

Abstract
We show that a targetron based on the Lactococcus lactis Ll.LtrB group II intron can be used for efficient chromosomal gene disruption in the human pathogen Staphylococcus aureus. Targetrons expressed from derivatives of vector pCN37, which uses a cadmium-inducible promoter, or pCN39, a derivative of pCN37 with a temperature-sensitive replicon, gave site-specific disruptants of the hsa and seb genes in 37%–100% of plated colonies without selection. To disrupt hsa, an essential gene, we used a group II intron that integrates in the sense orientation relative to target gene transcription and thus could be removed by RNA splicing, enabling the production of functional HSa protein. We show that because splicing of the Ll.LtrB intron by the intron-encoded protein is temperature-sensitive, this method yields a conditional hsa disruptant that grows at 32°C but not 43°C. The temperature sensitivity of the splicing reaction suggests a general means of obtaining one-step conditional disruptions in any organism. In nature, temperature sensitivity of group II intron splicing could limit the temperature range of an organism containing a group II intron inserted in an essential gene.
Keywords: functional genomics, gene targeting, retrotransposon, reverse transcriptase, ribozyme
INTRODUCTION
Staphylococcus aureus is a facultative pathogen, responsible for most of the infections suffered by hospital patients and increasingly implicated in serious community infections (Fridkin et al. 2005). Its danger as a pathogen reflects that S. aureus can adapt to widely varying conditions, including the hostile environment of infected animal tissues, and can develop resistance to any and all antibiotics. At present, there is an urgent need to understand the pathobiology of the organism and to develop new anti-bacterial agents to counter it. A major approach for both objectives is to identify targetable metabolic process by evaluating the function of specific genes through targeted gene inactivation. Unfortunately, gene targeting in S. aureus has been hampered by the rather low efficiency of homologous recombination. Beyond traditional strategies, such as chemical and transposon mutagenesis, recently developed methods for inactivating genes in S. aureus use antisense RNAs (Kernodle et al. 1997) or counterselections for rare recombination events, e.g., by using a dominant streptomycin-sensitivity allele (Chen and Novick 2006) or an antisense RNA blocking the expression of an essential gene, secA (Bae and Schneewind 2006). All these methods have advantages and disadvantages. Transposon mutagenesis is fairly easy, but transposons have hot spots and are useless for essential genes. Counterselection strategies require laborious gene-specific constructions and, again, are useless for essential genes. Antisense RNAs can target any gene but have a significant failure rate and, as they repress but do not eliminate gene function, may not always reveal essentiality.
A different strategy that overcomes many of these disadvantages is gene targeting with mobile group II introns (“targetrons”) (for review, see Lambowitz et al. 2005). A targetron based on the Lactococcus lactis Ll.LtrB intron has been used for targeted gene disruption and site-specific DNA insertion in Escherichia coli and other Gram-negative enterobacteria (Karberg et al. 2001; Perutka et al. 2004), as well as in the Gram-positive bacteria L. lactis and Clostridium perfringens (Frazier et al. 2003; Chen et al. 2005). The utility of group II introns for gene targeting derives from their unique DNA integration mechanism in which the intron RNA uses its ribozyme activity to insert (reverse splice) directly into a DNA target site and is then reverse-transcribed by the intron-encoded protein (IEP) (for review, see Lambowitz and Zimmerly 2004). The mobility reactions are carried out by a ribonucleoprotein (RNP) particle that is formed during RNA splicing and contains the IEP and excised intron lariat RNA. RNPs initiate mobility by using both the IEP and base-pairing of the intron RNA to recognize DNA target sequences. The base-pairing interactions span a 13- to 16-nt region of the DNA target site, and involve intron RNA sequences denoted exon-binding sites 1 and 2 (EBS1 and EBS2) and δ (a sequence adjacent to EBS1) and DNA target site sequences denoted intron-binding sites 1 and 2 (IBS1 and IBS2) in the 5′-exon, and δ′ in the 3′-exon (see below) (Eskes et al. 1997; Guo et al. 1997, 2000; Mohr et al. 2000). Because these base-pairing interactions contribute most of the DNA target specificity, this feature makes it possible to retarget group II introns to insert into desired sites simply by modifying the EBS1, EBS2, and δ sequences in the intron RNA (Lambowitz et al. 2005).
In bacteria, targetrons are generally expressed from a donor plasmid, and RNPs are assembled intracellularly (Guo et al. 2000; Karberg et al. 2001). The commonly used E. coli donor plasmid pACD3 specifies a precursor RNA containing a 0.9-kb Ll.LtrB-ΔORF intron and short flanking exon sequences, with the IEP (denoted LtrA protein) expressed from a position just downstream of the 3′-exon (Guo et al. 2000; Karberg et al. 2001). The IEP binds to the intron in the precursor RNA, promotes its splicing by stabilizing the catalytically active RNA structure, and remains bound to the excised intron lariat RNA in RNPs that promote mobility. For mobility, the Ll.LtrB intron reverse-splices into one strand of the target DNA, while the IEP cleaves the opposite strand and uses the 3′ DNA end at the cleavage site as a primer for reverse transcription of the inserted intron RNA. The resulting intron cDNA is integrated into the recipient DNA by using host DNA replication and repair enzymes (Matsuura et al. 1997; Cousineau et al. 1998; Smith et al. 2005).
The Ll.LtrB intron is targeted to desired sites with the aid of a computer algorithm, which scans the target sequence for the best matches for positions recognized by the IEP and then designs PCR primers for optimal base-pairing interactions between the intron RNA's EBS1, EBS2, and δ sequences and the IBS1, IBS2, and δ′ sequences in the DNA target site (Perutka et al. 2004). The positions recognized by the IEP are sufficiently few and flexible that the algorithm generally identifies multiple rank-order target sites in any gene. The required modifications are introduced into the donor plasmid via PCR (Guo et al. 2000; Karberg et al. 2001). In E. coli, targetrons commonly insert at frequencies of 1%–80%, and insertions are detected either by colony PCR or by incorporating a genetic marker within the intron (Zhong et al. 2003; Perutka et al. 2004). Thus, targetrons can potentially be used to disrupt any gene for which a sequence is available.
In addition to targeted gene disruption, targetrons have been used to insert (“knock in”) cargo genes site-specifically at desired chromosomal locations (Frazier et al. 2003) or to introduce targeted double-strand breaks that can be repaired by homologous recombination with a cotransformed DNA fragment, enabling the introduction of point mutations (Karberg et al. 2001). Additionally, a targetron with randomized target-site recognition sequences was used to generate libraries of random disruptants, analogous to global transposon mutagenesis (Zhong et al. 2003). Individual targetrons were then re-isolated from the library DNA by PCR and used without modification to obtain desired single disruptants (Yao et al. 2005).
In principle, targetrons can also be used to obtain either unconditional or conditional disruptions, including conditional disruptions of essential genes. This is so because the targetron can be made to integrate in either orientation relative to target gene transcription by selecting target sequences in the sense or antisense strands. A targetron inserted in the antisense orientation is transcribed into the complement of the intron RNA sequence, cannot be spliced, and yields an unconditional disruption. In contrast, a targetron inserted in the sense orientation is transcribed into a functional intron RNA and can potentially be spliced from a precursor RNA by the IEP, restoring the mRNA (Karberg et al. 2001; Frazier et al. 2003). Because the splicing reaction is dependent on the IEP, we suggested that conditional disruptants might be obtained by controlling expression of the IEP from a separate construct with an inducible promoter (Karberg et al. 2001; Frazier et al. 2003).
Here, we constructed targetron donor plasmids for S. aureus and used them to disrupt both the essential hsa gene and the nonessential seb gene at high frequencies without selection. The hsa gene was disrupted by using a targetron that inserts in the sense orientation relative to target gene transcription and could be spliced in an IEP-assisted manner from precursor RNA, resulting in the production of functional HSa protein. Further, we show that this IEP-assisted splicing reaction is inherently temperature-sensitive, and consequently, this procedure yields a conditional hsa disruptant that can grow at 32°C but not at 43°C. Our results demonstrate for the first time that targetrons can be used to disrupt essential genes, and they suggest a novel general strategy for obtaining one-step conditional gene disruptions in any organism. In nature, the temperature sensitivity of group II intron splicing reactions could limit the temperature range of organisms containing group II introns inserted within essential genes.
RESULTS
Use of the Ll.LtrB targetron for gene disruption in S. aureus
For gene targeting in S. aureus, we constructed intron-donor plasmids patterned after the intron-donor plasmid pACD3 used in E. coli (Guo et al. 2000; Karberg et al. 2001). pNL9161 contains a 0.9-kb Ll.LtrB-ΔORF intron and flanking exons cloned behind a cadmium-inducible promoter (Pcad) in the S. aureus vector pCN37 (Charpentier et al. 2004). The LtrA ORF is deleted from its normal location within the intron and inserted just downstream of the 3′-exon. In E. coli, the Ll.LtrB-ΔORF intron is less susceptible to nuclease degradation than is the full-length intron, leading to substantially higher integration frequencies (Guo et al. 2000).
Two candidate genes were chosen to test intron targeting in S. aureus: hsa, which encodes a member of the HU family of bacterial histone-like proteins (Viter et al. 1999), and seb, which encodes enterotoxin B (Shafer and Iandolo 1978). First, we analyzed the genes by a computer algorithm to identify potential insertions sites for the Ll.LtrB targetron (see Materials and Methods), and we selected single target sites from among eight potential target sites in seb and four potential target sites in hsa. Because two laboratories had independently failed to obtain an hsa knockout (A. Cheung, pers. comm.; Y. Fang and R.P. Novick, unpubl.), we suspected that this gene might be essential, and therefore selected an anti-Hsa targetron that inserts in the sense orientation and could potentially be removed by RNA splicing, enabling the production of functional HSa protein. An additional challenge of the hsa gene was its small size (270 bp) (Viter et al. 1999), which limits the choice of target sites.
Donor plasmids containing Ll.LtrB targetrons that insert into the selected sites in the hsa and seb genes were constructed via PCR using primers designed by the algorithm to modify the intron's EBS1, EBS2, and δ sequences to base-pair optimally to DNA target site sequences IBS1, IBS2, and δ′. The IBS1 and IBS2 sequences in the 5′-exon of the donor plasmid were also modified to be complementary to the intron's retargeted EBS1 and EBS2 sequences for efficient RNA splicing from the donor construct (see Fig. 1; Materials and Methods). The pCN37-based donor plasmids containing targetrons Hsa-24s and Seb-123a are denoted pNL9163 and pNL9165, respectively (Table 1). The predicted base-pairing interactions between these targetrons and their target sites are shown in Figure 2.
FIGURE 1.

Intron-donor plasmids for S. aureus gene targeting. (A) pNL9161 is a derivative of the S. aureus expression vector pCN37, which employs a cadmium-inducible promoter (Pcad) with the cadmium regulator (cadR) to express a precursor RNA containing the Ll.LtrB-ΔORF intron and flanking 5′- and 3′-exon sequences (E1 and E2, respectively), plus the LtrA ORF downstream of E2. The plasmid contains a staphylococcal pT181 replicon and a colE1 replicon, as well as erythromycin-resistance (ermC) and ampicillin-resistance (ampR) genes. (B) pNL9162 is a derivative of the S. aureus expression vector pCN39 and is similar to pNL9161 (above), except that it contains a temperature-sensitive replicon (pT181 cop-634ts repC4) in place of the wild-type pT181 replicon. (C) PCR reactions used to modify the targetron for insertion into different genes. For targeting, the EBS1, EB2, and δ sequences in the Ll.LtrB-ΔORF intron are made complementary to IBS1, IBS2, and δ′ in the DNA target site, and IBS1 and IBS2 in E1 of the donor plasmid are made complementary to the intron's retargeted EBS1 and EBS2 sequences for efficient RNA splicing. The modifications are introduced into the donor plasmid via a two-step PCR (PCR1 and 2), using primers p1 + p2 and p3 + p4, as diagrammed in the figure and described in detail in Materials and Methods. The PCRs yield a 353-bp HindIII/BsrGI fragment, which is swapped for the corresponding fragment of the donor plasmid.
TABLE 1.
S. aureus intron-donor plasmids used in this study
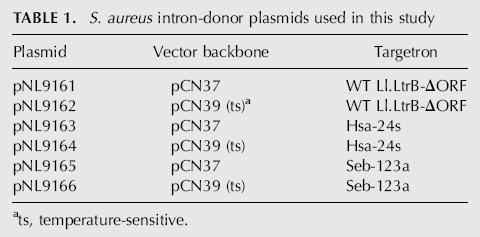
FIGURE 2.

DNA target sites and base-pairing interactions for the Hsa-24s and Seb-123a targetrons. The figure shows DNA target site sequences from positions −30 to +15 from the intron-insertion site and intron RNA/DNA target site base-pairing interactions for the wild-type (WT) Ll.LtrB intron and targetrons Hsa-24s and Seb-123a, designed to insert into the S. aureus hsa and seb genes, respectively. Targetrons are named by the nucleotide position 5′ to their insertion site in the target gene's coding sequence, followed by “s” or “a” indicating sense or antisense strands, respectively. The intron-insertion site in the top strand (IS) and the bottom-strand cleavage site of the IEP (CS) are indicated by arrowheads. Nucleotide residues in the DNA target site that match the wild-type Ll.LtrB intron target site are highlighted in gray.
S. aureus strains containing the donor plasmids were grown to early log phase and then either plated directly on BHI medium containing erythromycin or induced with CdCl2 prior to plating. After incubating the plates overnight at 37°C, colonies were screened for targetron insertion by PCR, using primers flanking the predicted insertion site in the target gene. The initial hsa disruption was done in the avirulent S. aureus strain RN4220. The seb disruption was done in strain RN8908, which contains the SaP13 pathogenicity island encoding SEB, after conditioning the donor plasmid by passage through RN4220, which does not contain SaP13 (see Materials and Method).
The Pcad promoter has a low basal level of transcription in the absence of cadmium (Charpentier et al. 2004), and at the outset, it was not clear whether cadmium induction would be necessary to obtain disruptants. Indeed, for hsa, we PCR-screened 10 colonies from cells without cadmium induction and found that all 10 had the desired disruption (Fig. 3A, left panel). Insertion of the targetron at the correct site was confirmed by sequencing the PCR products (data not shown). For seb, only one of the 24 colonies from cells that had not been induced with cadmium had the desired disruption, but after cadmium induction, nine of 24 colonies had the desired disruption (Fig. 4A; the left panel shows PCR data for a subset of colonies). The correct disruption was again confirmed by sequencing the colony PCR products (data not shown). For both hsa and seb, two randomly chosen disruptants analyzed by Southern hybridization using an intron probe showed just a single band of the size expected for integration at the desired target site, along with a larger band corresponding to the donor plasmid, and no additional bands due to nonspecific insertions (Figs. 3A, 4A, right panels and figure legends for molecular weight range spanned by the blot). In other experiments, the Hsa-24s targetron was also used to disrupt the hsa gene in S. aureus strain RN7206 and in several plasmid-carrying derivatives of RN4220 and RN7206, with the disruption again found in every one of a small number of colonies analyzed (data not shown).
FIGURE 3.

Use of the Ll.LtrB targetron to disrupt the S. aureus hsa gene. (A,B) Disruption of the hsa gene in S. aureus RN4220 using targetron Hsa-24s expressed from pNL9163 and pNL9164, respectively. For all steps, cells containing pNL9163 (pCN37-derivative) were grown at 37°C, and those containing pNL9164 (pCN39-derivative; temperature-sensitive replicon) were grown at 32°C. Donor plasmids were electroporated into RN4220, and cells were grown in BHI medium containing erythromycin for 1 h, then plated on the same medium. The left panels show colony PCR of wild-type (WT) RN4220 and disruptants obtained with each of the donor plasmids. The right panels show Southern blots of MfeI-digested donor plasmids pNL9163 and pNL9164 (left lane) and genomic DNA from randomly chosen hsa disruptants obtained with the corresponding donor plasmid (right lanes). The blots were hybridized with a 32P-labeled intron probe. The bands shown are the only ones visible on the blots (molecular weight range 100 bp–12 kb). The schematic at the bottom shows a diagram of the hsa gene, with (top) and without (bottom) the integrated targetron, including the location of primers pf and pr used to detect targetron integration by colony PCR, and the MfeI sites used for Southern hybridization analysis.
FIGURE 4.

Use of the Ll.LtrB targetron to disrupt the S. aureus seb gene. (A,B) Disruption of the seb gene in S. aureus RN8098 using targetron Seb-123a expressed from pNL9165 and pNL9166. For all steps, cells containing pNL9165 (pCN37-derivative) were grown at 37°C, and those containing pNL9166 (pCN39-derivative; temperature-sensitive replicon) were grown at 32°C. Donor plasmids were electroporated into S. aureus RN4220, then reisolated and electroporated into RN8098 (see Materials and Methods). Cells were grown in BHI medium containing erythromycin, then induced with 10 mM CdCl2 for 90 min, and plated on BHI medium containing erythromycin. The left panels show colony PCR of wild-type RN8098 (WT) and representative colonies obtained with each of the donor plasmids. The right panels show Southern blots of AseI-digested donor plasmids pNL9165 and pNL9166 (left lane) and randomly chosen seb disruptants obtained with the corresponding donor plasmid (right lane). The blots were hybridized with a 32P-labeled intron probe. The samples in the two rightmost lanes of the Southern blot in panel B are from cells in which pNL9166 was cured from the disruptants by overnight incubation at 43°C on BHI plates. The bands shown are the only ones visible on the blots (molecular weight range 100 bp–12 kb). The schematic at the bottom shows a diagram of the seb gene, with (top) and without (bottom) the integrated targetron, including the location of primers pf and pr used to detect targetron integration by colony PCR and the location of the AseI sites used for Southern hybridization analysis.
Ll.LtrB gene targeting in S. aureus using a temperature-sensitive vector
Because the donor plasmid expresses LtrA protein, which could potentially remobilize or splice the inserted Ll.LtrB-ΔORF intron, it is desirable to cure it from the disruptants prior to analysis. To facilitate curing of the donor plasmid, we recloned the Ll.LtrB targetron constructs into the vector backbone of pCN39, which has a temperature-sensitive replicon (Fig. 1B). The resulting donor plasmid containing the wild-type Ll.LtrB intron is denoted pNL9162, and the donor plasmids containing targetrons Hsa-24s and Seb-123a are denoted pNL9164 and pNL9166, respectively (Table 1). The latter donor plasmids gave targeted disruptions of the hsa and seb genes with the same high efficiency and specificity as pCN37-based donor plasmids (Figs. 3B, 4B). The Hsa-24s targetron again gave 100% disruptants (24/24) without cadmium induction, while the Seb-123a targetron gave 7/19 disruptants after cadmium induction (Figs. 3B, 4B; the left panels show PCR data for a subset of colonies). The pCN39-based donor plasmid was readily cured from the seb disruptants by plating cells at 43°C (Fig. 4B, Southern blot, right-hand lanes). However, the donor plasmid could not be cured from the hsa disruptants, presumably because the IEP-assisted splicing of the inserted intron is required to produce HSa protein. The inability to cure the donor plasmid confirms the essentiality of the hsa gene.
Targetron insertion creates a conditional hsa disruptant
We noticed that hsa disruptants containing either the pCN37- or pCN39-based donor plasmids were temperature-sensitive and could grow on plates at 32°–41°C but not at 43°C (Fig. 5A; the seb disruptants were not temperature-sensitive; data not shown). Because the temperature sensitivity of the hsa disruptants was observed whether or not the plates contained erythromycin to force retention of the donor plasmid that supplies the IEP, we suspected that it might reflect the temperature sensitivity of the IEP-assisted splicing reaction.
FIGURE 5.

hsa disruptants are temperature sensitive for growth and splicing of the Ll.LtrB-ΔORF intron. (A) Plating assay. Wild-type RN4220 and hsa disruptants obtained with pNL9163 and pNL9164 (ts) were streaked on TSA agar plates at temperatures between 32° and 43°C. The plates were incubated for 24–48 h and photographed. The temperature-sensitive plasmid pNL9164 expressing LtrA (pCN39-derivative) is present in higher copy number than is pNL9163 (pCN37-derivative) (Charpentier et al. 2004), likely resulting in somewhat more residual growth after the temperature shift to 43°C. (B) RT-PCR assay of Ll.LtrB-ΔORF intron splicing from the hsa gene. Cells were grown in BHI medium overnight, then diluted 1:20 into fresh medium, and incubated for 4 h at 32°C or for 2 h at 43°C. RNA was extracted and analyzed by RT-PCR, using primers pE1 and pE2 flanking the Ll.LtrB-ΔORF intron inserted in the hsa gene. The smaller RT-PCR product (0.1 kb) is derived from hsa mRNA without the inserted intron, and the larger product (1.0 kb) is derived from the precursor RNA containing the inserted intron. The identities of the RT-PCR products were confirmed by sequencing (data not shown). Controls showed no products if the initial reverse transcription step was omitted or if the reactions were carried out in the absence of RNA substrate (data not shown), indicating that the amplified bands in the experimental samples are derived from cellular RNA. The schematic below the gel shows the hsa precursor and mRNAs, including the location of primers pE1 and pE2 and the expected RT-PCR products.
To determine whether the splicing reaction is temperature-sensitive, the splicing of the Ll.LtrB-ΔORF intron inserted in the hsa gene was assayed by RT-PCR, using primers flanking the intron-insertion site (Fig. 5B). At 32°C, in the wild-type (WT) strain, the predominant product was a 0.1-kb band corresponding to hsa mRNA (Fig. 5B, lane 1), and this same band, corresponding to mRNA with spliced exons, was also the predominant product in the hsa disruptants obtained with donor plasmids pNL9163 and pNL9164 (Fig. 5B, lanes 2 and 3, respectively). The disruptants also show a lighter 1.0-kb band corresponding to unspliced precursor RNA, which was more prominent in cells containing pNL9163 (the pCN37-based donor plasmid) (Fig. 5B, lane 2) than in those containing pNL9164 (the pCN39-based donor plasmid) (Fig. 5B, lane 3). This difference may reflect different plasmid copy numbers (20–25 and 300–400 copies per cell for the vectors pCN37 and pCN39, respectively) (Charpentier et al. 2004), resulting in different levels of expression of the IEP needed for precursor RNA splicing.
Strikingly, when cells were grown at 32°C and shifted to 43°C for 2 h, the wild-type strain again showed a single band corresponding to hsa mRNA (Fig. 5B, lane 4), while the disruptants showed substantial accumulation of the larger band corresponding to unspliced precursor RNA, and concomitant reduction in the intensity of the band corresponding to the mRNA with spliced exons (Fig. 5B, lanes 5,6). Quantitation of precursor and mRNA levels by real-time RT-PCR confirmed the accumulation of precursor RNA and reduction in hsa mRNA levels in the disruptants after the shift to 43°C (Table 2). Together, these findings indicate that the temperature sensitivity of the hsa disruptants is due to the temperature sensitivity of the IEP-assisted splicing reaction of the inserted Ll.LtrB-ΔORF intron.
TABLE 2.
Real-time RT-PCR analysis of levels of hsa transcripts in S. aureus strains

IEP-assisted splicing of the wild-type Ll.LtrB-ΔORF intron is temperature-sensitive in both E. coli and S. aureus
To determine whether the IEP-assisted splicing reaction is inherently temperature-sensitive, we carried out similar RT-PCR analysis of wild-type Ll.LtrB-ΔORF intron transcripts expressed from donor plasmids pACD2X in E. coli and pNL9161 in S. aureus. As shown in Figure 6, we observed a similar inhibition of splicing within 1 or 1.5 h after temperature shift from 30°C (E. coli) or 32°C (S. aureus) to 37°C or 43°C, with further accumulation of unspliced precursor RNA and decrease in the level of spliced-exon RNA after longer incubation at either temperature. In other experiments, real-time RT-PCR showed that splicing of the full-length wild-type Ll.LtrB intron expressed from plasmid pACD-LtrB in E. coli HMS174(DE3) is also temperature-sensitive (unspliced precursor RNA, 2.57 ± 0.05 at 32°C and 11.1 ± 0.6 at 43°C; spliced exons, 4.2 ± 0.2 at 32°C and 2.10 ± 0.05 at 43°C, for overnight incubation at the indicated temperatures). Thus, the IEP-assisted splicing reaction of the Ll.LtrB group II intron appears to be inherently temperature-sensitive.
FIGURE 6.

Temperature sensitivity of splicing of the wild-type Ll.LtrB-ΔORF intron in E. coli and S. aureus. (A) E. coli HMS174(DE3) containing intron-donor plasmid pACD2X was grown to log phase, then 100 μM IPTG was added to induce intron expression, and the cultures were incubated for 1 h or overnight (O/N) at 30°, 37°, or 43°C. (B) S. aureus strain RN4220 containing pNL9161 was grown overnight, then diluted 1:20 into fresh BHI containing erythromycin. Then 10 μM CdCl2 was added to induce intron expression, and the cultures were incubated for 90 min or 6 h at 32°, 37°, or 43°C. RNA was extracted and analyzed by RT-PCR, using primers pE1 and pE2, which hybridize with 5′ (E1) or 3′ (E2) exon sequences flanking the Ll.LtrB-ΔORF intron in pACD2X or pNL9161. The schematic beneath the gel shows the precursor and mRNAs, with the location of primers pE1 and pE2 and the expected RT-PCR products.
DISCUSSION
We show here that the Ll.LtrB targetron can be used for efficient chromosomal gene disruption in the human pathogen S. aureus. Targetron Hsa-24s, used to disrupt the hsa gene, was exceptionally efficient, giving 100% disruptant colonies, even without cadmium induction of targetron transcription, while targetron Seb-123a, used to disrupt the seb gene encoding enterotoxin B, was somewhat less efficient, giving ~37% disruptant colonies only after cadmium induction. The difference in frequency for the Hsa-24s and Seb-123a targetrons likely reflects primarily how well the retargeted introns recognize their new target sites, as seen previously for different targetrons in E. coli (Perutka et al. 2004). The Ll.LtrB targetron was used previously for targeted gene disruption in two other Gram-positive bacteria, L. lactis and C. perfringens. In L. lactis, two different Ll.LtrB targetrons expressed from a nisin-inducible promoter gave disruptants of the mleS gene in 100% of screened colonies, and one of these targetrons was also used to site-specifically insert a cargo gene cloned within the intron (the abiD gene conferring phage resistance) (Frazier et al. 2003). In C. perfringens, an Ll.LtrB targetron expressed from a constitutive promoter gave disruptants of the α-toxin gene (plc) in two of 38 screened colonies (Chen et al. 2005). Based on these examples, it seems likely that the Ll.LtrB targetron will be generally useful for gene targeting in Gram-positive bacteria.
Our results show for the first time that a targetron that inserts in the sense orientation and can be removed from the precursor RNA by RNA splicing can be used to disrupt an essential gene. Further, we find that the IEP-assisted splicing reaction of the Ll.LtrB intron is inherently temperature-sensitive, thereby resulting in a one-step conditional disruption. We note that the ability to obtain a conditional disruptant is dependent on several factors, including the splicing efficiency of the inserted intron, the stability of the target gene mRNA, and the amount of gene product needed to sustain growth. These factors may differ for different genes in different organisms, and it may not be possible to obtain conditional disruptants in all cases. The splicing efficiency of the inserted targetron is dependent on the sequences flanking its insertion site, both because of the EBS/IBS and δ–δ′ interactions between the intron and flanking exon sequences and because the flanking exon sequences can potentially form obstructing RNA structures. Ultimately, it may be possible to assess these parameters systematically to obtain general rules for predicting the splicing efficiency of targetrons inserted at different sites.
In principle, the temperature sensitivity of the RNA splicing reaction could reflect temperature sensitivity of the IEP, the intron RNA, or some combination of the two. Previous studies showed that purified LtrA protein is rapidly and irreversibly denaturated when incubated in splicing reaction medium at 37°C in the absence of intron RNA and that lower temperatures favor the expression of active LtrA protein in E. coli (Saldanha et al. 1999). Additionally, immunoblot experiments with E. coli HMS174(DE3) expressing donor plasmid pACD2X showed decreased levels of soluble LtrA protein at 43°C (J. Yao and A.M. Lambowitz, unpubl.). Thus, the temperature sensitivity of the IEP is likely a major factor in the temperature sensitivity of the splicing reaction. Further, based on in vitro results, we expect this temperature sensitivity to be exacerbated when the IEP is expressed from an ectopic location where it cannot bind immediately to the intron RNA (see Saldanha et al. 1999; Cui et al. 2004). The thermal stability of both the IEP and the intron RNA structure may differ in different hosts, and the latter could also be influenced by the location of the intron-insertion site within the target gene. Consequently, it will be necessary to define the optimal temperature range for conditional disruption in each case. In the future, it may be possible to change the operational temperature range by genetically selecting variants of the IEP having greater or lesser temperature sensitivity.
Notably, some bacteria contain natural group II introns inserted into essential genes (e.g., groEL in Azotobacter) (Ferat et al. 2003). It will be of interest to determine whether these and other group II introns display temperature-sensitive splicing similar to that of the Ll.LtrB intron. If so, the temperature range at which some bacteria can grow may be limited by the inherent temperature sensitivity of the splicing reaction of the inserted group II intron. Moreover, sporadic insertion of group II introns in nature could rapidly change the fitness of organisms to grow at different temperatures. Nevertheless, at least some bacterial group II introns can splice at elevated temperatures, possibly reflecting adaptation of the intron to the host after insertion (Chee and Takami 2005).
The methods used for adapting targetrons for use in S. aureus (this study) or C. perfringens (Chen et al. 2005) are relatively straightforward. Principally, the targetron is recloned into a donor plasmid with a promoter suitable for expressing the intron in the desired host. In E. coli, targetrons have been expressed from a phage T7 promoter, using T7 RNA polymerase introduced via a λDE3 lysogen or from a separate plasmid (Guo et al. 2000; Karberg et al. 2001). However, the results for S. aureus and C. perfringens show that a native host promoter, either inducible or constitutive, is sufficient to express targetrons at levels that give highly efficient gene disruption. A caveat is that both these studies used the short Ll.LtrB-ΔORF intron without a selectable marker, and there is some indication that longer forms of the intron containing a genetic marker are not expressed efficiently from such promoters (J. Yao and A.M. Lambowitz, unpubl.). The efficiency of translation of the IEP, which is required for both RNA splicing and intron integration, is another parameter that could affect targetron expression in different hosts. This parameter is influenced by the Shine-Dalgarno and other sequences around the translation initiation site, as well as codon usage in the LtrA protein compared to that optimal in the host organism. However, neither in our work with S. aureus nor that of Chen et al. (2005) with C. perfringens was the Shine-Dalgarno sequence optimized for the specific host. Although thus far, targetrons have been introduced via donor plasmids that replicate in the desired host, it should also be possible to introduce them via conjugation (Belhocine et al. 2004) or nonreplicating DNA and perhaps even as pre-assembled RNPs. We anticipate that these procedures will be broadly applicable for the use of targetrons in a wide variety of prokaryotes.
MATERIALS AND METHODS
Bacterial strains and growth conditions
The S. aureus strains were RN4220 and RN8098. RN4220 is a restriction-defective, nonlysogenic derivative of NCTC8325 and is an efficient recipient for E. coli DNA (Kreiswirth et al. 1983). RN8098 contains the SaPI3 pathogenicity island that encodes the staphylococcal superantigen enterotoxin B (SEB) (Novick et al. 2001). S. aureus strains were grown in brain heart infusion (BHI) or trypticase soy agar (TSA) media, with erythromycin added at 10 μg/mL. Unless specified otherwise, cells containing pCN37-based plasmids were grown at 37°C, and those containing pCN39-based plasmids, which have a temperature-sensitive replicon, were grown at 32°C.
E. coli strain DH5α was used for cloning, and HMS174 (DE3) was used for RT-PCR analysis of Ll.LtrB intron splicing. E. coli strains were grown in LB medium, with ampicillin added at 100 μg/mL, chloramphenicol at 25 μg/mL, and erythromycin at 200 μg/mL.
Recombinant plasmids
pACD2X contains a 0.9-kb Ll.LtrB-ΔORF intron and flanking exons cloned downstream of a T7lac promoter in a CamR pACYC184-based vector (San Filippo and Lambowitz 2002). The ORF encoding the LtrA protein is expressed from a position just downstream of the 3′-exon, and the Ll.LtrB-ΔORF intron has an additional phage T7 promoter inserted in intron RNA domain IV for use in E. coli genetic assays of intron mobility (see Guo et al. 2000; Karberg et al. 2001). pACD-LtrB is a related plasmid that expresses the full-length wild-type Ll.LtrB intron with the LtrA ORF encoded in intron domain IV (Guo et al. 2000).
The S. aureus targetron donor plasmids used in this work are summarized in Table 1. pNL9161 contains the same Ll.LtrB targetron construct as pACD2X cloned downstream of a cadmium-inducible promoter (Pcad) in the S. aureus vector pCN37 (Charpentier et al. 2004). To construct this plasmid, a 3-kb XbaI/XhoI fragment of pACD2X, containing the Ll.LtrB-ΔORF intron and flanking exon sequences plus the downstream LtrA ORF, was gel-purified, blunt-ended with T4 DNA polymerase, and cloned into the SmaI site of pCN37. To facilitate the introduction of modifications needed for gene targeting, the XbaI site downstream of Pcad was replaced by an NheI site by cleaving the pCN37 multicloning site with PstI and EcoRI, then ligating in a DNA oligonucleotide containing an NheI site flanked by PstI and EcoRI sites. In addition, the HindIII site in the pT181 repC was eliminated without altering the amino acid sequence (AAGCTT to AAGCTA; Lys-Leu to Lys-Leu), and the BsrGI site downstream of repC was removed by BsrGI digestion, followed by fill-in with T4 DNA polymerase, and religation.
pNL9162 contains the same Ll.LtrB targetron construct cloned in pCN39 (Charpentier et al. 2004), which has a temperature-sensitive replicon. It was constructed as follows: First, the Pcad promoter was cut from pCN37 by digestion with SphI and PstI, and cloned between the SphI and PstI sites of pCN39. Then, the XbaI site downstream of Pcad was replaced by an NheI site and the HindIII and BsrGI sites were removed, as described above for pNL9161. Finally, pACD2X was digested with XbaI and EcoRV, and the 3-kb fragment containing the Ll.LtrB-ΔORF intron and flanking exons plus the downstream LtrA ORF was cloned between the NheI and SmaI sites of pCN39.
Retargeting the Ll.LtrB intron to insert into S. aureus genes
The Ll.LtrB targetron was retargeted to insert into S. aureus genes by using a computer algorithm that identifies potential insertion sites and designs PCR primers for modifying the intron RNA to base-pair to those sites (Perutka et al. 2004). Single target sites in hsa and seb were chosen from among potential target sites identified by the algorithm in each gene. Donor plasmids were then constructed in which the intron RNA's EBS1, EBS2, and δ sequences were modified to optimally base-pair to DNA target site sequences IBS1, IBS2, and δ′. The IBS2 and IBS1 sequences in the 5′-exon of the donor plasmid were also modified to be complementary to the retargeted EBS2 and EBS1 sequences for efficient RNA splicing (Perutka et al. 2004).
The required modifications were introduced into the donor plasmid via a two-step PCR diagrammed in Figure 1C (Karberg et al. 2001). In the first step, two segments of the donor plasmid were amplified by PCRs. One PCR used primers p1 (5′-exon positions −25 to +18 with modifications at positions −12 to −1 for IBS1 and IBS2; for Seb-123a donor plasmids, positions −25 to −13 were also changed to ATAATTATCCTTA to prevent self-targeting) plus p2 (5′-CGAAATTAGAAACTTGCGTTCAGTAAAC). The other PCR used primers p3 (intron positions +198 to +246 with modifications at positions +223 to +227 for EBS2) plus p4 (intron positions +259 to +326 with modifications at positions +276 to +285 for EBS1 and δ). In the second step, the two PCR products from the first step were gel-purified, then mixed and amplified with the outside primers p1 and p4 to generate a 353-bp PCR product corresponding to the 5′-exon and 5′-end of the intron (nucleotide positions E1–25 to I + 326), with modified IBS1/2, EBS2, and EBS1/δ sequences. The final PCR product was purified in a 0.8% agarose gel and cloned between the BsrGI and HindIII sites of the donor plasmid. In initial experiments for disruption of hsa and seb, the modifications were first introduced into pACD2X in order to validate the targetron, using an E. coli plasmid assay for targetron integration (Guo et al. 2000; Karberg et al. 2001). The modified pACD2X segment was then recloned into pCN37 (3-kb XbaI + XhoI fragment of pACD2X blunt-ended with T4 DNA polymerase and ligated into the SmaI site of pCN37) to generate the S. aureus donor plasmids pNL9163 and pNL9165, which contain targetrons Hsa-24s and Seb-123a, respectively (Table 1). For pCN39 derivatives, the same modifications targeting the intron to the hsa and seb genes were introduced directly into pNL9162 to generate donor plasmids pNL9164 and pNL9166, respectively (Table 1).
Gene targeting in S. aureus
The donor plasmids containing the retargeted introns were electroporated into S. aureus RN4220 (Schenk and Laddaga 1992). E. coli DNAs are difficult to transform directly into S. aureus RN8098 because of restriction. Consequently, the seb targetron plasmid was first propagated in S. aureus RN4220, re-isolated using a Qiaquick Miniprep Kit (Qiagen), and electroporated into RN8098. Cells were grown overnight in BHI containing erythromycin, diluted 1:100 into 5 mL of fresh medium, grown until early log phase (OD595 = 0.5), and then either plated directly on BHI containing erythromycin or induced with 10 μM CdCl2 for 90 min prior to plating. Targetron integration was assayed by colony PCR, using primers flanking the target gene and referred to generically as pf and pr. For hsa, the pf and pr primers were 5′-CGGAATCAGGAGGTGAATGTCTAATGA and 5′-CGGAATTCACTTAGAAGAACATTACAATTTAT; and for seb, the pf and pr primers were 5′-GGATAAAGGAGATAAAAAATGTATAAGAG and 5′-AGGTACTCTATAAGTGCCTGCCTTAATTAC.
Southern hybridization
Cells were grown up in BHI medium overnight, and DNA was isolated by using a Bacterial Genomic DNA Prep kit (QIAGEN). Southern hybridization was as described (Perutka et al. 2004), using a 32P-labeled intron probe generated by PCR of pACD2X with primers 5′-TCTTGCAAGGGTACGGAGTA and 5′-GTAGGGAGGTACCGCCTTGTTC. The probe was labeled using a High Prime DNA Labeling kit (Roche Diagnostics).
RT-PCR and qRT-PCR analysis of RNA splicing
S. aureus RN4220 strains containing pCN37- or pCN39-based plasmids were grown overnight in BHI medium containing erythromycin at 37°C or 32°C, respectively, then 200 μL of the overnight culture was inoculated into 20 mL of fresh medium and incubated under conditions specified for individual experiments in Figures 5 and 6. After the incubations, RNA was extracted from 1 mL of culture by using an RNeasy mini Kit (Qiagen). RT-PCR was carried out on 300 ng of RNA by using the SuperScript One-Step RT-PCR system (Invitrogen), with primers flanking the inserted intron. For the Ll.LtrB-ΔORF intron inserted in hsa (Fig. 5), the primers were 5′-AGGTGAATGTCTAATGAACAAAACAGA and 5′-CATCTACTGCTGAACCAGCTTCTT, and the RT-PCR was done at 50°C, 30 min; 94°C, 5 min; then 20 cycles of 94°C, 30 sec; 55°C, 30 sec; 72°C, 75 sec; followed by 72°C, 7 min. For the Ll.LtrB-ΔORF intron in pNL9161 (Fig. 6), the primers were 5′-CTAGCAGCACGCCATAGTG and 5′-TCGTGAACACATCCATAAC, and the RT-PCR was done at 52°C, 30 min; 94°C, 5 min; then 40 cycles of 94°C, 30 sec, 58°C, 30 sec, 72°C, 90 sec; followed by 72°C, 7 min. The PCR products were analyzed in a 2% agarose gel, which was stained with ethidium bromide.
For E. coli, strain HMS174 (DE3), which contains an IPTG-inducible phage T7 RNA polymerase, was transformed with pACD2X or pACD-LtrB and plated on LB medium containing chloramphenicol. A single colony was grown up in LB medium containing chloramphenicol until OD595 = 0.2–0.3, then 100 μM IPTG was added to induce T7 RNA polymerase transcription, and the culture was incubated at 30°, 37°, or 43°C for 1 h or overnight. RNA was then extracted and analyzed by RT-PCR as above, using the same primers and conditions used to analyze splicing of the Ll.LtrB-ΔORF intron from pNL9161.
Real-time reverse transcription PCR (qRT-PCR) of S. aureus RNA was carried out on 300 ng of cellular RNA samples by using a SYBR green RT-PCR kit (Applied Biosystems), with primers: 5′-ATGAACAAAACAGATTTAATCAATGCAG [PF(E1–E2)] plus 5′- CATCTACTGCTGAACCAGCTTCTT (PR) for ligated exons, and 5′-AGGCGGTACCTCCCTACTTC [PF(Pre)] plus PR for unspliced precursor RNA. The forward primer for precursor RNA corresponds to the 3′-end of the intron, ending 2 nt upstream of the 3′-splice site, while the forward primer for ligated exons spans the splice junction, with 24 nt in the 5′-exon and 4 nt in the 3′-exon, thereby ensuring that it will amplify only ligated exons and not precursor RNA. The common reverse primer is complementary to a sequence in the 3′-exon. The specificity of the primers for their intended RNAs was checked by RT-PCR (data not shown).
qRT-PCR was done using universal thermal cycling conditions, as specified by the manufacturer (48°C, 30 min; 95°C, 10 min; then 40 cycles of 95°C, 15 sec, 60°C, 1 min) in an ABI 7900HT fast real-time PCR system and analyzed by using SDS software (Applied Biosystems). Serial twofold dilutions (1:5–1:1280) of S. aureus wild-type RN4220 cellular RNA were used to generate a standard curve of fluorescence versus cycle number for the 16S rRNA using primers 5′-CTAATCAGAAAGCCACGGCTAAC and 5′-CGGATAACGCTTGCCACCTA. The amounts of hsa precursor RNA and mRNA were calculated by plotting the corresponding CT (cycle number at which the fluorescence crosses a threshold value set by the SDS program) on the standard curve and expressed as a percent of 16S rRNA. The CT values for 16S rRNA were similar in all RNA samples, and control reactions without addition of the RT or RNA substrate showed no nonspecific amplification.
qRT-PCR of E. coli RNAs was done similarly using primers: 5′-GATCGTGAACACATCCATAACCATA plus 5′-AAATTGTTTGCCAGTATAAAGATTCG for ligated exons; 5′-CAAGGCGGTACCTCCCTACTTC plus 5′-AAATTGTTTGCCAGTATAAAGATTCG for unspliced precursor RNA; and 5′-CGTGTTGTGAAATGTTGGGTTAA and 5′-CCGCTGGCAACAAAGGATA for 16S rRNA.
ACKNOWLEDGMENTS
This work was supported by NIH grant GM37949 and Welch Foundation grant F-1607 to A.M.L., and NIH grant AI30138 to R.P.N. We thank David Goerlitz, Hong Cui, and Sijiong Mou for their contributions to initial experiments on S. aureus gene targeting; Jiri Perutka for help with targetron design; and Greg Davis (Sigma-Aldrich) for helpful suggestions.
Footnotes
Article and publication are at http://www.rnajournal.org/cgi/doi/10.1261/rna.68706.
REFERENCES
- Bae T., Schneewind O. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid. 2006;55:58–63. doi: 10.1016/j.plasmid.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Belhocine K., Plante I., Cousineau B. Conjugation mediates transfer of the Ll.LtrB group II intron between different bacterial species. Mol. Microbiol. 2004;51:1459–1469. doi: 10.1111/j.1365-2958.2004.03923.x. [DOI] [PubMed] [Google Scholar]
- Charpentier E., Anton A.I., Barry P., Alfonso B., Fang Y., Novick R.P. Novel cassette-based shuttle vector system for Gram-positive bacteria. Appl. Environ. Microbiol. 2004;70:6076–6085. doi: 10.1128/AEM.70.10.6076-6085.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chee G.-J., Takami H. Housekeeping recA gene interrupted by group II intron in the thermophilic Geobacillus kaustophilus . Gene. 2005;363:211–220. doi: 10.1016/j.gene.2005.08.017. [DOI] [PubMed] [Google Scholar]
- Chen J., Novick R. SvrA, a multidrug exporter (MepA) does not regulate agr or staphylococcal virulence. J. Bacteriol. 2006 [Google Scholar]
- Chen Y., McClane B.A., Fisher D.J., Rood J.I., Gupta P. Construction of an α toxin gene knockout mutant of Clostridium perfringens type A by use of a mobile group II Intron. Appl. Environ. Microbiol. 2005;71:7542–7547. doi: 10.1128/AEM.71.11.7542-7547.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousineau B., Smith D., Lawrence-Cavanagh S., Mueller J.E., Yang J., Mills D., Manias D., Dunny G., Lambowitz A.M., Belfort M. Retrohoming of a bacterial group II intron: mobility via complete reverse splicing, independent of homologous DNA recombination. Cell. 1998;94:451–462. doi: 10.1016/s0092-8674(00)81586-x. [DOI] [PubMed] [Google Scholar]
- Cui X., Matsuura M., Wang Q., Ma H., Lambowitz A.M. A group II intron–encoded maturase functions preferentially in cis and requires both the reverse transcriptase and X domains to promote RNA splicing. J. Mol. Biol. 2004;340:211–231. doi: 10.1016/j.jmb.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Eskes R., Yang J., Lambowitz A.M., Perlman P.S. Mobility of yeast mitochondrial group II introns: Engineering a new site specificity and retrohoming via full reverse splicing. Cell. 1997;88:865–874. doi: 10.1016/s0092-8674(00)81932-7. [DOI] [PubMed] [Google Scholar]
- Ferat J.-L., Le Gouar M., Michel F. A group II intron has invaded the genus Azotobacter and is inserted within the termination codon of the essential groEL gene. Mol. Microbiol. 2003;49:1407–1423. doi: 10.1046/j.1365-2958.2003.03649.x. [DOI] [PubMed] [Google Scholar]
- Frazier C.L., San Filippo J., Lambowitz A.M., Mills D.A. Genetic manipulation of Lactococcus lactis by using targeted group II introns: Generation of stable insertions without selection. Appl. Environ. Microbiol. 2003;69:1121–1128. doi: 10.1128/AEM.69.2.1121-1128.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridkin S.K., Hageman J.C., Morrison M., Sanza L.T., Como-Sabetti K., Jernigan J.A., Harriman K., Harrison L.H., Lynfield R., Farley M.M., et al. Methicillin-resistant Staphylococcus aureus disease in three communities. N. Engl. J. Med. 2005;352:1436–1444. doi: 10.1056/NEJMoa043252. [DOI] [PubMed] [Google Scholar]
- Guo H., Zimmerly S., Perlman P.S., Lambowitz A.M. Group II intron endonucleases use both RNA and protein subunits for recognition of specific sequences in double-stranded DNA. EMBO J. 1997;16:6835–6848. doi: 10.1093/emboj/16.22.6835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H., Karberg M., Long M., Jones J.P., III, Sullenger B., Lambowitz A.M. Group II introns designed to insert into therapeutically relevant DNA target sites in human cells. Science. 2000;289:452–457. doi: 10.1126/science.289.5478.452. [DOI] [PubMed] [Google Scholar]
- Karberg M., Guo H., Zhong J., Coon R., Perutka J., Lambowitz A.M. Group II introns as controllable gene targeting vectors for genetic manipulation of bacteria. Nat. Biotechnol. 2001;19:1162–1167. doi: 10.1038/nbt1201-1162. [DOI] [PubMed] [Google Scholar]
- Kernodle D.S., Voladri R.K., Menzies B.E., Hager C.C., Edwards K.M. Expression of an antisense hla fragment in Staphylococcus aureus reduces α-toxin production in vitro and attenuates lethal activity in a murine model. Infect. Immun. 1997;65:179–184. doi: 10.1128/iai.65.1.179-184.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreiswirth B.N., Löfdahl S., Betley M.J., O'Reilly M., Schlievert P.M., Bergdoll M.S., Novick R.P. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature. 1983;305:709–712. doi: 10.1038/305709a0. [DOI] [PubMed] [Google Scholar]
- Lambowitz A.M., Zimmerly S. Mobile group II introns. Annu. Rev. Genet. 2004;38:1–35. doi: 10.1146/annurev.genet.38.072902.091600. [DOI] [PubMed] [Google Scholar]
- Lambowitz A.M., Mohr G., Zimmerly S. Group II intron homing endonucleases: Ribonucleoprotein complexes with programmable target specificity. In: Belfort M., et al., editors. In Homing endonucleases and inteins. Nucleic acids and molecular biology. Vol. 16. Springer-Verlag; Heidelberg, Germany: 2005. 1997. pp. 121–145. [Google Scholar]
- Matsuura M., Saldanha R., Ma H., Wank H., Yang J., Mohr G., Cavanagh S., Dunny G.M., Belfort M., Lambowitz A.M. A bacterial group II intron encoding reverse transcriptase, maturase, and DNA endonuclease activities: Biochemical demonstration of maturase activity and insertion of new genetic information within the intron. Genes & Dev. 1997;11:2910–2924. doi: 10.1101/gad.11.21.2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr G., Smith D., Belfort M., Lambowitz A.M. Rules for DNA target-site recognition by a lactococcal group II intron enable retargeting of the intron to specific DNA sequences. Genes & Dev. 2000;14:559–573. [PMC free article] [PubMed] [Google Scholar]
- Novick R.P., Schlievert P., Ruzin A. Pathogenicity and resistance islands of staphylococci. Microbes Infect. 2001;3:585–594. doi: 10.1016/s1286-4579(01)01414-9. [DOI] [PubMed] [Google Scholar]
- Perutka J., Wang W., Goerlitz D., Lambowitz A.M. Use of computer-designed group II introns to disrupt Escherichia coli DExH/D-box protein and DNA helicase genes. J. Mol. Biol. 2004;336:421–439. doi: 10.1016/j.jmb.2003.12.009. [DOI] [PubMed] [Google Scholar]
- Saldanha R., Chen B., Wank H., Matsuura M., Edwards J., Lambowitz A.M. RNA and protein catalysis in group II intron splicing and mobility reactions using purified components. Biochemistry. 1999;38:9069–9083. doi: 10.1021/bi982799l. [DOI] [PubMed] [Google Scholar]
- San Filippo J., Lambowitz A.M. Characterization of the C-terminal DNA-binding/DNA endonuclease region of a group II intron-encoded protein. J. Mol. Biol. 2002;324:933–951. doi: 10.1016/s0022-2836(02)01147-6. [DOI] [PubMed] [Google Scholar]
- Schenk S., Laddaga R.A. Improved method for electroporation of Staphylococcus aureus . FEMS Microbiol. Lett. 1992;94:133–138. doi: 10.1016/0378-1097(92)90596-g. [DOI] [PubMed] [Google Scholar]
- Shafer W.M., Iandolo J.J. Chromosomal locus for staphylococcal enterotoxin B. Infect. Immun. 1978;20:273–278. doi: 10.1128/iai.20.1.273-278.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D., Zhong J., Matsuura M., Lambowitz A.M., Belfort M. Recruitment of host functions suggests a repair pathway for late steps in group II intron retrohoming. Genes & Dev. 2005;19:2477–2487. doi: 10.1101/gad.1345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viter S., Shaw K.J., Gennaro M.L. HSa of Staphylococcus aureus, a new member of the HU family of bacterial histone-like proteins. Res. Microbiol. 1999;150:287–290. doi: 10.1016/s0923-2508(99)80054-0. [DOI] [PubMed] [Google Scholar]
- Yao J., Zhong J., Lambowitz A.M. Gene targeting using randomly inserted group II introns (targetrons) recovered from an Escherichia coli gene disruption library. Nucleic Acids Res. 2005;33:3351–3362. doi: 10.1093/nar/gki649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J., Karberg M., Lambowitz A.M. Targeted and random bacterial gene disruption using a group II intron (targetron) vector containing a retrotransposition-activated selectable marker. Nucleic Acids Res. 2003;31:1656–1664. doi: 10.1093/nar/gkg248. [DOI] [PMC free article] [PubMed] [Google Scholar]