

Abstract
All histidine tRNA molecules have an extra nucleotide, G-1, at the 5′ end of the acceptor stem. In bacteria, archaea, and eukaryotic organelles, G-1 base pairs with C73, while in eukaryotic cytoplasmic tRNAHis, G-1 is opposite A73. Previous studies of Escherichia coli histidyl-tRNA synthetase (HisRS) have demonstrated the importance of the G-1:C73 base pair to tRNAHis identity. Specifically, the 5′-monophosphate of G-1 and the major groove amine of C73 are recognized by E. coli HisRS; these individual atomic groups each contribute ∼4 kcal/mol to transition state stabilization. In this study, two chemically synthesized 24-nucleotide RNA microhelices, each of which recapitulates the acceptor stem of either E. coli or Saccharomyces cervisiae tRNAHis, were used to facilitate an atomic group “mutagenesis” study of the −1:73 base pair recognition by S. cerevisiae HisRS. Compared with E. coli HisRS, microhelixHis is a much poorer substrate relative to full-length tRNAHis for the yeast enzyme. However, the data presented here suggest that, similar to the E. coli system, the 5′ monophosphate of yeast tRNAHis is critical for aminoacylation by yeast HisRS and contributes ∼3 kcal/mol to transition state stability. The primary role of the unique −1:73 base pair of yeast tRNAHis appears to be to properly position the critical 5′ monophosphate for interaction with the yeast enzyme. Our data also suggest that the eukaryotic HisRS/tRNAHis interaction has coevolved to rely less on specific major groove interactions with base atomic groups than the bacterial system.
Keywords: yeast histidine-tRNA synthetase, tRNA recognition, acceptor stem microhelix, extra base pair, phosphate recognition, atomic group mutagenesis
INTRODUCTION
Efficient and accurate aminoacylation of transfer RNAs (tRNA) by aminoacyl-tRNA synthetases (aaRSs) is critical to protein translation. The fidelity of the aminoacylation reaction is governed by the synthetases’ ability to discriminate among the pool of cellular amino acids and tRNAs, and then join the appropriate partners together. Discrimination of tRNAs involves unique sets of RNA–protein interactions, which give rise to tRNA identity (Normanly and Abelson 1989). The tRNA elements most commonly recognized by synthetases include the tRNA acceptor stem, the anticodon loop, and the tRNA tertiary core (McClain 1993; Giegé et al. 1998). Among the interactions that stabilize tRNA–aaRS complexes are base-specific atomic group contacts or backbone contacts to the phosphate oxygens or ribose 2′-hydroxyl groups (Beuning and Musier-Forsyth 1999); many aaRSs also appear to recognize tRNAs based on their overall shape (Tukalo et al. 2005). The precise mechanism by which recognition of specific identity elements promotes efficient aminoacylation remains to be established, but it likely involves rate enhancement of catalytic steps (as indicated by effects on k cat), as opposed to stabilization of tRNA binding (Ebel et al. 1973).
Histidine tRNA represents one particular system for which the molecular basis of tRNA identity and recognition has been investigated in detail (Francklyn and Arnez 2005). All histidyl-tRNA molecules are distinguished by an extra nucleotide at the 5′ end of the acceptor stem, G-1, which is located opposite to the “discriminator” base, N73 (Crothers et al. 1972; Sprinzl and Vassilenko 2005). G-1 is encoded in the tRNAHis genes of bacteria (Sprinzl and Vassilenko 2005) but is added post-transcriptionally in eukarya (Williams et al. 1990) by a specific guanyl-transferase that is conserved in eukaryotes (Gu et al. 2003,2005). In bacteria, archaea, and eukaryotic organelles, N73 is a cytosine, which creates a canonical Watson-Crick G-1:C73 base pair (Sprinzl and Vassilenko 2005). However, in eukaryotic cytoplasmic histidine tRNAs, the extra base pair is a G-1:A73 mismatch.
Previous studies of Escherichia coli histidyl-tRNA synthetase (HisRS) have demonstrated the importance of the G-1:C73 base pair to tRNAHis identity (Himeno et al. 1989; Yan and Francklyn 1994; Yan et al. 1996). Specifically, the 5′-monophosphate of G-1 and the major groove amine of C73 are recognized by E. coli HisRS (Allen and Parsons 1977; Fromant et al. 2000; Rosen and Musier-Forsyth 2004); these individual atomic groups each contribute ∼4 kcal/mol to transition state stabilization (Rosen and Musier-Forsyth 2004). The requirement for a 5′ monophosphate appears to be unique to the interactions between HisRS and tRNAHis isoacceptors (Allen and Parsons 1977). Saccharomyces cerevisiae HisRS also recognizes the extra base pair (Rudinger et al. 1994, 1997; Nameki et al. 1995; Hawko and Francklyn 2001). Standard base substitutions at G-1:A73 of yeast tRNAHis result in reduced aminoacylation efficiency, but the effects are generally less severe than those observed in E. coli (Nameki et al. 1995). It was proposed that yeast HisRS does not make functional interactions with the G-1 or A73 bases (Nameki et al. 1995). However, deletion of G-1 resulted in a 470- to 740-fold decrease in activity relative to wild-type tRNAHis (Rudinger et al. 1994; Nameki et al. 1995).
Taken together, these data suggest that the backbone functional groups of G-1 may play a role in recognition by the yeast enzyme. To better understand how tRNA recognition properties of the yeast HisRS enzyme differ from those of the E. coli enzyme, the atomic group mutagenesis approach was applied to the yeast microhelixHis system. The results of these studies suggest that, although both yeast and E. coli HisRS exhibit specificity for the same regions of the tRNA, contacts to individual functional groups in the acceptor stem of the tRNA contribute less to stabilization of the transition state. In the yeast system, the acceptor stem does not make as large a contribution to the recognition of the entire tRNA as is observed in the prokaryotic system.
RESULTS
Initial characterization of yeast HisRS
Prior to aminoacylation studies, the yeast histidyl-tRNA synthetase was cloned and expressed in E. coli, as described in the Materials and Methods. The protein was purified by affinity purification, employing a His6 affinity tag appended to the N terminus of the cytoplasmic version of the protein. The cytoplasmic version of yeast HisRS lacks the 60-amino-acid leader sequence that is involved in mitochondrial import (Natsoulis et al. 1986). To perform an initial characterization and confirmation of the activity of this enzyme, the kinetics of the pyrophosphate exchange reaction were measured. As expected, the enzyme exhibited straightforward Michaelis Menten kinetics under conditions of constant histidine/variable ATP and constant ATP/variable histidine (data not shown). As shown in Table 1, the kinetic parameters for pyrophosphate exchange were as follows: K M His = 9.0 ± 1 μM; k cat = 40 ± 9 sec−1; K M ATP = 500 ± 94 μM. In comparison to the values for the E. coli enzyme reported previously (Augustine and Francklyn 1997; Francklyn et al. 1998), the yeast enzyme has an approximately threefold lower K M (histidine), and k cat is also reduced threefold (Table 1). This analysis suggests that the yeast enzyme purified from bacterial sources retains full activity, allowing further studies of its tRNA specificity.
TABLE 1.
Comparison of pyrophosphate exchange and aminoacylation activities for S. cerevisiae and E. coli histidyl-tRNA synthetases

Comparison of full-length and microhelix substrates based on tRNAHis
Previous studies involving yeast HisRS focused on a comparison of the role of specific nucleotides in the cognate tRNA (Nameki et al. 1995) but did not critically compare the activities of E. coli and yeast enzymes purified to homogeneity. In the work presented here, steady-state aminoacylation assays employing modified S. cerevisiae tRNA were performed, allowing the Michaelis constant for tRNA and k cat for aminoacylation to be determined. The results of these experiments (Table 1) showed that both enzymes exhibit comparable catalytic efficiencies (k cat/K M) in the aminoacylation of tRNA, with yeast HisRS exhibiting ∼2.4-fold and fourfold reductions in K M and k cat, respectively. This provides additional evidence that the two enzymes are of comparable overall activity, and that production of the yeast enzyme by expression in bacteria is not associated with the absence of a critical post-translational modification.
E. coli HisRS is notable for its ability to aminoacylate small helical RNAs (referred in previous literature as microhelixHis) that recapitulate the acceptor stem of tRNAHis at rates that are ∼700 fold slower than the full-length tRNA (Francklyn and Schimmel 1990; Francklyn et al. 1992; Augustine and Francklyn 1997). Prior to a detailed analysis of individual functional groups, the yeast enzyme was tested for aminoacylation of a 23-nucleotide, chemically synthesized microhelixHis that mimics the acceptor stem of S. cerevisiae tRNAHis (Fig. 1). As shown in Table 1, compared to the E. coli system, the k cat for aminoacylation of the yeast microhelix was 20-fold slower and the K M 32-fold higher, indicating that the yeast enzyme is considerably less active in the aminoacylation of small helical substrates than the E. coli enzyme. This is consistent with prior observations that the yeast enzyme relies to a greater extent on interactions outside of the acceptor stem than the E. coli enzyme (Nameki et al. 1995). To broaden the range of potential substrates, the activity of yeast HisRS on microhelices based on the acceptor stems of E. coli and yeast histidyl-tRNAs was compared. The sequences of these substrates are shown in Figure 1. Both RNA helices were substrates for the yeast enzyme, and the E. coli microhelix was only 3.3-fold reduced in aminoacylation efficiency relative to yeast microhelixHis. This result is in good agreement with the small 1.1-fold decrease observed upon substitution of G-1:C73 into yeast microhelixHis (Table 2), and with the fact that yeast HisRS aminoacylates yeast mitochondrial tRNAHis. The mitochondrial tRNA contains the same four base pairs at the top of the acceptor stem as the E. coli microhelix (Fig. 1). Thus, to facilitate identification of atomic group determinants for recognition by yeast HisRS, substitutions were made in the context of both the E. coli and yeast microhelices.
FIGURE 1.

Secondary structures of unmodified S. cerevisiae tRNAHis and the chemically synthesized yeast and E. coli microhelixHis substrates used in this work. The shaded boxes highlight the −1:73 base pair. Also depicted is the acceptor stem of the S. cerevisiae mitochondrial tRNAHis to show its similarity to E. coli microhelixHis.
TABLE 2.
Yeast HisRS aminoacylation of microhelixHis variants
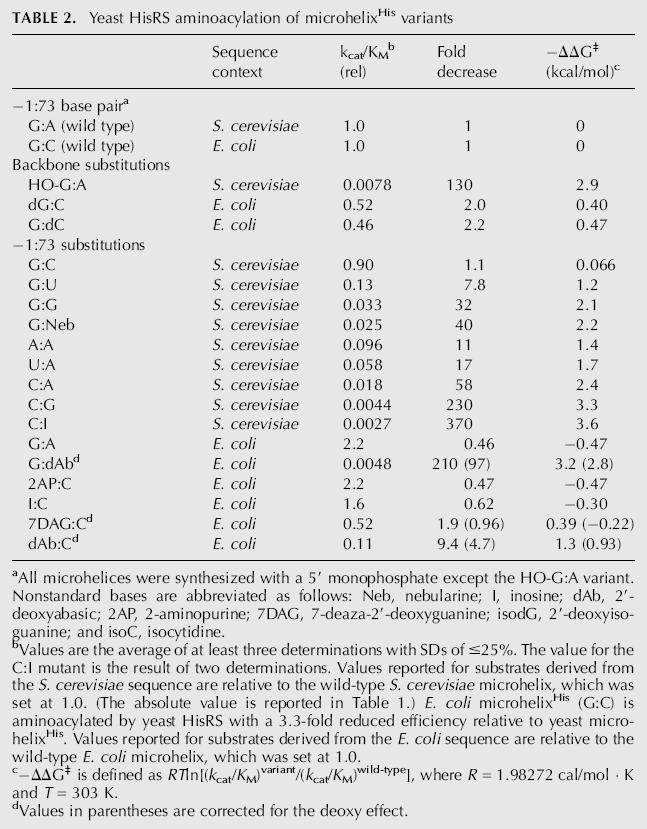
aAll microhelices were synthesized with a 5′ monophosphate except the HO-G:A variant. Nonstandard bases are abbreviated as follows: Neb, nebularine; I, inosine; dAb, 2′-deoxyabasic; 2AP, 2-aminopurine; 7DAG, 7-deaza-2′-deoxyguanine; isodG, 2′-deoxyisoguanine; and isoC, isocytidine.
bValues are the average of at least three determinations with SDs of ≤25%. The value for the C:I mutant is the result of two determinations. Values reported for substrates derived from the S. cerevisiae sequence are relative to the wild-type S. cerevisiae microhelix, which was set at 1.0. (The absolute value is reported in Table 1.) E. coli microhelixHis (G:C) is aminoacylated by yeast HisRS with a 3.3-fold reduced efficiency relative to yeast microhelixHis. Values reported for substrates derived from the E. coli sequence are relative to the wild-type E. coli microhelix, which was set at 1.0.
c−ΔΔG‡ is defined as RTln[(k cat/K M)variant/(k cat/K M)wild-type], where R = 1.98272 cal/mol ;· ;K and T = 303 K.
dValues in parentheses are corrected for the deoxy effect.
Functional group analysis of acceptor stem nucleotide groups in the context of microhelixHis
Previous studies indicated that the phosphodiester backbone and the 5′ phosphate in particular are important for tRNAHis recognition by E. coli HisRS in the context of both the full-length and microhelixHis (Fromant et al. 2000; Rosen and Musier-Forsyth 2004). To explore whether the yeast HisRS possesses comparable specificity, backbone recognition was probed by incorporating deoxynucleotide analogs at positions −1 and 73 of E. coli microhelixHis. Deletion of each of the 2′-hydroxyl groups resulted in a small, approximately twofold reduction in aminoacylation by yeast HisRS (−ΔG‡ = 0.40–0.47 kcal/mol) (Table 2). In contrast, the 5′ monophosphate of yeast microhelixHis contributes 2.9 kcal/mol to transition state stabilization (HO-G:A) (Table 2). Thus, as for E. coli HisRS, the 5′ monophosphate is a major recognition element, although the magnitude of its contribution to catalysis is smaller.
Standard base substitutions at the −1 position of yeast microhelixHis resulted in 11- to 58-fold decreases in k cat/K M, in good agreement with previous results (Table 2; Rudinger et al. 1994; Nameki et al. 1995). We next specifically probed the role of major and minor groove functional groups of the −1:73 base pair by incorporating modified bases into E. coli microhelixHis. The lack of an effect upon deletion of the N7 nitrogen (7DAG:C) and the increased activity observed upon deletion of the 6-keto oxygen (2AP:C) confirm that the major groove of G-1 is not directly recognized by yeast HisRS (Table 2; Fig. 2A). An inosine (I) substitution, which probed the importance of the minor groove amine at the −1 position (Fig. 2A), also resulted in a small increase in k cat/K M relative to the wild-type E. coli microhelix (I:C, Table 2). Deletion of the guanine base to generate a dAbasic (dAb):C variant (Fig. 2A) resulted in only a 4.7-fold decrease in aminoacylation efficiency. This substitution, which retains the critical 5′ monophosphate, demonstrates that the base moiety at G-1 is dispensable for efficient aminoacylation by S. cerevisiae HisRS.
FIGURE 2.

Proposed structures of the base pairs substituted at the −1:73 position of E. coli and S. cerevisiae microhelixHis. (A) Substitutions that were made at position G-1 in the context of E. coli microhelixHis. (B) Substitutions that were made at position A73 of S. cerevisiae microhelixHis, except for the G:dAb substitution, which was made in the E. coli microhelixHis. Numbers in parentheses are fold decrease in k cat/K M relative to wild type. Values indicated with a double dagger (‡) were corrected for a deoxy effect.
The base at position 73 of all histidyl-tRNAs possesses an evolutionarily conserved major groove amine (Sprinzl and Vassilenko 2005). We hypothesized that it might serve as a major recognition element for S. cerevisiae HisRS. However, in contrast to results obtained with E. coli HisRS (Rosen and Musier-Forsyth 2004) but in good agreement with data previously reported for full-length yeast tRNAHis (Nameki et al. 1995), a U73 substitution in the yeast microhelix only moderately reduced (7.8-fold) the catalytic efficiency of yeast HisRS (Table 2; Fig. 2B). This result suggests that the major groove amine at position 73 contributes at most 1.2 kcal/mol to transition state stability. Deletion of the cytosine base moiety at position 73 of E. coli microhelixHis contributed >4.2 kcal/mol to aminoacylation by E. coli HisRS. However, a G-1:dAb73 substitution in the E. coli microhelix resulted in only a ∼2.8 kcal/mol increase in the free energy of activation in the yeast system. G:nebularine and G:G variants of the yeast microhelix provide additional evidence for only modest recognition of the major groove at position 73 (Fig. 2B). These substitutions result in 40- and 32-fold decreases in aminoacylation activity, respectively (−ΔG‡ ≈ 2.2 kcal/mol) (Table 2). Substitution of a C-1:G73 base pair results in a large 230-fold decrease in aminoacylation of yeast microhelixHis, and a C-1:I73 substitution suggests that this decrease is not due to the negative effect of a minor groove amine at position 73 (Table 2). At least part of the negative effect of a C:G substitution can be compensated by changing the base pair orientation to purine:pyrimidine, as an isodG-1:isoC73 variant E. coli microhelixHis, which projects the same major and minor groove functional groups as C:G, was only 37-fold reduced relative to the dG:C E. coli microhelix. In contrast, the isodG:isoC microhelix was not a substrate for E. coli HisRS (Rosen and Musier-Forsyth 2004).
DISCUSSION
The HisRS/tRNAHis system is notable for the significant effect that a single base pair (G-1:C73 in prokaryotes, G-1:A73 in eukaryotes) exerts on tRNA recognition, both in the context of full length and small helical substrates. By use of chemically synthesized 23-nucleotide microhelices, we showed that S. cerevisiae HisRS is capable of aminoacylating a minimal substrate that mimics the tRNAHis acceptor stem. This suggests that, similar to the E. coli system (Francklyn and Schimmel 1990), the acceptor stem contributes significantly to recognition by the yeast synthetase. However, the microhelix proved to be a much poorer substrate for the S. cerevisiae enzyme than for the E. coli enzyme. This difference is not due to the absence of modified bases in the microhelix, as a yeast tRNAHis transcript displayed similar aminoacylation activity as fully modified S. cerevisiae tRNAHis (data not shown). Quantitatively, the yeast microhelixHis was 30,000-fold less active than the full-length yeast tRNA, while the E. coli microhelix is only 730-fold less active than its cognate tRNA. This indicates that in the yeast system, tRNA structural elements outside of the acceptor stem play a more important role in tRNA recognition than they do in the E. coli system. In energetic terms, the difference is significant: For the E. coli system, the microhelix provides 11.2 of 14.4 kcal/mol of free energy (Francklyn et al. 1992), but in the yeast system only 8.2 kcal/mol (this work). Notably, the decreased specificity for the acceptor stem observed in the yeast system was not accompanied by increased specificity for the anticodon, as substitutions of the anticodon nucleotides in yeast tRNAHis and viral RNA substrates resulted in relatively minor (≤70-fold) effects on aminoacylation efficiency in vitro (Nameki et al. 1995; Rudinger et al. 1997; Felden and Giegé 1998).
The amino group that projects into the major groove and is common to both cytosine and adenine can be considered to be an evolutionarily conserved functional group at position 73 of all tRNAHis molecules (Sprinzl and Vassilenko 2005). We hypothesized that it might serve as a major recognition element for S. cerevisiae HisRS, providing a rationale for the ability of the synthetase to aminoacylate both cytoplasmic G-1:A73 tRNAHis as well as mitochondrial G-1:C73 tRNAHis (Natsoulis et al. 1986; Chiu et al. 1992). However, in contrast to results obtained with the E. coli enzyme (Rosen and Musier-Forsyth 2004), the major groove amine at position 73 contributes at most ∼1 kcal/mol to catalysis. The decreased contribution made by the amine group to aminoacylation by yeast HisRS likely constitutes at least part of the basis of reduced activity of the yeast microhelixHis. As observed for the E. coli system, substitutions at position 73 were generally more detrimental to efficient aminoacylation by the yeast enzyme than changes at position −1. In fact, complete elimination of the −1 base moiety only resulted in a ∼1 kcal/mol effect on catalysis. Also, similar to the E. coli system (Rosen and Musier-Forsyth 2004), the 5′ monophosphate of yeast tRNAHis is critical and contributes ∼3 kcal/mol to transition state stability. Therefore, the primary role of the unique −1:73 base pair of yeast tRNAHis may be to properly position the critical 5′ monophosphate for interaction with the yeast enzyme. Our data also suggest that the eukaryotic HisRS/tRNAHis interaction has coevolved to rely less on specific major groove interaction with base atomic groups than the bacterial system.
The selection of tRNA by aaRSs involves discrimination at the level of initial binding (Lam and Schimmel 1975) and at the level of Vmax (Ebel et al. 1973). Previous studies indicate that substitutions of the G-1:C73 determinant in the histidine system generally impose little or no effect on K M in the kinetics of aminoacylation (Himeno et al. 1989; Yan et al. 1996), or on the apparent dissociation constant (KD), as determined by a competitive filter binding analysis (Bovee et al. 1999). While a crystallographic model of the HisRS:tRNAHis complex is not yet available, phylogenetics (Ardell and Andersson 2006), mutagenesis (Hawko and Francklyn 2001; Connolly et al. 2004), and comparative modeling with the AspRS:tRNAAsp complex all support the prediction that the motif 2 loop of HisRS provides critical side-chains for reading out the identity/recognition elements on the tRNAHis acceptor stem. By analogy to seryl-tRNA synthetase (Cusack et al. 1996), which belongs to the same subclass of synthetases as HisRS (i.e., class IIa) (Cusack et al. 1991), these side-chains are likely to undergo a small but functionally significant conformational change between the histidinol-ATP bound state and the tRNA bound state. On the basis of published mutational analyses (Hawko and Francklyn 2001; Connolly et al. 2004), and the observed covariation between the motif 2 loop and the −1:73 base pair (Ardell and Andersson 2006), the interactions most critical for specificity in the prokaryotic HisRS system appear to be between Arg123 and the 5′ phosphate, and between Gln118 and G-1:C73 (Connolly et al. 2004). The substitution of Gln118 by a tripeptide of hydrophobic residues in the yeast and other eukaryotic HisRS systems may indicate a diminished role for polar contacts to G-1:A73. Fine-tuning the relative contributions of the anticodon and the acceptor stem to discrimination could serve as an important component of the differentiation between prokaryotic and eukaryotic systems and could limit cross-species mischarging.
Interactions with acceptor stem determinants may exert their greatest effect on elementary steps that follow the initial tRNA binding steps, including the accommodation of the acceptor stem into the active site. Early biophysical work (for review, see Schimmel and Söll 1979) on the yeast seryl and phenylalanyl systems suggested that cognate complexes are distinguished by a two-step tRNA binding process in which the initial encounter is followed by a unimolecular conformational change that provides thermodynamic specificity at the level of binding. In contrast, noncognate complexes exhibited a binding event distinguished by a single relaxation that lacked the isomerization step. These fluorescence and temperature jump data are consistent with models describing the multistep nature of tRNA binding events in class II aaRSs, proposed on the basis of systematic analysis of homologous and heterologous complexes in the AspRS system (Eiler et al. 1999; Briand et al. 2000; Moulinier et al. 2001). In these models, an initial encounter complex that is dominated by interactions with the tRNA anticodon undergoes a progressive docking or “zippering” event to ultimately position the acceptor end for aminoacyl transfer. In heterologous complexes, the failure to complete this sequence of events prevents the acceptor end from achieving correct positioning in the active site and leaves the CCA end in a configuration incompatible with transfer (Moulinier et al. 2001). Recent studies indicate that for the wild-type E. coli histidine system at least, aminoacyl transfer proceeds faster than the overall rate of aminoacylation, suggesting that activation of the amino acid limits k cat (Guth et al. 2005). However, mutations in identity elements in the tRNA or in the side-chains that contact these elements alter reaction kinetics such that transfer becomes rate limiting (E. Guth and C.S. Francklyn, in prep.). A straightforward interpretation of these observations is that substitutions at identity elements compromise the mutual conformational changes in tRNA and aaRS required to bring the terminal (A76) ribose into position for transfer, such that orientation of CCA becomes the rate-limiting step. Future experiments that correlate the rates of these elementary steps with structural changes in both macromolecular partners will be necessary to complete the definition of the molecular basis of tRNA specificity.
MATERIALS AND METHODS
Chemicals
All RNA synthesis chemicals and standard bases were purchased from Glen Research. Modified bases from Glen Research included dAbasic, 7-deaza-3′-deoxyguanine, 2-aminopurine, and 2′-deoxyisoguanine. All other modified bases, including isoC, nebularine, and inosine, were from Chemgenes. L- [U-14C] histidine (314 mCi/mmol) was from Amersham Biosciences.
RNA preparation
Solid-phase synthesis of RNA microhelices was accomplished by using the phosphoramidite method on an Expedite 8909 Nucleic Acid Synthesis System (Applied Biosystems) (Scaringe et al. 1990; Sproat et al. 1995). All microhelices were synthesized using 2′-O-triisopropylsilyloxymethyl (TOM)-protected standard base phosphoramidites (Wu and Pitsch, 1998). The 2-aminopurine phosphoramidite was also TOM-protected. All microhelices were synthesized with a 5′ monophosphate unless otherwise noted. RNA oligonucleotides were purified on 16% polyacrylamide gels, eluted, and desalted as described (Scaringe et al. 1990; Sproat et al. 1995). Kinetic measurements involving full-length tRNAHis were performed by using a commercially available preparation of total tRNA from S. cerevisiae (Sigma). The concentration of histidine specific acceptor in the preparation was determined from aminoacylation plateau measurements employing [14C]-histidine as substrate.
Preparation of S. cerevisiae HisRS
Yeast HisRS was purified from an E. coli overexpression strain by use of Ni-NTA chromatography, using the basic approach described previously (Yan et al. 1996). The full details of the cloning, overexpression, and purification will be published elsewhere. Briefly, the HTS1 gene from S. cerevisiae (Natsoulis et al. 1986) was amplified from yeast genomic DNA by the polymerase chain reaction, using complementary primers to allow insertion of the fragment into the vector pET-Blue2 (Novagen). The resulting expression plasmid, which encodes His6 affinity tag at the N terminus of HisRS, was propagated in the BL-21 Rosetta background (Novagen) to provide rare tRNAs for heterologous expression. The purification of the protein by Ni-NTA chromatography was accomplished essentially as described (Yan et al. 1996). The enzyme was eluted from the nickel column by using a 30–200 mM imidazole gradient. Fractions containing the protein were identified by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, pooled, concentrated, and then stored in a final buffer of 20 mM Tris (pH 7.9), 100 mM NaCl, 5 mM dithiothreitol, and 50% glycerol.
Pyrophosphate exchange and aminoacylation assays
Pyrophosphate exchange assays were carried out by determining the rate of exchange of 32PPi label into ATP and then trapping the product on charcoal (Calendar and Berg 1966). The protocol for this assay was described previously (Francklyn et al. 1998) and was used without modification. Reactions were initiated by the addition of yeast HisRS to a final concentration of 8.2 nM. The HisRS concentrations were determined by active site titration (Fersht et al. 1975). RNA microhelices (5–15 μM) were annealed immediately before use by incubating for 2 min at 80°C, cooling for 2 min to 60°C, adding MgCl2 to 10 mM, cooling for 5 min to 25°C, and placing on ice. Aminoacylation assays were performed at 30°C as previously described (Francklyn and Schimmel 1990) in a buffer containing 50 mM HEPES buffer (pH 7.5), 20 mM β-mercaptoethanol, 20 mM KCl, 10 mM MgCl2, 4 mM ATP, and 26.2 μM [3H]histidine (Francklyn and Schimmel 1990). Microhelices and all other assay components were pre-equilibrated to 30°C before initiating the reaction by addition of 0.33 μM HisRS. Initial rates of aminoacylation are proportional to RNA concentration under the conditions used for these experiments. Thus, Vo/[S] is an accurate reflection of k cat/K M. The assays involving full-length tRNA used slightly different conditions (50 mM HEPES buffer at pH 7.5, 4 mM DTT, 20 mM KCl, 10 mM MgCl2, 2.5 mM ATP, 40 μM [3H]histidine) and were initiated by using 10 nM yeast HisRS.
ACKNOWLEDGMENTS
This work was supported by NIH grants GM49928 (K.M.-F.) and GM54899 (C.S.F.). A.E.R. and E.G. were supported by NIH Predoctoral Training Grants T32-GM08700 and T32-ES07122, respectively.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.78606.
REFERENCES
- Allen J.D., Parsons S.M. Properties of the 5′-terminus of tRNAHis: Kinetics of polynucleotide kinase catalyzed exchange and effect of dephosphorylation on the aminoacylation reaction. Biochem. Biophys. Res. Commun. 1977;78:28–35. doi: 10.1016/0006-291x(77)91217-7. [DOI] [PubMed] [Google Scholar]
- Ardell D.H., Andersson S.G. TFAM detects co-evolution of tRNA identity rules with lateral transfer of histidyl-tRNA synthetase. Nucleic Acids Res. 2006;34:893–904. doi: 10.1093/nar/gkj449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine J., Francklyn C. Design of an active fragment of a class II aminoacyl-tRNA synthetase and its significance for synthetase evolution. Biochemistry. 1997;36:3473–3482. doi: 10.1021/bi962395y. [DOI] [PubMed] [Google Scholar]
- Beuning P.J., Musier-Forsyth K. Transfer RNA recognition by aminoacyl-tRNA synthetases. Biopolymers. 1999;52:1–28. doi: 10.1002/(SICI)1097-0282(1999)52:1<1::AID-BIP1>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Bovee M.L., Yan W., Sproat B.S., Francklyn C.S. tRNA discrimination at the binding step by a class II aminoacyl-tRNA synthetase. Biochemistry. 1999;38:13725–13735. doi: 10.1021/bi991182g. [DOI] [PubMed] [Google Scholar]
- Briand C., Poterszman A., Eiler S., Webster G., Thierry J., Moras D. An intermediate step in the recognition of tRNA(Asp) by aspartyl-tRNA synthetase. J. Mol. Biol. 2000;299:1051–1060. doi: 10.1006/jmbi.2000.3819. [DOI] [PubMed] [Google Scholar]
- Calendar R., Berg P. The catalytic properties of tyrosyl ribonucleic acid synthases from Escherichia coli and Bacillus subtilis . Biochemistry. 1966;5:1690–1695. doi: 10.1021/bi00869a034. [DOI] [PubMed] [Google Scholar]
- Chiu M.I., Mason T.L., Fink G.R. HTS1 encodes both the cytoplasmic and mitochondrial histidyl-tRNA synthetase of Saccharomyces cerevisiae: Mutations alter the specificity of compartmentation. Genetics. 1992;132:987–1001. doi: 10.1093/genetics/132.4.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly S.A., Rosen A.E., Musier-Forsyth K., Francklyn C.S. G-1:C73 recognition by an arginine cluster in the active site of Escherichia coli histidyl-tRNA synthetase. Biochemistry. 2004;43:962–969. doi: 10.1021/bi035708f. [DOI] [PubMed] [Google Scholar]
- Crothers D.M., Seno T., Söll D.G. Is there a discriminator site in transfer RNA? Proc. Natl. Acad. Sci. 1972;69:3063–3067. doi: 10.1073/pnas.69.10.3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusack S., Hartlein M., Leberman R. Sequence, structural and evolutionary relationships between class 2 aminoacyl-tRNA synthetases. Nucleic Acids Res. 1991;19:3489–3498. doi: 10.1093/nar/19.13.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusack S., Yaremchuk A., Tukalo M. The crystal structure of the ternary complex of T. thermophilus seryl-tRNA synthetase with tRNASer and a seryl-adenylate analogue reveals a conformational switch in the active site. EMBO J. 1996;15:2834–2842. [PMC free article] [PubMed] [Google Scholar]
- Ebel J.P., Giegé R., Bonnet J., Kern D., Befort N., Bollack C., Fasiolo F., Gangloff J., Dirheimer G. Factors determining the specificity of the tRNA aminoacylation reaction: Non-absolute specificity of tRNA-aminoacyl-tRNA synthetase recognition and particular importance of the maximal velocity. Biochimie. 1973;55:547–557. doi: 10.1016/s0300-9084(73)80415-8. [DOI] [PubMed] [Google Scholar]
- Eiler S., Dock-Bregeon A., Moulinier L., Thierry J.C., Moras D. Synthesis of aspartyl-tRNA(Asp) in. A snapshot of the second step. EMBO J . Escherichia coli: 1999;18:6532–6541. doi: 10.1093/emboj/18.22.6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felden B., Giegé R. Resected RNA pseudoknots and their recognition by histidyl-tRNA synthetase. Proc. Natl. Acad. Sci. 1998;95:10431–10436. doi: 10.1073/pnas.95.18.10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fersht A.R., Ashford J.S., Bruton C.J., Jakes R., Koch G.L.E., Hartley B.S. Active site titration and aminoacyl adenylate binding stoichiometry of aminoacyl-tRNA synthetases. Biochemistry. 1975;14:1–4. doi: 10.1021/bi00672a001. [DOI] [PubMed] [Google Scholar]
- Francklyn C., Arnez J. Histidyl-tRNA synthetases. In: Ibba M., et al., editors. The aminoacyl-tRNA synthetases. Landes Bioscience; Georgetown, TX: 2005. [Google Scholar]
- Francklyn C., Schimmel P. Enzymatic aminoacylation of an eight-base-pair microhelix with histidine. Proc. Natl. Acad. Sci. 1990;87:8655–8659. doi: 10.1073/pnas.87.21.8655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francklyn C., Shi J.-P., Schimmel P. Overlapping nucleotide determinants for specific aminoacylation of RNA microhelices. Science. 1992;255:1121–1125. doi: 10.1126/science.1546312. [DOI] [PubMed] [Google Scholar]
- Francklyn C., Adams J., Augustine J. Catalytic defects in mutants of class II histidyl-tRNA synthetase from Salmonella typhimurium previously linked to decreased control of histidine biosynthesis regulation. J. Mol. Biol. 1998;280:847–858. doi: 10.1006/jmbi.1998.1902. [DOI] [PubMed] [Google Scholar]
- Fromant M., Plateau P., Blanquet S. Function of the extra 5′-phosphate carried by histidine tRNA. Biochemistry. 2000;39:4062–4067. doi: 10.1021/bi9923297. [DOI] [PubMed] [Google Scholar]
- Giegé R., Sissler M., Florentz C. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 1998;26:5017–5035. doi: 10.1093/nar/26.22.5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W., Jackman J.E., Lohan A.J., Gray M.W., Phizicky E.M. tRNAHis maturation: An essential yeast protein catalyzes addition of a guanine nucleotide to the 5′ end of tRNAHis . Genes & Dev. 2003;17:2889–2901. doi: 10.1101/gad.1148603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W., Hurto R.L., Hopper A.K., Grayhack E.J., Phizicky E.M. Depletion of Saccharomyces cerevisiae tRNAHis guanylyltransferase Thg1p leads to uncharged tRNAHis with additional m5C. Mol. Cell. Biol. 2005;25:8191–8201. doi: 10.1128/MCB.25.18.8191-8201.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guth E., Connolly S.H., Bovee M.L., Francklyn C. A substrate-assisted concerted mechanism for aminoacylation by a class II aminoacyl-tRNA synthetase. Biochemistry. 2005;44:3785–3794. doi: 10.1021/bi047923h. [DOI] [PubMed] [Google Scholar]
- Hawko S.A., Francklyn C.S. Covariation of a specificity-determining structural motif in an aminoacyl-tRNA synthetase and a tRNA identity element. Biochemistry. 2001;40:1930–1936. doi: 10.1021/bi0025316. [DOI] [PubMed] [Google Scholar]
- Himeno H., Hasegawa T., Ueda T., Watanabe K., Miura K.-I., Shimizu M. Role of the extra G-C pair at the end of the acceptor stem of tRNAHis in aminoacylation. Nucleic Acids Res. 1989;17:7855–7863. doi: 10.1093/nar/17.19.7855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam S.S., Schimmel P.R. Equilibrium measurements of cognate and noncognate interactions between aminoacyl transfer RNA synthetases and transfer RNA. Biochemistry. 1975;14:2775–2780. doi: 10.1021/bi00683a034. [DOI] [PubMed] [Google Scholar]
- McClain W.H. Rules that govern tRNA identity in protein synthesis. J. Mol. Biol. 1993;234:257–280. doi: 10.1006/jmbi.1993.1582. [DOI] [PubMed] [Google Scholar]
- Moulinier L., Eiler S., Eriani G., Gangloff J., Thierry J.C., Gabriel K., McClain W.H., Moras D. The structure of an AspRS–tRNAAsp complex reveals a tRNA-dependent control mechanism. EMBO J. 2001;20:5290–5301. doi: 10.1093/emboj/20.18.5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nameki N., Asahara H., Shimizu M., Okada N., Himeno H. Identity elements of Saccharomyces cerevisiae tRNAHis . Nucleic Acids Res. 1995;23:389–394. doi: 10.1093/nar/23.3.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsoulis G., Hilger F., Fink G.R. The HTS1 gene ecodes both the cytoplasmic and mitochondrial histidine tRNA synthetases of S. cerevisiae . Cell. 1986;46:235–243. doi: 10.1016/0092-8674(86)90740-3. [DOI] [PubMed] [Google Scholar]
- Normanly J., Abelson J.1989. tRNA identity Annu. Rev. Biochem. 581029–1049. [DOI] [PubMed] [Google Scholar]
- Rosen A.E., Musier-Forsyth K. Recognition of G-1:C73 atomic groups by Escherichia coli histidyl-tRNA synthetase. J. Am. Chem. Soc. 2004;126:64–65. doi: 10.1021/ja0381609. [DOI] [PubMed] [Google Scholar]
- Rudinger J., Florentz C., Giegé R. Histidylation by yeast HisRS of tRNA or tRNA-like structure relies on residues −1 and 73 but is dependent on the RNA context. Nucleic Acids Res. 1994;22:5031–5037. doi: 10.1093/nar/22.23.5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudinger J., Felden B., Florentz C., Giegé R. Strategy for RNA recognition by yeast histidyl-tRNA synthetase. Bioorg. Med. Chem. Lett. 1997;5:1001–1009. doi: 10.1016/s0968-0896(97)00061-8. [DOI] [PubMed] [Google Scholar]
- Scaringe S.A., Francklyn C., Usman N. Chemical synthesis of biologically active oligoribonucleotides using β-cyanoethyl protected ribonucleoside phosphoramidites. Nucleic Acids Res. 1990;18:5433–5441. doi: 10.1093/nar/18.18.5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimmel P.R., Söll D. Aminoacyl-tRNA synthetases: General features and recognition of transfer RNAs. Annu. Rev. Biochem. 1979;48:601–648. doi: 10.1146/annurev.bi.48.070179.003125. [DOI] [PubMed] [Google Scholar]
- Sprinzl M., Vassilenko K.S. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 2005;33:D139–D140. doi: 10.1093/nar/gki012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sproat B., Colonna F., Mullah B., Tsou D., Andrus A., Hampel A., Vinayak R. An efficient method for the isolation and purification of oligoribonucleotides. Nucleosides Nucleotides. 1995;14:255–273. [Google Scholar]
- Tukalo M., Yaremchuck A., Fukunaga R., Yokoyama S., Cusack S. The crystal structure of leucyl-tRNA synthetase complexed with tRNALeu in the post-transfer-editing conformation. Nat. Struct. Mol. Biol. 2005;12:923–930. doi: 10.1038/nsmb986. [DOI] [PubMed] [Google Scholar]
- Williams J.B., Cooley L., Söll D. Enzymatic addition of guanylate to histidine transfer RNA. Methods Enzymol. 1990;181:451–462. doi: 10.1016/0076-6879(90)81143-i. [DOI] [PubMed] [Google Scholar]
- Wu X., Pitsch S. Synthesis and pairing properties of oligoribonucleotide analogues containing a metal-binding site attached to β-D-allofuranosyl cytosine. Nucleic Acids Res. 1998;26:4315–4323. doi: 10.1093/nar/26.19.4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W., Francklyn C. Cytosine 73 is a discriminator nucleotide in vivo for histidyl-tRNA in Escherichia coli . J. Biol. Chem. 1994;269:1–7. [PubMed] [Google Scholar]
- Yan W., Augustine J., Francklyn C. A tRNA identity switch mediated by the binding interaction between a tRNA anticodon and the accessory domain of a class II aminoacyl-tRNA synthetase. Biochemistry. 1996;35:6559–6568. doi: 10.1021/bi952889f. [DOI] [PubMed] [Google Scholar]