

Abstract
In the yeast Saccharomyces cerevisiae, Mer1p is expressed only during meiosis, and its expression is linked to the splicing of at least three mRNAs: MER2, MER3, and AMA1. Previous evidence suggests that Mer1p activates splicing by directly recruiting snRNPs or stabilizing intermediate splicing complexes formed on pre-mRNA that contains an intronic Mer1p enhancer element. However, some splicing factors, especially accessory/non-snRNP factors, have critical roles in retaining unspliced pre-mRNAs in the nucleus. We tested if Mer1p may indirectly regulate splicing by preventing the export of pre-mRNAs to the cytoplasm and also demonstrated that a second subunit of the Retention and Splicing (RES) complex, Bud13p, has transcript-specific effects on Mer1p-activated splicing. The results indicated that Mer1p can retain unspliced pre-mRNA in the nucleus; however, nuclear retention could not be uncoupled from splicing activation. In the absence of Mer1p, the AMA1 pre-mRNA is exported to the cytoplasm, translated, but not subjected to nonsense-mediated decay (NMD) despite a premature stop codon in the intron. These data imply that Mer1p can retain pre-mRNAs in the nucleus only by facilitating their interaction with the spliceosome and that two subunits of the RES complex modulate Mer1p function on two of the three Mer1p-dependent introns. The results also support models for cytoplasmic degradation of unspliced pre-mRNAs that fail to assemble into spliceosomes in yeast.
Keywords: splicing, nuclear retention, Mer1p, RES complex, nonsense-mediated decay
INTRODUCTION
Precursor messenger RNAs (pre-mRNAs) produced by RNA polymerase II in the yeast Saccharomyces cerevisiae undergo several processing events before they are exported to the cytoplasm for translation. One of these processing events, splicing, removes intervening sequences, or introns, from pre-mRNAs to produce mature mRNAs that have uninterrupted translational reading frames. Splicing occurs by two sequential transesterification reactions and uses three conserved intronic elements: the 5′ splice site sequence, the branchpoint sequence, and the 3′ splice site sequence. The reactions are catalyzed by the spliceosome, a macromolecular complex consisting of five small nuclear ribonucleoprotein particles (snRNPs) and several accessory proteins (for reviews, see Staley and Guthrie 1998; Brow 2002; Jurica and Moore 2003; Butcher and Brow 2005). In yeast, the splicing process is initiated when the U1 snRNP binds to the 5′ splice site region of a pre-mRNA to form a commitment complex (CC) (Seraphin and Rosbash 1989). This complex is stabilized by base-pairing between U1 snRNA and the 5′ splice site sequence (Seraphin et al. 1988; Siliciano and Guthrie 1988) and by several U1 snRNP protein–mRNA interactions (Puig et al. 1999; Zhang and Rosbash 1999). After the CC has formed, the U2 snRNP binds, and base pairs form between U2 snRNA and the branchpoint sequence of the intron (Parker et al. 1987; Wu and Manley 1989). The remaining U4, U5, and U6 snRNPs bind as a tri-snRNP to the above pre-spliceosome (Cheng and Abelson 1987), and several conformational changes ensue that lead to the displacement of the U1 and U4 snRNPs and formation of a catalytically active spliceosome (for reviews, see Staley and Guthrie 1998; Brow 2002; Butcher and Brow 2005).
The above accretion model for spliceosome assembly is based on numerous in vitro studies and was called into question with the isolation of a functional “penta-snRNP” spliceosome holoenzyme from cells (Stevens et al. 2002). However, recent studies measuring spliceosome assembly in vivo support the accretion model (Gornemann et al. 2005; Lacadie and Rosbash 2005). Regardless of whether the spliceosome binds to pre-mRNA sequentially as individual snRNPs or simultaneously as a holoenzyme, significant RNA and snRNP rearrangements must occur prior to and during both chemical reactions. For example, U6, which also forms base pairs with the 5′ splice site (Kandels-Lewis and Seraphin 1993), cannot pair with the 5′ splice site until U1 has been displaced by Prp28p (Staley and Guthrie 1999), and U4 must unwind from U6 before U6 can form base pairs with U2 to form the catalytic core of the spliceosome (Hausner et al. 1990; Raghunathan and Guthrie 1998). This dynamic nature of spliceosome assembly provides ample opportunities for splicing regulators to affect the formation of the spliceosome and alter selection of splice sites.
Mer1p is expressed only during meiosis (Engebrecht et al. 1991), and its expression corresponds to increases in splicing of at least three genes required for meiosis: MER2/REC107, MER3/HFM1, and AMA1/SPO70 (Engebrecht et al. 1991; Nakagawa and Ogawa 1999; Davis et al. 2000). Although evidence suggests that Mer1p activates splicing by affecting the formation or stability of the earliest splicing complexes on pre-mRNAs that contain the Mer1p intronic enhancer element (Nandabalan et al. 1993; Nandabalan and Roeder 1995; Spingola and Ares 2000; Spingola et al. 2004), an in vitro demonstration of Mer1p's effects on spliceosome assembly and splicing has been elusive. An alternative hypothesis for Mer1p's function is that its major role is to prevent unspliced enhancer-containing pre-mRNAs from escaping the nucleus or from being degraded before being spliced in the nucleus. Indeed, several retention factors have been described and fall into two categories: retention factors that modulate spliceosome activity and retention factors that do not modulate spliceosome activity. The latter category includes Pml1p, a component of the RES complex (Dziembowski et al. 2004), and M1p1p and M1p2p, which line the nuclear pore complex (NPC), prevent pre-mRNAs from exporting to the cytoplasm, down-regulate transcription, and may link the NPC to euchromatin (Galy et al. 2004; Casolari et al. 2005; Vinciguerra et al. 2005). While loss of Mlp1p or Pml1p has no effect on splicing, their loss is accompanied by the export and translation of unspliced pre-mRNAs (Dziembowski et al. 2004; Galy et al. 2004). In contrast, the loss of several accessory splicing factors and early-acting snRNP proteins has been shown to have small to moderate reductions on splicing but large increases in the export and translation of unspliced pre-mRNAs. Chief among these proteins are Branchpoint Binding Protein (Bbp1p), and Mud2p, two non-snRNP accessory factors that bind to the commitment complex (Rain and Legrain 1997; Rutz and Seraphin 2000). It has been proposed that the essential role of Bbp1p is nuclear retention and not splicing because extracts depleted of Bbp1p (the homolog of mammalian SF1) show no splicing defects with a model pre-mRNA in vitro, temperature-sensitive bbp1 alleles allow pre-mRNAs to export to the cytoplasm and be translated while only showing a slight reduction in splicing with sensitive splicing reporters that have nonconsensus splicing signals, and a bbp1 allele is synthetic lethal with the deletion of a nonsense-mediated decay gene, UPF1 (Rutz and Seraphin 1999, 2000).
Our analysis of pre-mRNA export to the cytoplasm indicates that Mer1p, like many splicing factors that act early during the splicing process (Legrain and Rosbash 1989), can help retain unspliced pre-mRNA in the nucleus, but that this retention effect cannot be separated or uncoupled from splicing. At a minimum, retention by Mer1p requires a functional 5′ splice site, the Mer1p intronic enhancer element, the U1 snRNP protein Nam8p, and the domains of Mer1p that interact with the U1 snRNP and enhancer. Furthermore, AMA1 pre-mRNA is readily exported to the cytoplasm if Mer1p is not present to activate its splicing, and unlike the MER2 and MER3 unspliced pre-mRNAs that leak to cytoplasm, the AMA1 pre-mRNA that is exported to the cytoplasm is not degraded by the nonsense-mediated decay process.
Recently a trimeric complex involved in nuclear retention and splicing, the RES complex, was purified from yeast (Dziembowski et al. 2004). Two of its components have been described as splicing and retention factors, Snu17p/Ist3p and Bud13p, and one as a retention factor that does not affect splicing, Pml1p. Snu17p is also a subunit of the U2 snRNP (Wang and Rymond 2003; Wang et al. 2005). Since Snu17p is required for Mer1p function (Spingola et al. 2004), we tested if the remaining two subunits of the RES complex were critical for Mer1p function. The results show that loss of Bud13p abolishes Mer1p-activated splicing of AMA1, impairs Mer1p-activated splicing of MER2, and has no effect on Mer1p-activated splicing of MER3. Loss of Pml1p had little effect on Mer1p-activated splicing. These results suggest that one function of the RES complex may be to regulate differential splicing during meiosis by modulating Mer1p's activity on specific transcripts. Furthermore, our data support the model that Mer1p activates splicing by stabilizing or promoting the formation of early splicing complexes on pre-mRNA. Our data also support the model proposing that unspliced pre-mRNAs in yeast that are poorly spliced and do not efficiently assemble into spliceosomes are exported to the cytoplasm and not degraded in the nucleus (Hilleren and Parker 2003).
RESULTS
The Bud13p subunit of the RES complex has transcript-specific effects on Mer1p-activated splicing
Snu17p is required for Mer1p-activated splicing of AMA1 (Spingola et al. 2004) and is a subunit of the U2 snRNP (Wang et al. 2005) and the RES complex, which also includes Bud13p and Pml1p (Dziembowski et al. 2004). Bud13p and Snu17p are thought to be splicing factors, while Pml1p has been characterized as a retention factor. Since Bud13p and Pml1p form a complex with a protein that is required for Mer1p-activated splicing, we determined if these subunits of the RES complex are also important for Mer1p function. Strains deleted of either of the two remaining genes were obtained and transformed with a MER1 expression plasmid and splicing reporter plasmids. RNA was isolated from these cells and analyzed for Mer1p-activated splicing by primer extension. The results (Fig. 1) indicate that Bud13p, like Snu17p, is critical for Mer1p-activated splicing of AMA1 reporter pre-mRNA. Furthermore, loss of Bud13p causes a reduction in the constitutive splicing that occurs without Mer1p for AMA1 mRNA (from 31% spliced to 14% spliced) (Fig. 1; Table 1). The third component of the RES complex, Pml1p, is not required for Mer1p to activate AMA1 splicing, but its loss slightly reduced the AMA1 splicing levels.
FIGURE 1.

Primer extension analysis of AMA1, MER2, and MER3 splicing in wild-type or RES deletion strains with or without constitutive expression of MER1. U represents cDNAs from unspliced RNA; S represents cDNA from spliced mRNA. In A, a primer complementary to AMA1 exon 2 was used. In B and C, a CUP1 primer was used, which also primed reverse transcription on endogenous CUP1 RNA. Splicing efficiencies are reported below each lane using the formula S/(S + U) × 100. Primer extension of a polymerase III transcript, 7S RNA, was performed as a loading control.
TABLE 1.
Splicing efficiencies for Mer1p-dependent introns in RES deletion strains
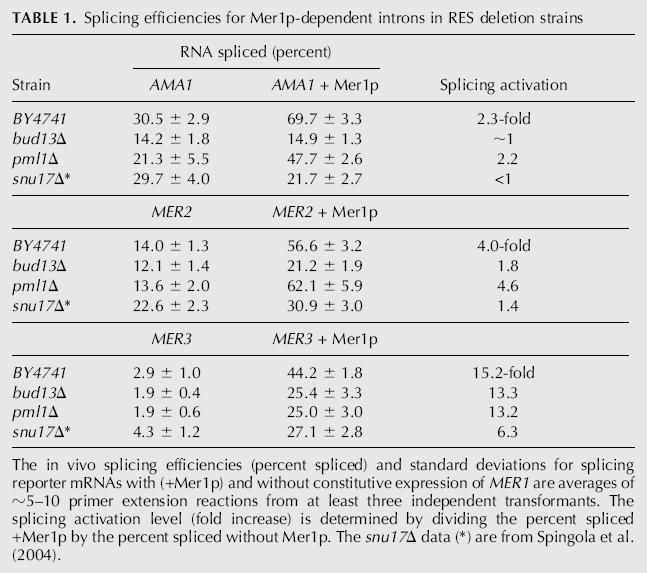
We extended our splicing studies with the RES deletion strains to MER2 and MER3 splicing reporters and surprisingly found dramatically different requirements for each pre-mRNA. Mer1p could not activate splicing of MER2 to the wild-type level when Bud13p is deleted. When Bud13p is present, there is approximately a fourfold activation of splicing by Mer1p, but only a twofold activation when Bud13p is deleted. In contrast, loss of Bud13p did not at all hinder the ability of Mer1p to activate the splicing of MER3 (see Fig. 1; Table 1). Thus, Bud13p is essential for the Mer1p-activated splicing of AMA1, helpful but not essential for Mer1p-activated splicing of MER2, and not necessary or helpful for the Mer1p-activated splicing of MER3. We conclude that Bud13p modulates the activity of Mer1p on certain transcripts.
Mer1p prevents pre-mRNAs containing the intronic splicing enhancer from exporting to the cytoplasm
Several factors first isolated as splicing factors have been shown to prevent pre-mRNA “leakage” to the cytoplasm (Legrain and Rosbash 1989; Rain and Legrain 1997; Rutz and Seraphin 2000; Dziembowski et al. 2004). Null or conditional alleles of these genes often had minimal effects on splicing but allowed pre-mRNAs to export out of the nucleus into the cytoplasm, suggesting that while these factors may enhance splicing, they have critical roles in retaining pre-mRNAs in the nucleus. Mer1p, with the ability to bind pre-mRNA containing a Mer1p enhancer element (Spingola and Ares 2000), could potentially block export of pre-mRNAs and retain them in the nucleus for splicing. To address this possibility, we tested if Mer1p affects export of enhancer-containing pre-mRNAs by constructing a reporter gene (CUP1) that is fused to a portion of the Mer1p-activatable genes in two different translational frames (see Fig. 2). For example, the AMA1-CUP1 splicing reporter only produces reporter protein, which allows cells to grow in the presence of copper, if the intron is removed by splicing. Unless MER1 is expressed, the level of spliced reporter mRNA is insufficient to support growth on copper (Spingola and Ares 2000). The AMA1-CUP1 export reporter only produces reporter protein if the intron is not removed, and the pre-mRNA is exported to the cytoplasm and translated. Analogous MER2-CUP1 and MER3-CUP1 splicing and export reporters were also constructed and tested. When the export reporter plasmids are expressed in cup1Δ yeast, cells constitutively expressing MER1 do not survive on media containing copper (Fig. 3A). In contrast, strains that do not express MER1 grow on media containing copper. These results indicate that (1) unspliced reporter pre-mRNAs are exported to the cytoplasm and translated, and (2) that Mer1p inhibits this process, either by facilitating the conversion of pre-mRNA into mRNA or by physically preventing pre-mRNA from exporting to the cytoplasm.
FIGURE 2.

Design of splicing reporter and export reporter plasmids. The numbers indicate the nucleotides of AMA1 (nucleotides 900–1320) fused to CUP1 and mark the first nucleotide of the intron (nucleotide 1184) and the first nucleotide of exon 2 (nucleotide 1277). The octagonal stop sign indicates the location of premature stop codons in the constructs. Analogous plasmids were constructed for MER2 and MER3 and included the entire exon 1, intron, and ∼25 nt of exon 2.
FIGURE 3.

Growth/export assays and splicing assays. (A) Growth of KH46 yeast (cup1Δ) containing the AMA1-CUP1 export reporter plasmid and either a MER1 expression vector or a control vector on media containing 150 μM copper. (B) Primer extension analysis of export and splicing reporter RNAs isolated from KH46 yeast. Splicing efficiencies are reported below each lane using the formula S/(S + U) × 100. (C) Growth at 30°C of AMA1-, MER2-, and MER3-CUP1 export (-E) and splicing (-S) reporters on copper in strains KH46 (wild type) and KH46-bud13Δ with (+) and without (−) MER1 expression. A variety of copper concentrations was used to best demonstrate differences due to Mer1p. The micromolar concentration of copper is printed on each panel.
Analysis of AMA1-CUP1 export reporter RNA by primer extension indicates that Mer1p does not affect the abundance of RNA (spliced plus unspliced); neither an increase nor a decrease in total reporter RNA is apparent (Fig. 3B). Thus it is unlikely that Mer1p causes a down-regulation of transcription of the export reporter that is ultimately responsible for the copper-sensitive phenotype. Rather, an increase in spliced product is measured with a concomitant decrease in unspliced RNA. This suggests that the major reason for a copper-sensitive phenotype when Mer1p is expressed with the export reporter is the conversion of pre-mRNA into mRNA by splicing.
We extended our analysis of splicing and export reporters to MER2 and MER3. The growth patterns on media containing copper largely correlate to their splicing efficiencies. Cells with MER2 and MER3 export reporters grew readily on media containing copper, and the amount of growth was reduced by the expression of Mer1p (Fig. 3C). Furthermore in the KH46-bud13Δ strain, Mer1p did not change the level of growth for either the AMA1 splicing or export reporter, and Mer1p led to only subtle changes in growth for the MER2 reporters. In the KH46-bud13Δ strain, Mer1p had the same effect on MER3 as in the wild-type strain: It inhibited growth for cells containing the MER3 export reporter and stimulated growth for cells containing the splicing reporter. We conclude that Mer1p inhibits the export of unspliced AMA1, MER2, and MER3 pre-mRNAs, and that nuclear retention of AMA1 by Mer1p, like splicing activation, requires Bud13p.
Past research has suggested that the primary role for some splicing factors, in particular, Bbp1p, may actually be to retain unspliced pre-mRNA in the nucleus (Rutz and Seraphin 2000). By measuring the activity of export reporters, unspliced pre-mRNAs are exported to the cytoplasm and translated in strains with temperature-sensitive bbp1 alleles or deletions of Mud2p, Snu17p, or Bud13p (Rain and Legrain 1997; Rutz and Seraphin 2000; Dziembowski et al. 2004). As an additional test to determine if Mer1p might also have a role in retaining pre-mRNAs in the nucleus, we measured β-galactosidase enzyme activity produced from AMA1 export and splicing reporters that have the LacZ gene in place of CUP1 in a variety of strains including BY4741 (wild-type), snu17Δ, KH46-bud13Δ, pml1Δ, mud2Δ, and KH46-nam8Δ. In the wild-type strain, Mer1p has similar effects on both reporters; Mer1p reduces the amount of o-nitrophenol produced by β-galactosidase from the export reporter by about twofold and increases by ∼2.5-fold with the splicing reporter (see Fig. 4). In snu17Δ, bud13Δ, and nam8Δ, very little β-galactosidase is produced from the splicing reporter, as indicated by the low levels of o-nitrophenol produced, and the levels do not change with MER1 expression, consistent with the observation that these proteins are needed for Mer1p-activated splicing of AMA1. With the export reporter, much more β-galactosidase is produced in snu17Δ, bud13Δ, and nam8Δ, and the levels of o-nitrophenol produced approach that of the wild-type strain. Again, Mer1p has little effect on the amount of β-galactosidase produced from the export reporter in these strains. There is a small difference in the amount of β-galactosidase activity in the snu17Δ strain when Mer1p is produced. However, the large standard deviations of these samples imply that these differences are not significant, and moreover, this difference is not as large as the differences seen in strains that support Mer1p-activated splicing (BY4741, mud2Δ, and pml1Δ). These results are consistent with the conclusion that AMA1 pre-mRNAs are best retained in the nucleus by Mer1p only if splicing activation can occur. The loss of Nam8p, Snu17p, or Bud13p has only a minimal effect, if any, on the basal level of AMA1 splicing without Mer1p. Consistent with a minimal effect on splicing, the loss of these factors has only a minimal effect on export as well.
FIGURE 4.

Splicing and pre-mRNA export analysis with the AMA1-LacZ reporter plasmids. The nanomoles of o-nitrophenol produced by β-galactosidase cleavage of o-nitrophenyl-β-D-galactopyranoside are averages of duplicates on at least two independent transformants. Error bars represent the standard deviation. The strains used include the wild type (BY4741), KH46-nam8Δ, KH46-bud13Δ, snu17Δ, mud2Δ, and pml1Δ.
The AMA1 reporter pre-mRNA, which is poorly spliced (∼30% is spliced), seems to efficiently leak to the cytoplasm. In the absence of Mer1p, the β-galactosidase activity from the ∼30% spliced mRNA from the splicing reporter is nearly equal to the signal generated from the ∼30% unspliced pre-mRNA from the export reporter in the presence of Mer1p; this suggests that most of the unspliced AMA1 pre-mRNA is leaking to the cytoplasm and being translated. In the pml1Δ strain, relative to wild-type, there is a slight reduction in β-galactosidase activity with the splicing reporter and a small increase in activity for the export reporter, consistent with a role for Pml1p in export and not in splicing. The deletion of Mud2p severely inhibits the activity from the splicing reporter when Mer1p is not produced, but Mer1p restores β-galactosidase levels to ∼60% of the wild type. Previously we have shown that the deletion of Mud2p severely reduces the splicing of AMA1, but that Mer1p can activate splicing without Mud2p (Spingola and Ares 2000). With the export reporter, loss of Mud2p increases the levels of β-galactosidase more than the deletion of the factors required for Mer1p-activated splicing (Snu17p, Bud13p, and Nam8p) and to levels greater than the wild type. This observation is consistent with Mud2p playing a role in both splicing and nuclear retention.
Nuclear retention by Mer1p cannot be uncoupled from splicing activation
If Mer1p has a role in retaining pre-mRNAs in the nucleus that is distinct from its role in activating splicing, it may be possible to uncouple the two activities. We attempted this with (1) mutant alleles of MER1 that cannot activate splicing but still contain the RNA-binding KH domain (KH in Fig. 5A), (2) strains of yeast that do not support Mer1p-activated splicing (nam8Δ), and (3) mutant introns that cannot splice because the 5′ splice site sequence has been altered from GUACGU to AUACGU (a G1A mutation). In each case, splicing activation could not be uncoupled from nuclear retention, and cells containing the CUP1 export reporter grew on media containing copper. For example, the KH domain fragment of Mer1p contains the structural motif (Siomi et al. 1993) that binds to RNA, but it does not activate splicing (Spingola and Ares 2000). If binding to the intronic enhancer were sufficient to elicit nuclear retention, perhaps by preventing export factors from binding, then the KH fragment should cause retention, and the yeast will not grow on copper. However, the KH fragment does not elicit retention, and yeast continue to export and translate the pre-mRNA, which allows growth on media containing copper (Fig. 5A). Primer extension analysis verifies that the KH fragment does not activate splicing (Fig. 5D, lane 4). Secondly, Mer1p requires a nonessential U1 snRNP protein, Nam8p, to activate splicing (Fig. 5D, lanes 8,9). If Mer1p has a role in retention that is independent of splicing, then it should not require a splicing factor for retention. In nam8Δ cells, Mer1p does not activate splicing (Fig. 5D; Spingola and Ares 2000); however, cells containing the export reporter grow on copper, indicating that Mer1p also fails to retain pre-mRNA and demonstrating that Nam8p is necessary for this retention effect (Fig. 5B). Thirdly, we also tested if a cis-acting mutation to the 5′ splice site (G1A) that abolishes splicing would uncouple splicing from nuclear retention. Once again, cells with the export reporter grew on copper; thus, Mer1p did not retain the pre-mRNA in the nucleus (Fig. 5C). Primer extension results verify that Mer1p does not activate the splicing of the G1A intron (Fig. 5D, lanes 10,11).
FIGURE 5.

Growth and export assays for (A) yeast containing the AMA1-CUP1 export reporter and MER1 alleles that do not activate splicing, including the activation domain (MER1 AD) or KH domain (MER1 KH) fragments; (B) nam8Δ yeast carrying the AMA1-CUP1 export reporter; and (C) yeast carrying the AMA1-CUP1 export reporter with a G1A mutation in the intron, which abolishes its splicing. Media contain 150 μM copper, and cells were grown for 3 d at 30°C. (D) Primer extension assay for splicing of RNAs from cells containing various export reporters and MER1 alleles described in the text. Splicing efficiencies are reported below each lane using the formula S/(S + U) × 100. (E) Growth of yeast with and without Mer1p on 100 μM copper for strains containing the AMA1 export reporter or export reporter variant containing mutations to the enhancer element (ME), which abolish Mer1p-activated splicing.
Included in Figure 5D are AMA1 splicing data for miscellaneous MER1 alleles that do not activate splicing including the activation domain fragment of MER1 (AD), a C-terminal deletion of MER1 (ΔC) that lacks a short peptide adjacent to the KH domain, and an allele (GAQE) with mis-sense mutations in the codons for a highly conserved GXXG peptide element of the KH domain that contacts RNA (Lewis et al. 2000). Cells carrying the above alleles and the export reporter grow readily on copper (Fig. 5A; data not shown), and these alleles cannot facilitate nuclear retention of the AMA1 export reporter pre-mRNAs. Retention by Mer1p also relies on a functional enhancer element in the pre-mRNA. When the enhancer sequence is altered to one that does not support Mer1p-activated splicing (Fig. 5E), Mer1p can no longer retain the pre-mRNA in the nucleus. Nuclear retention of pre-mRNA by Mer1p requires at a minimum a functional 5′ splice site, the Mer1p enhancer element, the domains of Mer1p that interact with the U1 snRNP and bind to the enhancer, and an integral component of the U1 snRNP, Nam8p. We conclude that the ability of Mer1p to retain unspliced AMA1 pre-mRNA in the nucleus is due solely to its ability to activate splicing.
Exported AMA1 pre-mRNA is not subjected to nonsense-mediated decay
Unspliced pre-mRNAs that are exported to the cytoplasm can be degraded rapidly before ribosomes can initiate multiple rounds of translation on them (Maquat 2004). This quality-control system prevents unspliced pre-mRNAs from being translated into truncated proteins that may be lethal or harmful to the organism, besides being energetically wasteful. Yet AMA1 export reporter pre-mRNA is exported to the cytoplasm and readily translated in the absence of Mer1p (see Fig. 3A). One possible explanation for the translation of unspliced AMA1 pre-mRNA is that wild-type AMA1 pre-mRNA is degraded before translation in the cytoplasm by NMD, while the AMA1 export reporter is not. In fact, an in-frame premature stop codon had to be deleted from the intron of the export reporter to make its pre-mRNA reading frame open (see Materials and Methods). It is possible that by abolishing this stop codon the RNA is rendered resistant to NMD, and thus can be translated. To test this possibility, the half-lives of AMA1-CUP1 splicing reporter RNA and full-length AMA1 RNA, which both contain the intronic stop codon, were measured in isogenic strains of yeast that differ only by the deletion of the UPF1 gene (Kebaara et al. 2003), which is critical to NMD. These yeast also contain a temperature-sensitive lesion in a polymerase II subunit (rpb1-1) that allows for the inactivation of polymerase II transcription by increasing the temperature to 37°C. RNA was extracted from yeast after shifting to the restrictive temperature and measured by primer extension (Fig. 6A). A significant reduction in the half-life of the AMA1-CUP1 splicing reporter pre-mRNA is not apparent when UPF1 is deleted. Instead, the RNA has a slightly longer half-life in the presence of Upf1p: 37 min in the UPF1 strain and 26 min in the upf1Δ strain (Fig. 6B). Since the splicing reporter construct contains only a small portion of exon 2 of AMA1, it was possible that a downstream element important for NMD (Zhang et al. 1995) was not included. A full-length AMA1 clone was used to address this possibility, and less than a twofold difference in pre-mRNA stability was observed when Upf1p was deleted (Fig. 6C). The half-life for full-length AMA1 pre-mRNA was ∼42 min in upf1Δ cells and 35 min in UPF1. The 35-min T 1/2 is very close to a previously reported T 1/2 of 31 min from a genome-wide microarray measurement of RNA decay (Wang et al. 2002). We conclude that although AMA1 pre-mRNA contains a premature stop codon in its intron, it is not subjected to NMD.
FIGURE 6.

(A) Primer extension assay of pre-mRNA stability following the inactivation of RNA polymerase II in isogenic strains containing UPF1 or upf1Δ. Bands corresponding to unspliced pre-mRNA from the splicing reporter were quantified and normalized to a polymerase III transcript, 7S RNA, and plotted as the percent RNA remaining from time zero in B from five separate experiments. (C) A plot for the decay of full-length AMA1 RNA after inactivation of RNA polymerase II in isogenic strains containing or lacking UPF1. The data are averages from two independent trials.
DISCUSSION
Mer1p is a splicing regulator that prevents export of pre-mRNAs by facilitating their splicing
It has been proposed that several factors first characterized as splicing factors, in particular, Bbp1p and Mud2p, may have critical roles in retaining pre-mRNAs in the nucleus that cannot be attributed to their roles in splicing (Rain and Legrain 1997; Rutz and Seraphin 2000; Dziembowski et al. 2004). The deletion or inactivation of these genes has little effect on the splicing of various reporters but induces the export of the same reporter pre-mRNA. If Mer1p also has a role in preventing export of enhancer-containing pre-mRNAs, then the observed increases in splicing of these pre-mRNAs attributed to Mer1p could be the indirect result of nuclear retention. By retaining the pre-mRNA in the nucleus, the pre-mRNAs would have more opportunities to interact with snRNPs, which might account for the increase in splicing when Mer1p is expressed. We addressed whether Mer1p might also have a role in retaining unspliced pre-mRNAs in the nucleus by measuring the effect of Mer1p on splicing reporters and export reporters. We found that unspliced AMA1 reporter pre-mRNA is exported to the cytoplasm and translated, and that Mer1p reduces the amount of pre-mRNA that is exported and translated. Thus, in the broadest sense, Mer1p is a retention factor.
To further support the above claim, we attempted to uncouple splicing from nuclear retention with a mutant allele of MER1 that does not activate splicing, and with cis- and trans-acting mutants that disrupt splicing. In none of the cases above were we able to uncouple splicing activation from nuclear retention. The KH-domain fragment of Mer1p lacks a splicing activation domain but contains the RNA-binding domain. Presumably, the KH domain could bind to enhancer-containing pre-mRNA and block the binding of export factors, thus causing retention. Since the KH-domain fragment cannot activate splicing but can bind pre-mRNA, it could possibly uncouple retention from splicing activation. However, the KH domain failed to retain unspliced AMA1 export reporter pre-mRNA. Mer1p-mediated retention also failed to occur in cells missing a nonessential snRNP splicing factor protein, Nam8p, which is required for Mer1p function. Lastly, Mer1p-mediated retention did not occur if the 5′ splice site sequence was altered to one that abolishes splicing. Any nuclear retention by Mer1p required a functional 5′ splice site, a functional Mer1p intronic splicing enhancer element, the domains of Mer1p that interact with snRNPs and the enhancer, and the U1 snRNP protein Nam8p. Although we attempted only a few means of uncoupling nuclear retention from splicing, and by no means have exhausted a search for alleles that could uncouple splicing from retention, the results strongly suggest that Mer1p's ability to retain pre-mRNA in the nucleus is functionally linked to activating splicing and that Mer1p does not increase splicing indirectly by preventing export of pre-mRNA.
AMA1 pre-mRNA is not a substrate for nonsense-mediated decay
Several quality-control mechanisms that degrade aberrantly spliced and unspliced pre-mRNAs have been described that function in the nucleus or in the cytoplasm (Bousquet-Antonelli et al. 2000; Das et al. 2003; Hilleren and Parker 2003; Conti and Izaurralde 2005). These quality-control systems would seem to be critical in preventing aberrant or unspliced pre-mRNAs from being translated into proteins that are truncated and could have deleterious effects on the organism. Surprisingly, unspliced AMA1 pre-mRNA seems to evade these quality-control mechanisms and is translated. AMA1 pre-mRNA contains a premature stop codon in its intron, but no differences in its degradation rate were measured when NMD was functional or nonfunctional. In contrast, both MER2 and MER3 pre-mRNAs are substrates for NMD (He et al. 1993, 2003), and when NMD is disabled, their pre-mRNAs accumulate 10-fold and fivefold, respectively (He et al. 2003). Two important requirements for NMD in yeast are the position of the premature stop codon and the presence of an element downstream of the premature stop codon (Hagan et al. 1995). In the AMA1-CUP1 reporter pre-mRNA and the full-length AMA1 pre-mRNA, the premature stop codons are found at nucleotide 330 in a 612-nt transcript and at nucleotide 1230 in a 2283-nt transcript, respectively (positions are relative to the annotated start and stop codons). Hence, the first 54% of the AMA1 pre-mRNA could be translated before the premature stop codon is encountered. In contrast, only the first 38% of MER2 and 2% of MER3 pre-mRNAs would be translated before the premature stop codon is encountered (stop codons at nucleotide 396 of 1025 total and at 75 of 3716 total, respectively). With other model transcripts, a premature stop codon found in the last third of the transcript does not elicit NMD, and the NMD response diminishes as a larger fraction of the transcript is translated prior to encountering the stop codon (Hagan et al. 1995). Although the position of the AMA1 stop codon is not in the C-terminal third of the protein-coding region, a larger fraction and many more codons of AMA1 pre-mRNA would be translated before the stop codon is encountered relative to MER2, and this may circumvent any NMD. Moreover, MER2 and MER3 may have strong NMD downstream elements and AMA1 may not, although sequence analysis indicates that none of the three genes has a perfect match to the consensus.
Hilleren and Parker (2003) have proposed that “the vast majority of pre-mRNAs that are unable to assemble into spliceosomes degrade by the cytoplasmic mRNA turnover enzymes.” Our results for AMA1 pre-mRNA, which is poorly spliced in the absence of Mer1p, support Hilleren and Parker's model that pre-mRNAs that do not assemble into spliceosomes and do not undergo the first step of splicing are exported to the cytoplasm and not degraded in the nucleus. Other pre-mRNAs that undergo regulated splicing are also degraded in the cytoplasm. For example, the splicing of the RPL30 pre-mRNA can be inhibited when concentrations of Rpl30p are higher than needed for ribosome assembly. The U1 snRNP binds to the RPL30 pre-mRNA, but U2 snRNP is blocked from binding if Rpl30p is bound to the pre-mRNA (Vilardell and Warner 1994). Eventually, U1 snRNP dissociates, the Rpl30p-bound pre-mRNA is exported to the cytoplasm, and, upon dissociation of Rpl30p, the pre-mRNA is subjected to NMD (Vilardell et al. 2000). The remaining Mer1p-regulated pre-mRNAs, MER2 and MER3, suffer a similar fate in the cytoplasm. In the absence of Mer1p, their pre-mRNAs are very poorly spliced (Engebrecht et al. 1991; Nakagawa and Ogawa 1999), and their unspliced pre-mRNAs are exported to the cytoplasm and subjected to NMD (He et al. 1993, 2003).
A role for the RES complex in Mer1p-regulated splicing and meiosis
Snu17p has been described as a U2 snRNP protein (Wang et al. 2005) that is necessary for Mer1p-activated splicing and as a subunit of the RES complex, which also includes Bud13p and Pml1p (Dziembowski et al. 2004). Bud13p, a splicing factor, is also essential for Mer1p activity on specific transcripts, but Pml1p, a retention factor, is not. Although the basis for the requirement of Bud13p and Snu17p in the Mer1p-activated splicing has yet to be determined, our data imply that the RES complex, or two of its subunits, are critical to regulating splicing of a subset of Mer1p-regulated transcripts during meiosis. Based on the observations that loss of Bud13p has the same transcript-specific effects on Mer1p-activated splicing as loss of Snu17p, it is possible that only one of these subunits is needed, but that the loss of the other affects the stability or expression of the required factor. Purifications of the SF3b particle of U2 by the TAP tag method demonstrate that if the tag is on U2 snRNP protein Cus1p, Snu17p copurifies with U2, but only a minor trace of a peptide the size of Bud13p copurifies with U2 (Wang et al. 2005). This suggests that at least a fraction of Snu17p is stable when it is not associated with Bud13p. However, additional experimentation is needed to determine if loss of Snu17p affects the stability of Bud13p or vice versa.
Further experimentation is needed to determine the cis-acting features that make some Mer1p-regulated introns require the Bud13p and Snu17p. Of note, AMA1 is most obviously different from MER2 and MER3 by 5′ exon size. AMA1 has a very large 5′ exon (1183 nt from the start codon), whereas MER2 and MER3 have much shorter 5′ exons (317 nt and 58 nt, respectively). Large 5′ exons have been shown to reduce splicing efficiency, perhaps by destabilizing interactions between the cap-binding complex (CBC) and the commitment complex (CC) (Lewis et al. 1996a, b; Spingola and Ares 2000). A stable CBC–CC interaction may occur with the shorter 5′ exon of MER3, and to a lesser extent with MER2, but not with AMA1. We are currently testing if the RES complex may stabilize commitment complexes formed on mRNAs with large 5′ exons or whether Bud13p and Snu17p stabilize the binding of U2 to commitment complexes formed on pre-mRNAs with large 5′ exons.
MATERIALS AND METHODS
Strains and plasmids
The construction of many of the plasmids and strains used for splicing analysis in this study was described before (Spingola and Ares 2000; Spingola et al. 2004). Strains KH46 and BY4741 or gene deletions in BY4741 (Invitrogen) were used for isolating RNA. KH46 is cup1Δ and was used for copper-sensitivity assays. Strains AAY334 and AAY335 (Kebaara et al. 2003) were used for the mRNA transcription shutoff experiments and have the genotypes MATa ura3- his3-11,15 trp1-1 leu2-3, 112 rpb1-1 and MATa ura3- his3-11,15 trp1-1 leu2-3, 112 rpb1-1 upf1-Δ2 (URA3), respectively. The nonessential splicing gene deletion strains bud13Δ and pml1Δ, were purchased from Invitrogen, Inc. and are derived from strain BY4741 (MATa his3Δ1 leu2Δ 0 met15Δ 0 ura3Δ 0). A bud13Δ::HIS4 strain was produced in the KH46 background (cup1Δ) by the PCR product integration method (Longtine et al. 1998). To distinguish this strain from the bud13Δ strain in the BY4741 background, it is referred to as KH46-bud13Δ.
The splicing reporter CUP1 fusion plasmids, pRS316AMA1-CUP1, pRS316MER2-CUP1, and pRS316MER3-CUP1 were described previously (Spingola and Ares 2000; Spingola et al. 2004). The AMA1-CUP1 export reporter, pRS316CF7B, was constructed from pRS316AMA1-CUP1 using oligonucleotide site-directed mutagenesis (Kunkel et al. 1991). The synthetic oligonucleotide 5′-TTTTCTGGTATA_CGCTTATTTTTTCATTATGAAAAA-3′ deletes the G (the _ in the sequence above) from the in-frame stop codon in the intron. In addition to deleting the intronic stop codon, the translational frame was altered to ensure that only unspliced mRNA would be in the correct frame for production of reporter protein. Using the mutagenic oligonucleotide 5′-TACTAACAAATATTTTCTACAGGGTATTTCTCTGAA-3′, a single nucleotide was inserted (underlined above) at the beginning of the second exon, which disrupts the reading frame for spliced RNA and creates the correct frame for translation of unspliced RNA. The export reporter pRS316CF7B-G1A was created from pRS316CF7B by making a G-to-A substitution at the first nucleotide of the 5′ splice site using site-directed mutagenesis. Plasmid R1070 (constitutive MER1 expression) and its parental vector R1130 were gifts from G.S. Roeder (Yale University) and are described in Engebrecht et al. (1991). Plasmids pGHAMER1 (HA-tagged MER1), pGAD (MER1 splicing activation domain), and pGKH (MER1 RNA-binding KH domain) were derived from pGAC14 as previously described (Spingola et al. 2004). Plasmid pGAQE was derived by subcloning the constitutive G3PD promoter and MER1 open reading frame fragment from pGHAMER1 into pRS426 and subsequently performing site-directed mutagenesis to alter the signature KH element GXXG (Siomi et al. 1993) from GAKG to GAQE. Plasmid pGMER1ΔC lacks the C-most terminal peptide (22 residues) adjacent to the KH domain and was constructed by PCR amplifying the gene with primers that amplify all but the last 22 codons of MER1. The export reporter plasmid with a nonfunctional MER1 enhancer element was constructed by oligonucleotide mutagenesis of pRS316CF7b and alters the ATACCCTT enhancer element to CATGGCTT.
The MER2 export reporter was constructed by oligonucleotide mutagenesis of the MER2 splicing reporter and removes an intronic stop codon. Using the oligonucleotide 5′-CATTTACTAACAACTGTAGTACAGgGAAACGTGAAAACCTTAATAAAGG-3′, an in-frame stop codon (at the 3′ splice site sequence) was altered from TAG to CAG, and an additional G nucleotide was inserted in exon 2 (lowercase g) to make the pre-mRNA reading frame open and spliced mRNA out of frame for CUP1 translation.
The MER3 export reporter plasmid was created by mutagenesis of the MER3 splicing reporter using oligonucleotides 5′-CCAAATAGTAGTAACGAAGCTT**CAACACCCTTATCAGTTTACACC-3′, where ** represents the deletion of AG, and 5′-GGTTTTTCTGGAC#AGAATTTCAGAGGACTTACAGAaTATTGACTTTAACG-3′, where # represents the deletion of a T. Additionally, the 3′ splice site (an in-frame stop) was altered from TAG to CAG, and an A (lowercase) was inserted into exon 2 to make the unspliced reading frame open and spliced mRNA in an incorrect frame for production of Cup1p.
LacZ export and splicing reporters were produced by amplifying the LacZ gene from a plasmid by PCR with Vent DNA polymerase and primers containing KpnI sites at the ends. After digestion with KpnI, the LacZ PCR product was ligated into the AMA1-CUP1 reporters in which the CUP1 fragment had been removed by KpnI digestion.
RNA, splicing assays, and export assays
Five-milliliter overnight cultures were centrifuged, and the pellets were washed once with 1 mL of H2O and resuspended in 150 μL of LET (25 mM Tris at pH 8.0, 100 mM LiCl, 20 mM EDTA) and 150 μL of phenol equilibrated with LET. Glass beads (Sigma) were added and vortexed vigorously for 5 min. After addition of 250 μL of H2O and 250 μL of phenol/CHCl3 (1:1), tubes were again vortexed vigorously. Following centrifugation, the aqueous phase was transferred to a new tube containing 450 μL of phenol heated to 65°C and repeatedly vortexed and incubated at 65°C for five 1-min intervals. The aqueous phase was re-extracted with 450 μL of phenol/CHCl3 and 250 μL of H2O. The aqueous phase was extracted a final time with 450 μL of CHCl3 and ETOH precipitated.
Primer extension analysis was described before (Spingola et al. 2004) and performed on no fewer than two independent transformants. Primer extension products representing spliced and unspliced RNAs were quantified by PhosphorImaging. The formula S/(S + U) × 100, where S is spliced product and U is unspliced pre-mRNA, was used to calculate splicing efficiency. Primers were designed to anneal to the second exon.
Splicing and mRNA export were also assessed by growth of yeast containing CUP1 fusion plasmids and various MER1 or control vectors by streaking transformants on selective media containing 150 μM cupric sulfate and incubating at 30°C for 3–5 d. Alternatively, 4 μL of cultures grown in selective media to an O.D. of 1.0 at 600 nm was spotted on plates containing 50–800 μM cupric sulfate.
β-Galactosidase assays for the LacZ reporters were performed in duplicate on at least two independent transformants. Cells were grown in selective media to an O.D. of 1.0 at 600 nm prior to assaying and harvested prior to reaching stationary phase. One milliliter of cells was centrifuged, and the pellets were resuspended in 800 μL of Z buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCL, 1 mM MgSO4, 50 mM β-mercaptoethanol). Fifteen microliters (15 μL) of 0.1% SDS and 30 μL of CHCl3 were added to each sample, which was then vortexed vigorously for 3 min. Two hundred microliters of ONPG (o-nitrophenyl-β-D-galactopyranoside) was then added to each sample, and the samples were quickly mixed and incubated at 37°C for 5 min. The reactions were stopped by adding 500 μL of 1.0 M Na2CO3 and centrifuged briefly to pellet the cell debris and separate the chloroform from the aqueous supernatant. The O.D. of the supernatant was measured at 420 nm.
RNA polymerase II inactivation and RNA stability assays
Transcription arrest assays were performed as in Parker et al. (1991) following incubation at the restrictive temperature with the exceptions that RNA was isolated as above and analyzed by primer extension to more clearly distinguish between spliced and unspliced forms. Decay rate constants (k) were calculated by curve fitting using an exponential function, and half-life values were calculated by ln 2/k.
ACKNOWLEDGMENTS
We thank Audrey Atkin for providing the rpb1-1 strains and Leodis “Champ” Gupton for constructing GAQE mutant MER1 plasmid. This work was supported by Mission Enhancement funds from the University of Missouri–St. Louis.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2276806.
REFERENCES
- Bousquet-Antonelli C., Presutti C., Tollervey D. Identification of a regulated pathway for nuclear pre-mRNA turnover. Cell. 2000;102:765–775. doi: 10.1016/s0092-8674(00)00065-9. [DOI] [PubMed] [Google Scholar]
- Brow D.A. Allosteric cascade of spliceosome activation. Annu. Rev. Genet. 2002;36:333–360. doi: 10.1146/annurev.genet.36.043002.091635. [DOI] [PubMed] [Google Scholar]
- Butcher S.E., Brow D.A. Towards understanding the catalytic core structure of the spliceosome. Biochem. Soc. Trans. 2005;33:447–449. doi: 10.1042/BST0330447. [DOI] [PubMed] [Google Scholar]
- Casolari J.M., Brown C.R., Drubin D.A., Rando O.J., Silver P.A. Developmentally induced changes in transcriptional program alter spatial organization across chromosomes. Genes & Dev. 2005;19:1188–1198. doi: 10.1101/gad.1307205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S.C., Abelson J. Spliceosome assembly in yeast. Genes & Dev. 1987;1:1014–1027. doi: 10.1101/gad.1.9.1014. [DOI] [PubMed] [Google Scholar]
- Conti E., Izaurralde E. Nonsense-mediated mRNA decay: Molecular insights and mechanistic variations across species. Curr. Opin. Cell Biol. 2005;17:316–325. doi: 10.1016/j.ceb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Das B., Butler J.S., Sherman F. Degradation of normal mRNA in the nucleus of Saccharomyces cerevisiae . Mol. Cell. Biol. 2003;23:5502–5515. doi: 10.1128/MCB.23.16.5502-5515.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis C.A., Grate L., Spingola M., Ares M., Jr. Test of intron predictions reveals novel splice sites, alternatively spliced mRNAs and new introns in meiotically regulated genes of yeast. Nucleic Acids Res. 2000;28:1700–1706. doi: 10.1093/nar/28.8.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziembowski A., Ventura A.P., Rutz B., Caspary F., Faux C., Halgand F., Laprevote O., Seraphin B. Proteomic analysis identifies a new complex required for nuclear pre-mRNA retention and splicing. EMBO J. 2004;23:4847–4856. doi: 10.1038/sj.emboj.7600482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engebrecht J.A., Voelkel-Meiman K., Roeder G.S. Meiosis-specific RNA splicing in yeast. Cell. 1991;66:1257–1268. doi: 10.1016/0092-8674(91)90047-3. [DOI] [PubMed] [Google Scholar]
- Galy V., Gadal O., Fromont-Racine M., Romano A., Jacquier A., Nehrbass U. Nuclear retention of unspliced mRNAs in yeast is mediated by perinuclear Mlp1. Cell. 2004;116:63–73. doi: 10.1016/s0092-8674(03)01026-2. [DOI] [PubMed] [Google Scholar]
- Gornemann J., Kotovic K.M., Hujer K., Neugebauer K.M. Cotranscriptional spliceosome assembly occurs in a stepwise fashion and requires the cap binding complex. Mol. Cell. 2005;19:53–63. doi: 10.1016/j.molcel.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Hagan K.W., Ruiz-Echevarria M.J., Quan Y., Peltz S.W. Characterization of cis-acting sequences and decay intermediates involved in nonsense-mediated mRNA turnover. Mol. Cell. Biol. 1995;15:809–823. doi: 10.1128/mcb.15.2.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausner T.P., Giglio L.M., Weiner A.M. Evidence for base-pairing between mammalian U2 and U6 small nuclear ribonucleoprotein particles. Genes & Dev. 1990;4:2146–2156. doi: 10.1101/gad.4.12a.2146. [DOI] [PubMed] [Google Scholar]
- He F., Peltz S.W., Donahue J.L., Rosbash M., Jacobson A. Stabilization and ribosome association of unspliced pre-mRNAs in a yeast upf1-mutant. Proc. Natl. Acad. Sci. 1993;90:7034–7038. doi: 10.1073/pnas.90.15.7034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F., Li X., Spatrick P., Casillo R., Dong S., Jacobson A. Genome-wide analysis of mRNAs regulated by the nonsense-mediated and 5′ to 3′ mRNA decay pathways in yeast. Mol. Cell. 2003;12:1439–1452. doi: 10.1016/s1097-2765(03)00446-5. [DOI] [PubMed] [Google Scholar]
- Hilleren P.J., Parker R. Cytoplasmic degradation of splice-defective pre-mRNAs and intermediates. Mol. Cell. 2003;12:1453–1465. doi: 10.1016/s1097-2765(03)00488-x. [DOI] [PubMed] [Google Scholar]
- Jurica M.S., Moore M.J. Pre-mRNA splicing: Awash in a sea of proteins. Mol. Cell. 2003;12:5–14. doi: 10.1016/s1097-2765(03)00270-3. [DOI] [PubMed] [Google Scholar]
- Kandels-Lewis S., Seraphin B. Involvement of U6 snRNA in 5′ splice site selection. Science. 1993;262:2035–2039. doi: 10.1126/science.8266100. [DOI] [PubMed] [Google Scholar]
- Kebaara B., Nazarenus T., Taylor R., Atkin A.L. Genetic background affects relative nonsense mRNA accumulation in wild-type and upf mutant yeast strains. Curr. Genet. 2003;43:171–177. doi: 10.1007/s00294-003-0386-3. [DOI] [PubMed] [Google Scholar]
- Kunkel T.A., Bebenek K., McClary J. Efficient site-directed mutagenesis using uracil-containing DNA. Methods Enzymol. 1991;204:125–139. doi: 10.1016/0076-6879(91)04008-c. [DOI] [PubMed] [Google Scholar]
- Lacadie S.A., Rosbash M. Cotranscriptional spliceosome assembly dynamics and the role of U1 snRNA: 5′ss base pairing in yeast. Mol. Cell. 2005;19:65–75. doi: 10.1016/j.molcel.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Legrain P., Rosbash M. Some cis- and trans-acting mutants for splicing target pre-mRNA to the cytoplasm. Cell. 1989;57:573–583. doi: 10.1016/0092-8674(89)90127-x. [DOI] [PubMed] [Google Scholar]
- Lewis J.D., Gorlich D., Mattaj I.W. A yeast cap binding protein complex (yCBC) acts at an early step in pre-mRNA splicing. Nucleic Acids Res. 1996a;24:3332–3336. doi: 10.1093/nar/24.17.3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J.D., Izaurralde E., Jarmolowski A., McGuigan C., Mattaj I.W. A nuclear cap-binding complex facilitates association of U1 snRNP with the cap-proximal 5′ splice site. Genes & Dev. 1996b;10:1683–1698. doi: 10.1101/gad.10.13.1683. [DOI] [PubMed] [Google Scholar]
- Lewis H.A., Musunuru K., Jensen K.B., Edo C., Chen H., Darnell R.B., Burley S.K. Sequence-specific RNA binding by a Nova KH domain: Implications for paraneoplastic disease and the fragile X syndrome. Cell. 2000;100:323–332. doi: 10.1016/s0092-8674(00)80668-6. [DOI] [PubMed] [Google Scholar]
- Longtine M.S., McKenzie A., III, Demarini D.J., Shah N.G., Wach A., Brachat A., Philippsen P., Pringle J.R. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae . Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Maquat L.E. Nonsense-mediated mRNA decay: Splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell Biol. 2004;5:89–99. doi: 10.1038/nrm1310. [DOI] [PubMed] [Google Scholar]
- Nakagawa T., Ogawa H. The Saccharomyces cerevisiae MER3 gene, encoding a novel helicase-like protein, is required for crossover control in meiosis. EMBO J. 1999;18:5714–5723. doi: 10.1093/emboj/18.20.5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandabalan K., Roeder G.S. Binding of a cell-type-specific RNA splicing factor to its target regulatory sequence. Mol. Cell. Biol. 1995;15:1953–1960. doi: 10.1128/mcb.15.4.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandabalan K., Price L., Roeder G.S. Mutations in U1 snRNA bypass the requirement for a cell type-specific RNA splicing factor. Cell. 1993;73:407–415. doi: 10.1016/0092-8674(93)90239-m. [DOI] [PubMed] [Google Scholar]
- Parker R., Siliciano P.G., Guthrie C. Recognition of the TACTAAC box during mRNA splicing in yeast involves base pairing to the U2-like snRNA. Cell. 1987;49:229–239. doi: 10.1016/0092-8674(87)90564-2. [DOI] [PubMed] [Google Scholar]
- Parker R., Herrick D., Peltz S.W., Jacobson A. Measurement of mRNA decay rates in Saccharomyces cerevisiae . Methods Enzymol. 1991;194:415–423. doi: 10.1016/0076-6879(91)94032-8. [DOI] [PubMed] [Google Scholar]
- Puig O., Gottschalk A., Fabrizio P., Seraphin B. Interaction of the U1 snRNP with nonconserved intronic sequences affects 5′ splice site selection. Genes & Dev. 1999;13:569–580. doi: 10.1101/gad.13.5.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghunathan P.L., Guthrie C. RNA unwinding in U4/U6 snRNPs requires ATP hydrolysis and the DEIH-box splicing factor Brr2. Curr. Biol. 1998;8:847–855. doi: 10.1016/s0960-9822(07)00345-4. [DOI] [PubMed] [Google Scholar]
- Rain J.C., Legrain P. In vivo commitment to splicing in yeast involves the nucleotide upstream from the branch site conserved sequence and the Mud2 protein. EMBO J. 1997;16:1759–1771. doi: 10.1093/emboj/16.7.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutz B., Seraphin B. Transient interaction of BBP/ScSF1 and Mud2 with the splicing machinery affects the kinetics of spliceosome assembly. RNA. 1999;5:819–831. doi: 10.1017/s1355838299982286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutz B., Seraphin B. A dual role for BBP/ScSF1 in nuclear pre-mRNA retention and splicing. EMBO J. 2000;19:1873–1886. doi: 10.1093/emboj/19.8.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seraphin B., Rosbash M. Identification of functional U1 snRNA–pre-mRNA complexes committed to spliceosome assembly and splicing. Cell. 1989;59:349–358. doi: 10.1016/0092-8674(89)90296-1. [DOI] [PubMed] [Google Scholar]
- Seraphin B., Kretzner L., Rosbash M. A U1 snRNA:pre-mRNA base pairing interaction is required early in yeast spliceosome assembly but does not uniquely define the 5′ cleavage site. EMBO J. 1988;7:2533–2538. doi: 10.1002/j.1460-2075.1988.tb03101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siliciano P.G., Guthrie C. 5′ splice site selection in yeast: Genetic alterations in base-pairing with U1 reveal additional requirements. Genes & Dev. 1988;2:1258–1267. doi: 10.1101/gad.2.10.1258. [DOI] [PubMed] [Google Scholar]
- Siomi H., Matunis M.J., Michael W.M., Dreyfuss G. The pre-mRNA binding K protein contains a novel evolutionarily conserved motif. Nucleic Acids Res. 1993;21:1193–1198. doi: 10.1093/nar/21.5.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spingola M., Ares M., Jr. A yeast intronic splicing enhancer and Nam8p are required for Mer1p-activated splicing. Mol. Cell. 2000;6:329–338. doi: 10.1016/s1097-2765(00)00033-2. [DOI] [PubMed] [Google Scholar]
- Spingola M., Armisen J., Ares M., Jr. Mer1p is a modular splicing factor whose function depends on the conserved U2 snRNP protein Snu17p. Nucleic Acids Res. 2004;32:1242–1250. doi: 10.1093/nar/gkh281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley J.P., Guthrie C. Mechanical devices of the spliceosome: Motors, clocks, springs, and things. Cell. 1998;92:315–326. doi: 10.1016/s0092-8674(00)80925-3. [DOI] [PubMed] [Google Scholar]
- Staley J.P., Guthrie C. An RNA switch at the 5′ splice site requires ATP and the DEAD box protein Prp28p. Mol. Cell. 1999;3:55–64. doi: 10.1016/s1097-2765(00)80174-4. [DOI] [PubMed] [Google Scholar]
- Stevens S.W., Ryan D.E., Ge H.Y., Moore R.E., Young M.K., Lee T.D., Abelson J. Composition and functional characterization of the yeast spliceosomal penta-snRNP. Mol. Cell. 2002;9:31–44. doi: 10.1016/s1097-2765(02)00436-7. [DOI] [PubMed] [Google Scholar]
- Vilardell J., Warner J.R. Regulation of splicing at an intermediate step in the formation of the spliceosome. Genes & Dev. 1994;8:211–220. doi: 10.1101/gad.8.2.211. [DOI] [PubMed] [Google Scholar]
- Vilardell J., Chartrand P., Singer R.H., Warner J.R. The odyssey of a regulated transcript. RNA. 2000;6:1773–1780. doi: 10.1017/s135583820000145x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinciguerra P., Iglesias N., Camblong J., Zenklusen D., Stutz F. Perinuclear Mlp proteins downregulate gene expression in response to a defect in mRNA export. EMBO J. 2005;24:813–823. doi: 10.1038/sj.emboj.7600527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Rymond B.C. Rds3p is required for stable U2 snRNP recruitment to the splicing apparatus. Mol. Cell. Biol. 2003;23:7339–7349. doi: 10.1128/MCB.23.20.7339-7349.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Liu C.L., Storey J.D., Tibshirani R.J., Herschlag D., Brown P.O. Precision and functional specificity in mRNA decay. Proc. Natl. Acad. Sci. 2002;99:5860–5865. doi: 10.1073/pnas.092538799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., He J., Lynn B., Rymond B.C. Interactions of the yeast SF3b splicing factor. Mol. Cell. Biol. 2005;25:10745–10754. doi: 10.1128/MCB.25.24.10745-10754.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J., Manley J.L. Mammalian pre-mRNA branch site selection by U2 snRNP involves base pairing. Genes & Dev. 1989;3:1553–1561. doi: 10.1101/gad.3.10.1553. [DOI] [PubMed] [Google Scholar]
- Zhang D., Rosbash M. Identification of eight proteins that cross-link to pre-mRNA in the yeast commitment complex. Genes & Dev. 1999;13:581–592. doi: 10.1101/gad.13.5.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Ruiz-Echevarria M.J., Quan Y., Peltz S.W. Identification and characterization of a sequence motif involved in nonsense-mediated mRNA decay. Mol. Cell. Biol. 1995;15:2231–2244. doi: 10.1128/mcb.15.4.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]