfleiderer and Zwilling 1972), (2) the reported low pI (3.5) of astacin (Stöcker and Zwilling 1995), (3) the reported high pI (> 8.2) of Argiope protease B and Argiope protease D (Kavanagh 1979:113), and (4) our observation of a pI > 8.3 for at least two lower-molecular-mass Argiope peptidases (p16, p18) (Fig. 2B). Like astacin, meprin A has a considerably lower pI (4–5) (Butler et al. 1987) than the bulk of the peptidases in SDF. It remains to be established which peptidases, by the designations used in this paper, correspond to the four Argiope peptidase fractions (A-D) of Kavanagh (1979) and Kavanagh and Tillinghast (1983), and in particular to the apparently homogeneous Argiope protease B and Argiope protease D. Similarities with respect to molecular mass, pI, and relative abundance make it likely that p16 is one of these two peptidases. It also remains to be established if, as with many other enzymes including astacin (Zwilling and Neurath 1981; Stöcker et al. 1991b), α-amylase (Mommsen 1978b,d), and N-acetyl-β-glucosaminidase (Mommsen 1978b), any of the more minor Argiope peptidases (observed in SDS-PAGE gels in the vicinity of p35, p28, and p16 (Figs. 1, 4, 6A)) represent isozymes of one of the major peptidases. The small differences in N-terminal sequence between p16 and p18 recommend them as candidate isozymes.
4.2. SDF serine peptidase inhibitors
We found 10 serine peptidase inhibitors in SDF separated by non-denaturing PAGE (Fig. 7). When three of these (C, E, F) were isolated and subjected to SDS-PAGE, each separated into two bands of inhibition (Fig. 8). The additional bands may simply be the result of pairs of inhibitors essentially co-migrating on the native gels. Alternatively, there is the possibility that inhibitors C, E, and/or F are non-covalently bonded heterodimers with each subunit being inhibitory. In any case no inhibitors were observed when 2-mercaptoethanol was used during electrophoresis, suggesting that disulfide bonds are essential to their structure and function.
Based solely on the molecular mass range they occupy, the SDF serine peptidase inhibitors (about 15–32 kDa) would seem to be too small to be members of the serpin superfamily [about 40–100 kDa (Kanost 1990; Schulze et al. 1994) with most 40–60 kDa (Carrell and Boswell 1986; Worrall et al. 1999; Jiang and Kanost 2000; Gan et al. 2001)]. They are, on the other hand, larger than many of the non-serpin serine peptidase inhibitors, which include at least nine families of small ‘canonical’ or ‘standard mechanism’ serine peptidase inhibitors (Bode and Huber 1992), hirudins, the nematode smapin family (Zang and Maizels 2001), and others (see, e.g., the review by Kanost 1999). Nevertheless, in their heat stability they are reminiscent of some of these non-serpin serine peptidase inhibitors (e.g., Yamashita and Eguchi 1987; Ramesh et al. 1988; Matsui and Eguchi 1991; Broadway 1993; Fröbius et al. 2000; Zang and Maizels 2001). It should also be recalled that our molecular mass estimates were made under non-reducing conditions and there is evidence that some non-serpin serine peptidase inhibitors, including certain members of the bovine pancreatic trypsin inhibitor (Kunitz) (Ramesh et al. 1988) and smapin (Cappello et al. 1995) families, migrate as dimers by non-reducing SDS-PAGE.
One function of smapins in parasitic nematodes appears to be protecting the parasite from proteolysis by the host (Martzen et al. 1985). In like manner, the serine peptidase inhibitors in SDF may, during digestion of prey, protect spider digestive enzymes from prey serine peptidases.
Acknowledgments
We thank Lauren Keil and Max Diem for their contributions during the early stages of the research. Thanks also to Dr. Cathy Tugmon (Augusta College, Augusta GA) for help in collecting some of the SDF used in this study. Grant R15DK/OD51291 from the National Institutes of Health provided support for this study.
References
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernfeld P. Amylases, α and β. Meth Enzymol. 1955;1:149–158. [Google Scholar]
- Bode W, Huber R. Natural protein proteinase inhibitors and their interaction with proteinases. Eur J Biochem. 1992;204:433–451. doi: 10.1111/j.1432-1033.1992.tb16654.x. [DOI] [PubMed] [Google Scholar]
- Broadway RM. Purification and partial characterization of trypsin/chymotrypsin inhibitors from cabbage foliage. Phytochemistry. 1993;33:21–27. [Google Scholar]
- Butler PE, McKay MJ, Bond JS. Characterization of meprin, a membrane-bound metalloendopeptidase from mouse kidney. Biochem J. 1987;241:229–235. doi: 10.1042/bj2410229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappello M, Vlasuk GP, Bergum PW, Huang S, Hotez PJ. Ancylostoma caninum anticoagulant peptide: A hookworm-derived inhibitor of human coagulation factor Xa. Proc Natl Acad Sci USA. 1995;92:6152–6156. doi: 10.1073/pnas.92.13.6152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrell, R.W., Boswell, D.R., 1986. Serpins: the superfamily of plasma serine proteinase inhibitors. In: Barrett, A.J., Salvesen, G. (Eds.), Proteinase Inhibitors. Elsevier, Amsterdam, pp. 403–420.
- Coughlin SA, Green DM. Two-dimensional zymogram analysis of nucleases in Bacillus subtilis. Anal Biochem. 1983;133:322–329. doi: 10.1016/0003-2697(83)90091-x. [DOI] [PubMed] [Google Scholar]
- da Silva PH, da Silveira RB, Appel MH, Mangili OC, Gremski W, Veiga SS. Brown spiders and loxoscelism. Toxicon. 2004;44:693–709. doi: 10.1016/j.toxicon.2004.07.012. [DOI] [PubMed] [Google Scholar]
- Foradori MJ, Keil LM, Wells RE, Diem M, Tillinghast EK. An examination of the potential role of spider digestive proteases as a causative factor in spider bite necrosis. Comp Biochem Physiol C. 2001;130:209–218. doi: 10.1016/s1532-0456(01)00239-3. [DOI] [PubMed] [Google Scholar]
- Foradori MJ, Kovoor J, Moon MJ, Tillinghast EK. Relation between the outer cover of the egg case of Argiope aurantia (Araneae: Araneidae) and the emergence of its spiderlings. J Morphol. 2002;252:218–226. doi: 10.1002/jmor.1100. [DOI] [PubMed] [Google Scholar]
- Fröbius AC, Kanost MR, Götz P, Vilcinskas A. Isolation and characterization of novel inducible serine protease inhibitors from larval hemolymph of the greater wax moth Galleria mellonella. Eur J Biochem. 2000;267:2046–2053. doi: 10.1046/j.1432-1327.2000.01207.x. [DOI] [PubMed] [Google Scholar]
- Gan H, Wang Y, Jiang H, Mita K, Kanost MR. A bacteria-induced, intracellular serpin in granular hemocytes of Manduca sexta. Insect Biochem Mol Biol. 2001;31:887–898. doi: 10.1016/s0965-1748(01)00034-0. [DOI] [PubMed] [Google Scholar]
- Geren, C.R., Odell, G.V., 1984. The biochemistry of spider venoms. In: Tu, A.T. (Ed.), Handbook of Natural Toxins, vol. 2. Insect Poisons, Allergens, and Other Invertebrate Venoms. Marcel Dekker, New York, pp. 441–481.
- Gorbea CM, Marchand P, Jiang W, Copeland NG, Gilbert DJ, Jenkins NA, Bond JS. Cloning, expression, and chromosomal localization of the mouse meprin β subunit. J Biol Chem. 1993;268:21035–21043. [PubMed] [Google Scholar]
- Hames, B.D., 1981. An introduction to polyacrylamide gel electrophoresis. In: Hames, B.D., Rickwood, D. (Eds.), Gel Electrophoresis of Proteins: A Practical Approach. IRL Press, London, pp. 1–91.
- Jackson, J.R.H., Krapcho, K.J., Johnson, J.H., Kral, R.M., Jr., 1995. Insecticidally effective spider toxin. US Patent 5457178.
- Jiang H, Kanost MR. The clip-domain family of serine proteinases in arthropods. Insect Biochem Mol Biol. 2000;30:95–105. doi: 10.1016/s0965-1748(99)00113-7. [DOI] [PubMed] [Google Scholar]
- Kanost MR. Isolation and characterization of four serine proteinase inhibitors (serpins) from hemolymph of Manduca sexta. Insect Biochem. 1990;20:141–147. [Google Scholar]
- Kanost MR. Serine proteinase inhibitors in arthropod immunity. Dev Comp Immunol. 1999;23:291–301. doi: 10.1016/s0145-305x(99)00012-9. [DOI] [PubMed] [Google Scholar]
- Kavanagh, E.J., 1979. The structure of the orb web and its degradation by spider digestive fluid proteases. Ph.D. Dissertation, University of New Hampshire.
- Kavanagh EJ, Tillinghast EK. The alkaline proteases of Argiope–II. Fractionation of protease activity and isolation of a silk fibroin digesting protease. Comp Biochem Physiol B. 1983;74:365–372. [Google Scholar]
- Kuhn-Nentwig L, Schaller J, Nentwig W. Purification of toxic peptides and the amino acid sequence of CSTX-1 from the multicomponent venom of Cupiennius salei (Araneae: Ctenidae) Toxicon. 1994;32:287–302. doi: 10.1016/0041-0101(94)90082-5. [DOI] [PubMed] [Google Scholar]
- Kunitz M. Crystalline soybean trypsin inhibitor–II. General properties J Gen Physiol. 1947;30:291–310. doi: 10.1085/jgp.30.4.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeGendre, N., Matsudaira, P., 1989. Purification of proteins and peptides by SDS-PAGE. In: Matsudaira, P.T. (Ed.), A Practical Guide to Protein and Peptide Purification for Microsequencing. Academic Press, San Diego, pp. 49–69.
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Marchler-Bauer A, Bryant SH. CD-Search: protein domain annotations on the fly. Nucleic Acids Res. 2004;32 (Web Server issue):W327–W331. doi: 10.1093/nar/gkh454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martzen MR, Geise GL, Hogan BJ, Peanasky RJ. Ascaris suum: Localization by immunochemical and fluorescent probes of host proteases and parasite proteinase inhibitors in cross-sections. Exp Parasitol. 1985;60:139–149. doi: 10.1016/0014-4894(85)90016-5. [DOI] [PubMed] [Google Scholar]
- Matsui Y, Eguchi M. Purification and relationship of chymotrypsin inhibitors from the fat body and haemolymph of the silkworm, Bombyx mori. Comp Biochem Physiol B. 1991;99:851–857. [Google Scholar]
- Mebs D. Proteolytische Aktivität eines Vogelspinnengiftes. Naturwissenschaften. 1970;57:308. doi: 10.1007/BF00609387. [DOI] [PubMed] [Google Scholar]
- Mebs, D., 1972. Proteolytic activity of a spider venom. In: DeVries, A., Kochova, E. (Eds.), Toxins of Animal and Plant Origin. Gordon & Breach, New York, pp. 493–497.
- Meyer KH, Fischer EH, Bernfeld P. Sur les enzymes amylolytiques (I). L’isolement de l’α-amylase de pancreas. Helvet Chim Acta. 1947;30:64–78. doi: 10.1002/hlca.19470300111. [DOI] [PubMed] [Google Scholar]
- Mommsen TP. Digestive enzymes of a spider (Tegenaria atrica Koch)–I. General remarks, digestion of proteins. Comp Biochem Physiol A. 1978a;60:365–370. [Google Scholar]
- Mommsen TP. Digestive enzymes of a spider (Tegenaria atrica Koch)–II. Carbohydrases Comp Biochem Physiol A. 1978b;60:371–375. [Google Scholar]
- Mommsen TP. Digestive enzymes of a spider (Tegenaria atrica Koch)–III. Esterases, phosphatases, nucleases. Comp Biochem Physiol A. 1978c;60:377–382. [Google Scholar]
- Mommsen TP. Comparison of digestive α-amylases from two species of spiders (Tegenaria atrica and Cupiennius salei) J Comp Physiol B. 1978d;127:355–361. [Google Scholar]
- Mommsen TP. Chitinase and β-N-acetylglucosaminidase from the digestive fluid of the spider, Cupiennius salei. Biochim Biophys Acta. 1980;612:361–372. doi: 10.1016/0005-2744(80)90119-9. [DOI] [PubMed] [Google Scholar]
- Perret BA. Proteolytic activity of tarantula venoms due to contamination with saliva. Toxicon. 1977;15:505–510. doi: 10.1016/0041-0101(77)90101-5. [DOI] [PubMed] [Google Scholar]
- Peters G, Saborowski R, Mentlein R, Buchholz F. Isoforms of an N-acetyl-β-d-glucosaminidase from the Antarctic krill, Euphausia superba: purification and antibody production. Comp Biochem Physiol B. 1998;120:743–751. doi: 10.1016/s0305-0491(98)10073-1. [DOI] [PubMed] [Google Scholar]
- Peterson GL. A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal Biochem. 1977;83:346–356. doi: 10.1016/0003-2697(77)90043-4. [DOI] [PubMed] [Google Scholar]
- Pfleiderer G, Zwilling R, Sonneborn HH. Eine Protease vom Molekulargewicht 11000 und eine trypsinähnliche Fraktion aus Astacus fluviatilis Fabr. Hoppe-Seyler’s. Z Physiol Chem. 1967;348:1319–1331. [PubMed] [Google Scholar]
- Pfleiderer G, Zwilling R. Die molekulare Evolution proteolytischer Enzyme. Naturwissenschaften. 1972;59:396–405. doi: 10.1007/BF00623129. [DOI] [PubMed] [Google Scholar]
- Pickford GE. Studies on the digestive enzymes of spiders. Trans Conn Acad Arts Sci. 1942;35:33–72. [Google Scholar]
- Ramesh N, Sugumaran M, Mole JE. Purification and characterization of two trypsin inhibitors from the hemolymph of Manduca sexta larvae. J Biol Chem. 1988;263:11523–11527. [PubMed] [Google Scholar]
- Rawlings ND, Barrett AJ. Evolutionary families of metallopeptidases. Meths Enzymol. 1995;248:183–228. doi: 10.1016/0076-6879(95)48015-3. [DOI] [PubMed] [Google Scholar]
- Rawlings ND, Tolle DP, Barrett AJ. MEROPS: the peptidase database. Nucleic Acids Res. 2004;32 (Database issue):D160–D164. doi: 10.1093/nar/gkh071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze AJ, Huber R, Bode W, Engh RA. Structural aspects of serpin inhibition. FEBS Letts. 1994;344:117–124. doi: 10.1016/0014-5793(94)00369-6. [DOI] [PubMed] [Google Scholar]
- Schumacher GFB, Schill WB. Radial diffusion in gel for micro determination of enzymes. II Plasminogen activator, elastase, and nonspecific proteases. Anal Biochem. 1972;48:9–26. doi: 10.1016/0003-2697(72)90165-0. [DOI] [PubMed] [Google Scholar]
- Shi, Q., Jackowski, G., 1998. One-dimensional polyacrylamide gel electrophoresis. In: Hames, B.D. (Ed.), Gel Electrophoresis of Proteins: A Practical Approach, 3rd ed. Oxford University Press, Oxford, pp. 1–52.
- Stöcker W, Breit S, Sottrup-Jensen L, Zwilling R. α2-Macroglobulin from the haemolymph of the freshwater crayfish Astacus astacus. Comp Biochem Physiol B. 1991a;98:501–509. doi: 10.1016/0305-0491(91)90244-8. [DOI] [PubMed] [Google Scholar]
- Stöcker W, Sauer B, Zwilling R. Kinetics of nitroanilide cleavage by astacin. Biol Chem Hoppe-Seyler. 1991b;372:385–392. doi: 10.1515/bchm3.1991.372.1.385. [DOI] [PubMed] [Google Scholar]
- Stöcker W, Zwilling R. Astacin. Meths Enzymol. 1995;248:305–325. doi: 10.1016/0076-6879(95)48021-8. [DOI] [PubMed] [Google Scholar]
- Tatusova TA, Madden TL. BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol Lett. 1999;174:247–250. doi: 10.1111/j.1574-6968.1999.tb13575.x. [DOI] [PubMed] [Google Scholar]
- Tillinghast EK, Kavanagh EJ. The alkaline proteases of Argiope and their possible role in web digestion. J Exp Zool. 1977;202:213–222. [Google Scholar]
- Townley MA, Tillinghast EK. Orb web recycling in Araneus cavaticus (Araneae, Araneidae) with an emphasis on the adhesive spiral component, GABamide. J Arachnol. 1988;16:303–319. [Google Scholar]
- Tugmon CR, Tillinghast EK. Proteases and protease inhibitors of the spider Argiope aurantia (Araneae, Araneidae) Naturwissenschaften. 1995;82:195–197. [Google Scholar]
- Uriel J, Berges J. Characterization of natural inhibitors of trypsin and chymotrypsin by electrophoresis in acrylamide-agarose gels. Nature. 1968;218:578–580. doi: 10.1038/218578b0. [DOI] [PubMed] [Google Scholar]
- Williams, K.R., Stone, K.L., 1995. In gel digestion of SDS PAGE-separated proteins: Observations from internal sequencing of 25 proteins. In: Crabb, J.W. (Ed.), Techniques in Protein Chemistry VI. Academic Press, Orlando, pp. 143–152.
- Wolz RL, Bond JS. Meprins A and B. Meths. Enzymol. 1995;248:325–345. doi: 10.1016/0076-6879(95)48022-6. [DOI] [PubMed] [Google Scholar]
- Worrall DM, Blacque OE, Barnes RC. The expanding superfamily of serpins: searching for the real targets. Biochem Soc Trans. 1999;27:746–750. doi: 10.1042/bst0270746. [DOI] [PubMed] [Google Scholar]
- Yamashita M, Eguchi M. Comparison of six genetically defined inhibitors from the silkworm haemolymph against fungal protease. Comp Biochem Physiol B. 1987;86:201–208. [Google Scholar]
- Young AR, Pincus SJ. Comparison of enzymatic activity from three species of necrotising arachnids in Australia: Loxosceles rufescens, Badumna insignis and Lampona cylindrata. Toxicon. 2001;39:391–400. doi: 10.1016/s0041-0101(00)00145-8. [DOI] [PubMed] [Google Scholar]
- Zang X, Maizels RM. Serine proteinase inhibitors from nematodes and the arms race between host and pathogen. Trends Biochem Sci. 2001;26:191–197. doi: 10.1016/s0968-0004(00)01761-8. [DOI] [PubMed] [Google Scholar]
- Zwilling, R., 1997. Astacin and the astacins: A re-view. In: Zwilling, R., Stöcker, W. (Eds.), The Astacins: Structure and Function of a New Protein Family. Verlag Dr. Kovac, Hamburg, pp. 11–28.
- Zwilling R, Neurath H. Invertebrate Proteases. Meths Enzymol. 1981;80(C):633–664. [Google Scholar]